
Senescence-Associated Secretory Phenotype of Cardiovascular System Cells and Inflammaging: Perspectives of Peptide Regulation

 ,
,
Abstract
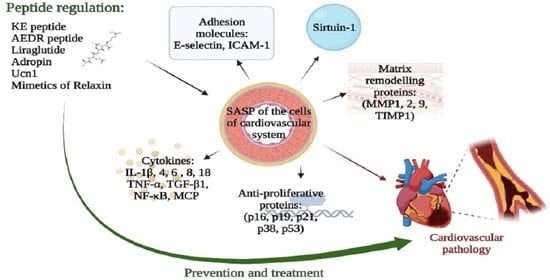
:
1. Introduction
2. SASP: Role in the Pathogenesis of Age-Related Cardiovascular Pathology
2.1. SASP of Endothelial Cells
2.2. SASP of Vascular Smooth Muscle Cells
2.3. SASP of Cardiomyocytes
3. Inflammaging: Role in the Pathogenesis of Age-Related Pathology of the Cardiovascular System
4. SASP and Inflammaging Molecules as Possible Targets for Pharmacotherapy of Cardiovascular Pathology
5. Peptides: Prospects for the Regulation of the Cardiovascular System Functions during the Formation of SASP and Inflammaging
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kuilman, T.; Michaloglou, C.; Mooi, W.J.; Peeper, D.S. The Essence of Senescence. Genes Dev. 2010, 24, 2463–2479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balistreri, C.R.; Candore, G.; Accardi, G.; Colonna-Romano, G.; Lio, D. NF-ΚB Pathway Activators as Potential Ageing Biomarkers: Targets for New Therapeutic Strategies. Immun. Ageing 2013, 10, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, D.J.; Wijshake, T.; Tchkonia, T.; LeBrasseur, N.K.; Childs, B.G.; van de Sluis, B.; Kirkland, J.L.; van Deursen, J.M. Clearance of P16Ink4a-Positive Senescent Cells Delays Ageing-Associated Disorders. Nature 2011, 479, 232–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khavinson, V.K.; Malinin, V.V. Gerontological Aspects of Genome Peptide Regulation; Karger AG: Basel, Switzerland, 2005; Volume 104. [Google Scholar]
- Khavinson, V.K. Peptides and Ageing. Neuroendocrinol. Lett. 2002, 23 (Suppl. 3), 11–144. [Google Scholar] [PubMed]
- Anisimov, V.N.; Khavinson, V.K. Peptide bioregulation of aging: Results and prospects. Biogerontology 2010, 11, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Khavinson, V.K.; Popovich, I.G.; Linkova, N.S.; Mironova, E.S.; Ilina, A.R. Peptide Regulation of Gene Expression: A Systematic Review. Molecules 2021, 26, 7053. [Google Scholar] [CrossRef]
- Ivanov, V.T.; Karelin, A.A.; Philippova, M.M.; Nazimov, I.V.; Pletnev, V.Z. Hemoglobin as a Source of Endogenous Bioactive Peptides: The Concept of Tissue-Specific Peptide Pool. Biopolymers 1997, 43, 171–188. [Google Scholar] [CrossRef]
- Ritschka, B.; Storer, M.; Mas, A.; Heinzmann, F.; Ortells, M.C.; Morton, J.P.; Sansom, O.J.; Zender, L.; Keyes, W.M. The Senescence-Associated Secretory Phenotype Induces Cellular Plasticity and Tissue Regeneration. Genes Dev. 2017, 31, 172–183. [Google Scholar] [CrossRef] [Green Version]
- Birch, J.; Gil, J. Senescence and the SASP: Many Therapeutic Avenues. Genes Dev. 2020, 34, 1565–1576. [Google Scholar] [CrossRef]
- Ohtani, N. The Roles and Mechanisms of Senescence-Associated Secretory Phenotype (SASP): Can It Be Controlled by Senolysis? Inflamm. Regen. 2022, 42, 11. [Google Scholar] [CrossRef]
- Watanabe, S.; Kawamoto, S.; Ohtani, N.; Hara, E. Impact of Senescence-associated Secretory Phenotype and Its Potential as a Therapeutic Target for Senescence-associated Diseases. Cancer Sci. 2017, 108, 563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Storer, M.; Mas, A.; Robert-Moreno, A.; Pecoraro, M.; Ortells, M.C.; Di Giacomo, V.; Yosef, R.; Pilpel, N.; Krizhanovsky, V.; Sharpe, J.; et al. Senescence Is a Developmental Mechanism That Contributes to Embryonic Growth and Patterning. Cell 2013, 155, 1119–1130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, S.G.; Jackson, J.G. SASP: Tumor Suppressor or Promoter? Yes! Trends Cancer 2016, 2, 676–687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terlecki-Zaniewicz, L.; Lämmermann, I.; Latreille, J.; Bobbili, M.R.; Pils, V.; Schosserer, M.; Weinmüllner, R.; Dellago, H.; Skalicky, S.; Pum, D.; et al. Small Extracellular Vesicles and Their MiRNA Cargo Are Anti-Apoptotic Members of the Senescence-Associated Secretory Phenotype. Aging. Albany N. Y. 2018, 10, 1103–1132. [Google Scholar] [CrossRef] [PubMed]
- Prašnikar, E.; Borišek, J.; Perdih, A. Senescent Cells as Promising Targets to Tackle Age-Related Diseases. Ageing Res. Rev. 2021, 66, 101251. [Google Scholar] [CrossRef]
- Salminen, A.; Kauppinen, A.; Kaarniranta, K. Emerging Role of NF-ΚB Signaling in the Induction of Senescence-Associated Secretory Phenotype (SASP). Cell Signal. 2012, 24, 835–845. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Wang, X.; Liu, T.; Zhu, X.; Pan, X. The Multifaceted Role of the SASP in Atherosclerosis: From Mechanisms to Therapeutic Opportunities. Cell Biosci. 2022, 12, 74. [Google Scholar] [CrossRef]
- Kopacz, A.; Kloska, D.; Targosz-Korecka, M.; Zapotoczny, B.; Cysewski, D.; Personnic, N.; Werner, E.; Hajduk, K.; Jozkowicz, A.; Grochot-Przeczek, A. Keap1 Governs Ageing-Induced Protein Aggregation in Endothelial Cells. Redox Biol. 2020, 34, 101572. [Google Scholar] [CrossRef]
- Jia, G.; Aroor, A.R.; Jia, C.; Sowers, J.R. Endothelial Cell Senescence in Aging-Related Vascular Dysfunction. Biochim. Biophys. Acta Mol. Basis. Dis. 2019, 1865, 1802–1809. [Google Scholar] [CrossRef]
- Sabbatinelli, J.; Prattichizzo, F.; Olivieri, F.; Procopio, A.D.; Rippo, M.R.; Giuliani, A. Where Metabolism Meets Senescence: Focus on Endothelial Cells. Front. Physiol. 2019, 10, 1523. [Google Scholar] [CrossRef]
- Sun, X.; Feinberg, M.W. Vascular Endothelial Senescence: Pathobiological Insights, Emerging Long Noncoding RNA Targets, Challenges and Therapeutic Opportunities. Front. Physiol. 2021, 12, 693067. [Google Scholar] [CrossRef] [PubMed]
- Hampel, B.; Fortschegger, K.; Ressler, S.; Chang, M.W.; Unterluggauer, H.; Breitwieser, A.; Sommergruber, W.; Fitzky, B.; Lepperdinger, G.; Jansen-Dürr, P.; et al. Increased Expression of Extracellular Proteins as a Hallmark of Human Endothelial Cell in Vitro Senescence. Exp. Gerontol. 2006, 41, 474–481. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.-R.; Shen, W.-L.; Yan, C.; Gao, P.-J. Downregulation of Dynamin-Related Protein 1 Contributes to Impaired Autophagic Flux and Angiogenic Function in Senescent Endothelial Cells. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1413–1422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, H.J.; Lee, Y.-R.; Kang, D.; Lee, H.C.; Seo, H.R.; Ryu, J.-K.; Kim, Y.-N.; Ko, Y.-G.; Park, H.J.; Lee, J.-S. Endothelial Cells under Therapy-Induced Senescence Secrete CXCL11, Which Increases Aggressiveness of Breast Cancer Cells. Cancer Lett. 2020, 490, 100–110. [Google Scholar] [CrossRef] [PubMed]
- Dominic, A.; Banerjee, P.; Hamilton, D.J.; Le, N.-T.; Abe, J.-I. Time-Dependent Replicative Senescence vs. Disturbed Flow-Induced Pre-Mature Aging in Atherosclerosis. Redox Biol. 2020, 37, 101614. [Google Scholar] [CrossRef] [PubMed]
- Barinda, A.J.; Ikeda, K.; Nugroho, D.B.; Wardhana, D.A.; Sasaki, N.; Honda, S.; Urata, R.; Matoba, S.; Hirata, K.-I.; Emoto, N. Endothelial Progeria Induces Adipose Tissue Senescence and Impairs Insulin Sensitivity through Senescence Associated Secretory Phenotype. Nat. Commun. 2020, 11, 481. [Google Scholar] [CrossRef] [Green Version]
- Khan, S.Y.; Awad, E.M.; Oszwald, A.; Mayr, M.; Yin, X.; Waltenberger, B.; Stuppner, H.; Lipovac, M.; Uhrin, P.; Breuss, J.M. Premature Senescence of Endothelial Cells upon Chronic Exposure to TNFα Can Be Prevented by N-Acetyl Cysteine and Plumericin. Sci. Rep. 2017, 7, 39501. [Google Scholar] [CrossRef] [Green Version]
- Venturini, W.; Olate-Briones, A.; Valenzuela, C.; Méndez, D.; Fuentes, E.; Cayo, A.; Mancilla, D.; Segovia, R.; Brown, N.E.; Moore-Carrasco, R. Platelet Activation Is Triggered by Factors Secreted by Senescent Endothelial HMEC-1 Cells In Vitro. Int. J. Mol. Sci. 2020, 21, 3287. [Google Scholar] [CrossRef]
- Ma, Y.; Chiao, Y.A.; Clark, R.; Flynn, E.R.; Yabluchanskiy, A.; Ghasemi, O.; Zouein, F.; Lindsey, M.L.; Jin, Y.-F. Deriving a Cardiac Ageing Signature to Reveal MMP-9-Dependent Inflammatory Signalling in Senescence. Cardiovasc. Res. 2015, 106, 421–431. [Google Scholar] [CrossRef]
- Spinetti, G.; Wang, M.; Monticone, R.; Zhang, J.; Zhao, D.; Lakatta, E.G. Rat Aortic MCP-1 and Its Receptor CCR2 Increase with Age and Alter Vascular Smooth Muscle Cell Function. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1397–1402. [Google Scholar] [CrossRef]
- Merz, A.A.; Cheng, S. Sex Differences in Cardiovascular Ageing. Heart 2016, 102, 825–831. [Google Scholar] [CrossRef] [PubMed]
- DuPont, J.J.; Kim, S.K.; Kenney, R.M.; Jaffe, I.Z. Sex Differences in the Time Course and Mechanisms of Vascular and Cardiac Aging in Mice: Role of the Smooth Muscle Cell Mineralocorticoid Receptor. Am. J. Physiol. Heart Circ. Physiol. 2021, 320, H169–H180. [Google Scholar] [CrossRef] [PubMed]
- Casella, G.; Munk, R.; Kim, K.M.; Piao, Y.; De, S.; Abdelmohsen, K.; Gorospe, M. Transcriptome Signature of Cellular Senescence. Nucleic Acids Res. 2019, 47, 7294–7305. [Google Scholar] [CrossRef] [Green Version]
- Lanigan, F.; Geraghty, J.G.; Bracken, A.P. Transcriptional Regulation of Cellular Senescence. Oncogene 2011, 30, 2901–2911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mijit, M.; Caracciolo, V.; Melillo, A.; Amicarelli, F.; Giordano, A. Role of P53 in the Regulation of Cellular Senescence. Biomolecules 2020, 10, 420. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Kim, C.-S.; Hoffman, T.A.; Naqvi, A.; Dericco, J.; Jung, S.-B.; Lin, Z.; Jain, M.K.; Irani, K. P53 Impairs Endothelial Function by Transcriptionally Repressing Kruppel-Like Factor 2. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 133–141. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.-J.; Kho, J.-H.; Kang, M.-R.; Um, S.-J. Active Regulator of SIRT1 Cooperates with SIRT1 and Facilitates Suppression of P53 Activity. Mol. Cell 2007, 28, 277–290. [Google Scholar] [CrossRef]
- Yokoyama, M.; Shimizu, I.; Nagasawa, A.; Yoshida, Y.; Katsuumi, G.; Wakasugi, T.; Hayashi, Y.; Ikegami, R.; Suda, M.; Ota, Y.; et al. P53 Plays a Crucial Role in Endothelial Dysfunction Associated with Hyperglycemia and Ischemia. J. Mol. Cell Cardiol. 2019, 129, 105–117. [Google Scholar] [CrossRef]
- Northcott, J.M.; Czubryt, M.P.; Wigle, J.T. Vascular Senescence and Ageing: A Role for the MEOX Proteins in Promoting Endothelial Dysfunction. Can. J. Physiol. Pharmacol. 2017, 95, 1067–1077. [Google Scholar] [CrossRef] [Green Version]
- Brühl, T.; Heeschen, C.; Aicher, A.; Jadidi, A.S.; Haendeler, J.; Hoffmann, J.; Schneider, M.D.; Zeiher, A.M.; Dimmeler, S.; Rössig, L. P21Cip1 Levels Differentially Regulate Turnover of Mature Endothelial Cells, Endothelial Progenitor Cells, and in Vivo Neovascularization. Circ. Res. 2004, 94, 686–692. [Google Scholar] [CrossRef]
- Wang, Y.; Boerma, M.; Zhou, D. Ionizing Radiation-Induced Endothelial Cell Senescence and Cardiovascular Diseases. Radiat Res. 2016, 186, 153–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosso, A.; Balsamo, A.; Gambino, R.; Dentelli, P.; Falcioni, R.; Cassader, M.; Pegoraro, L.; Pagano, G.; Brizzi, M.F. P53 Mediates the Accelerated Onset of Senescence of Endothelial Progenitor Cells in Diabetes. J. Biol. Chem. 2006, 281, 4339–4347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, G.H.-H.; Chan, E.; Kwok, C.T.-K.; Leung, G.P.-H.; Lee, S.M.-Y.; Seto, S.-W. The Role of P53 in the Alternation of Vascular Functions. Front. Pharmacol. 2022, 13, 981152. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Huang, X.; Halicka, D.; Brodsky, S.; Avram, A.; Eskander, J.; Bloomgarden, N.A.; Darzynkiewicz, Z.; Goligorsky, M.S. Contribution of P16INK4a and P21CIP1 Pathways to Induction of Premature Senescence of Human Endothelial Cells: Permissive Role of P53. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H1575–H1586. [Google Scholar] [CrossRef] [Green Version]
- Yin, H.; Pickering, J.G. Cellular Senescence and Vascular Disease: Novel Routes to Better Understanding and Therapy. Can. J. Cardiol. 2016, 32, 612–623. [Google Scholar] [CrossRef]
- Rubio-Ruiz, M.E.; Pérez-Torres, I.; Soto, M.E.; Pastelín, G.; Guarner-Lans, V. Aging in Blood Vessels. Medicinal Agents FOR Systemic Arterial Hypertension in the Elderly. Ageing. Res. Rev. 2014, 18, 132–147. [Google Scholar] [CrossRef]
- Stojanović, S.D.; Fiedler, J.; Bauersachs, J.; Thum, T.; Sedding, D.G. Senescence-Induced Inflammation: An Important Player and Key Therapeutic Target in Atherosclerosis. Eur. Heart J. 2020, 41, 2983–2996. [Google Scholar] [CrossRef] [Green Version]
- Gardner, S.E.; Humphry, M.; Bennett, M.R.; Clarke, M.C.H. Senescent Vascular Smooth Muscle Cells Drive Inflammation Through an Interleukin-1α-Dependent Senescence-Associated Secretory Phenotype. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1963–1974. [Google Scholar] [CrossRef] [Green Version]
- Morgan, M.J.; Liu, Z. Crosstalk of Reactive Oxygen Species and NF-ΚB Signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef] [Green Version]
- Monk, B.A.; George, S.J. The Effect of Ageing on Vascular Smooth Muscle Cell Behaviour—A Mini-Review. GER 2015, 61, 416–426. [Google Scholar] [CrossRef]
- Wang, M.; Khazan, B.; Lakatta, E.G. Central Arterial Aging and Angiotensin II Signaling. Curr. Hypertens. Rev. 2010, 6, 266–281. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Khalil, R.A. Chapter Eight-Matrix Metalloproteinases, Vascular Remodeling, and Vascular Disease. In Advances in Pharmacology; Khalil, R.A., Ed.; Vascular Pharmacology: Cytoskeleton and Extracellular Matrix; Academic Press: Cambridge, MA, USA, 2018; Volume 81, pp. 241–330. [Google Scholar]
- Jiang, L.; Zhang, J.; Monticone, R.E.; Telljohann, R.; Wu, J.; Wang, M.; Lakatta, E.G. Calpain-1 Regulation of Matrix Metalloproteinase 2 Activity in Vascular Smooth Muscle Cells Facilitates Age-Associated Aortic Wall Calcification and Fibrosis. Hypertension 2012, 60, 1192–1199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pohlers, D.; Brenmoehl, J.; Löffler, I.; Müller, C.K.; Leipner, C.; Schultze-Mosgau, S.; Stallmach, A.; Kinne, R.W.; Wolf, G. TGF-β and Fibrosis in Different Organs—Molecular Pathway Imprints. Biochim. Et Biophys. Acta (BBA)-Mol. Basis Dis. 2009, 1792, 746–756. [Google Scholar] [CrossRef] [Green Version]
- Weigert, C.; Brodbeck, K.; Klopfer, K.; Häring, H.U.; Schleicher, E.D. Angiotensin II Induces Human TGF-Beta 1 Promoter Activation: Similarity to Hyperglycaemia. Diabetologia 2002, 45, 890–898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, G.; Mccue, H.M.; Mastrangelo, L.; Millis, A.J.T. Endogenous TGF-β Activity Is Modified during Cellular Aging: Effects on Metalloproteinase and TIMP-1 Expression. Exp. Cell Res. 1996, 228, 271–276. [Google Scholar] [CrossRef]
- Fleenor, B.S.; Marshall, K.D.; Durrant, J.R.; Lesniewski, L.A.; Seals, D.R. Arterial Stiffening with Ageing Is Associated with Transforming Growth Factor-Β1-Related Changes in Adventitial Collagen: Reversal by Aerobic Exercise. J. Physiol. 2010, 588, 3971–3982. [Google Scholar] [CrossRef] [PubMed]
- Acosta, J.C.; Banito, A.; Wuestefeld, T.; Georgilis, A.; Janich, P.; Morton, J.P.; Athineos, D.; Kang, T.-W.; Lasitschka, F.; Andrulis, M.; et al. A Complex Secretory Program Orchestrated by the Inflammasome Controls Paracrine Senescence. Nat. Cell Biol. 2013, 15, 978–990. [Google Scholar] [CrossRef] [PubMed]
- Kunieda, T.; Minamino, T.; Nishi, J.-I.; Tateno, K.; Oyama, T.; Katsuno, T.; Miyauchi, H.; Orimo, M.; Okada, S.; Takamura, M.; et al. Angiotensin II Induces Premature Senescence of Vascular Smooth Muscle Cells and Accelerates the Development of Atherosclerosis via a P21-Dependent Pathway. Circulation 2006, 114, 953–960. [Google Scholar] [CrossRef]
- Grootaert, M.O.J.; Moulis, M.; Roth, L.; Martinet, W.; Vindis, C.; Bennett, M.R.; De Meyer, G.R.Y. Vascular Smooth Muscle Cell Death, Autophagy and Senescence in Atherosclerosis. Cardiovasc. Res. 2018, 114, 622–634. [Google Scholar] [CrossRef] [Green Version]
- Burton, D.G.A.; Matsubara, H.; Ikeda, K. Pathophysiology of Vascular Calcification: Pivotal Role of Cellular Senescence in Vascular Smooth Muscle Cells. Exp. Gerontol. 2010, 45, 819–824. [Google Scholar] [CrossRef]
- Baeuerle, P.A.; Baichwal, V.R. NF-Kappa B as a Frequent Target for Immunosuppressive and Anti-Inflammatory Molecules. Adv. Immunol. 1997, 65, 111–137. [Google Scholar] [PubMed]
- Davis, M.E.; Grumbach, I.M.; Fukai, T.; Cutchins, A.; Harrison, D.G. Shear Stress Regulates Endothelial Nitric-Oxide Synthase Promoter Activity through Nuclear Factor KappaB Binding. J. Biol. Chem. 2004, 279, 163–168. [Google Scholar] [CrossRef] [Green Version]
- Helenius, M.; Hänninen, M.; Lehtinen, S.K.; Salminen, A. Aging-Induced up-Regulation of Nuclear Binding Activities of Oxidative Stress Responsive NF-KB Transcription Factor in Mouse Cardiac Muscle. J. Mol. Cell Cardiol. 1996, 28, 487–498. [Google Scholar] [CrossRef] [PubMed]
- Sung, J.Y.; Kim, S.G.; Kim, J.-R.; Choi, H.C. Prednisolone Suppresses Adriamycin-Induced Vascular Smooth Muscle Cell Senescence and Inflammatory Response via the SIRT1-AMPK Signaling Pathway. PLoS ONE 2020, 15, e0239976. [Google Scholar] [CrossRef] [PubMed]
- Herranz, N.; Gil, J. Mechanisms and Functions of Cellular Senescence. J. Clin. Investig. 2018, 128, 1238–1246. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Chen, Z.; Shen, W.; Huang, G.; Sedivy, J.M.; Wang, H.; Ju, Z. Inflammation, Epigenetics, and Metabolism Converge to Cell Senescence and Ageing: The Regulation and Intervention. Signal. Transduct. Target. Ther. 2021, 6, 245. [Google Scholar] [CrossRef]
- Boyer, M.J.; Kimura, Y.; Akiyama, T.; Baggett, A.Y.; Preston, K.J.; Scalia, R.; Eguchi, S.; Rizzo, V. Endothelial Cell-Derived Extracellular Vesicles Alter Vascular Smooth Muscle Cell Phenotype through High-Mobility Group Box Proteins. J. Extracell. Vesicles 2020, 9, 1781427. [Google Scholar] [CrossRef]
- Badi, I.; Burba, I.; Ruggeri, C.; Zeni, F.; Bertolotti, M.; Scopece, A.; Pompilio, G.; Raucci, A. MicroRNA-34a Induces Vascular Smooth Muscle Cells Senescence by SIRT1 Downregulation and Promotes the Expression of Age-Associated Pro-Inflammatory Secretory Factors. J. Gerontol. A Biol. Sci. Med. Sci. 2015, 70, 1304–1311. [Google Scholar] [CrossRef]
- Gonzalo, S.; Kreienkamp, R.; Askjaer, P. Hutchinson-Gilford Progeria Syndrome: A Premature Aging Disease Caused by LMNA Gene Mutations. Ageing. Res. Rev. 2017, 33, 18–29. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.M.; Nobumori, C.; Tu, Y.; Choi, C.; Yang, S.H.; Vickers, T.A.; Rigo, F.; Bennett, C.F.; Young, S.G.; Fong, L.G. Modulation of LMNA Splicing as a Strategy to Treat Prelamin A Diseases. J. Clin. Investig. 2016, 126, 1592–1602. [Google Scholar] [CrossRef]
- Ragnauth, C.D.; Warren, D.T.; Liu, Y.; McNair, R.; Tajsic, T.; Figg, N.; Shroff, R.; Skepper, J.; Shanahan, C.M. Prelamin A Acts to Accelerate Smooth Muscle Cell Senescence and Is a Novel Biomarker of Human Vascular Aging. Circulation 2010, 121, 2200–2210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitry, M.A.; Laurent, D.; Keith, B.L.; Sira, E.; Eisenberg, C.A.; Eisenberg, L.M.; Joshi, S.; Gupte, S.; Edwards, J.G. Accelerated Cardiomyocyte Senescence Contributes to Late-Onset Doxorubicin-Induced Cardiotoxicity. Am. J. Physiol. Cell Physiol. 2020, 318, C380–C391. [Google Scholar] [CrossRef] [PubMed]
- Meijles, D.N.; Sahoo, S.; Al Ghouleh, I.; Amaral, J.H.; Bienes-Martinez, R.; Knupp, H.E.; Attaran, S.; Sembrat, J.C.; Nouraie, S.M.; Rojas, M.M.; et al. The Matricellular Protein TSP1 Promotes Human and Mouse Endothelial Cell Senescence through CD47 and Nox1. Sci. Signal. 2017, 10, eaaj1784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matthews, C.; Gorenne, I.; Scott, S.; Figg, N.; Kirkpatrick, P.; Ritchie, A.; Goddard, M.; Bennett, M. Vascular Smooth Muscle Cells Undergo Telomere-Based Senescence in Human Atherosclerosis: Effects of Telomerase and Oxidative Stress. Circ. Res. 2006, 99, 156–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, X.; Li, P.-H.; Chen, H.-Z. Cardiomyocyte Senescence and Cellular Communications Within Myocardial Microenvironments. Front. Endocrinol. 2020, 11, 280. [Google Scholar] [CrossRef]
- Maejima, Y.; Adachi, S.; Ito, H.; Hirao, K.; Isobe, M. Induction of Premature Senescence in Cardiomyocytes by Doxorubicin as a Novel Mechanism of Myocardial Damage. Aging. Cell 2008, 7, 125–136. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, C.; Liu, C.; Wang, Y.; Zhang, W.; Xing, Y. Trimetazidine and L-carnitine Prevent Heart Aging and Cardiac Metabolic Impairment in Rats via Regulating Cardiac Metabolic Substrates. Exp. Gerontol. 2019, 119, 120–127. [Google Scholar] [CrossRef]
- Bogazzi, F.; Raggi, F.; Ultimieri, F.; Russo, D.; D’Alessio, A.; Manariti, A.; Brogioni, S.; Manetti, L.; Martino, E. Regulation of Cardiac Fatty Acids Metabolism in Transgenic Mice Overexpressing Bovine GH. J. Endocrinol. 2009, 201, 419–427. [Google Scholar] [CrossRef] [Green Version]
- He, L.; Kim, T.; Long, Q.; Liu, J.; Wang, P.; Zhou, Y.; Ding, Y.; Prasain, J.; Wood, P.A.; Yang, Q. Carnitine Palmitoyltransferase-1b (CPT1b) Deficiency Aggravates Pressure-Overload-Induced Cardiac Hypertrophy Due to Lipotoxicity. Circulation 2012, 126, 1705–1716. [Google Scholar] [CrossRef] [Green Version]
- Franceschi, C.; Bonafè, M.; Valensin, S.; Olivieri, F.; De Luca, M.; Ottaviani, E.; De Benedictis, G. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann. N. Y. Acad. Sci. 2000, 908, 244–254. [Google Scholar] [CrossRef]
- Schroder, K.; Tschopp, J. The Inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennett, M.R.; Sinha, S.; Owens, G.K. Vascular Smooth Muscle Cells in Atherosclerosis. Circ. Res. 2016, 118, 692–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cafueri, G.; Parodi, F.; Pistorio, A.; Bertolotto, M.; Ventura, F.; Gambini, C.; Bianco, P.; Dallegri, F.; Pistoia, V.; Pezzolo, A.; et al. Endothelial and Smooth Muscle Cells from Abdominal Aortic Aneurysm Have Increased Oxidative Stress and Telomere Attrition. PLoS ONE 2012, 7, e35312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komaravolu, R.K.; Waltmann, M.D.; Konaniah, E.; Jaeschke, A.; Hui, D.Y. ApoER2 (Apolipoprotein E Receptor-2) Deficiency Accelerates Smooth Muscle Cell Senescence via Cytokinesis Impairment and Promotes Fibrotic Neointima After Vascular Injury. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 2132–2144. [Google Scholar] [CrossRef]
- Ferrucci, L.; Fabbri, E. Inflammageing: Chronic Inflammation in Ageing, Cardiovascular Disease, and Frailty. Nat. Rev. Cardiol. 2018, 15, 505–522. [Google Scholar] [CrossRef]
- Childs, B.G.; Baker, D.J.; Wijshake, T.; Conover, C.A.; Campisi, J.; van Deursen, J.M. Senescent Intimal Foam Cells Are Deleterious at All Stages of Atherosclerosis. Science 2016, 354, 472–477. [Google Scholar] [CrossRef] [Green Version]
- Stojanović, S.D.; Fuchs, M.; Kunz, M.; Xiao, K.; Just, A.; Pich, A.; Bauersachs, J.; Fiedler, J.; Sedding, D.; Thum, T. Inflammatory Drivers of Cardiovascular Disease: Molecular Characterization of Senescent Coronary Vascular Smooth Muscle Cells. Front. Physiol. 2020, 11, 520. [Google Scholar] [CrossRef]
- Vazquez-Padron, R.I.; Lasko, D.; Li, S.; Louis, L.; Pestana, I.A.; Pang, M.; Liotta, C.; Fornoni, A.; Aitouche, A.; Pham, S.M. Aging Exacerbates Neointimal Formation, and Increases Proliferation and Reduces Susceptibility to Apoptosis of Vascular Smooth Muscle Cells in Mice. J. Vasc. Surg. 2004, 40, 1199–1207. [Google Scholar] [CrossRef] [Green Version]
- Gorenne, I.; Kavurma, M.; Scott, S.; Bennett, M. Vascular Smooth Muscle Cell Senescence in Atherosclerosis. Cardiovasc. Res. 2006, 72, 9–17. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Uryga, A.K.; Reinhold, J.; Figg, N.; Baker, L.; Finigan, A.; Gray, K.; Kumar, S.; Clarke, M.; Bennett, M. Vascular Smooth Muscle Cell Senescence Promotes Atherosclerosis and Features of Plaque Vulnerability. Circulation 2015, 132, 1909–1919. [Google Scholar] [CrossRef]
- Wang, M.; Kim, S.H.; Monticone, R.E.; Lakatta, E.G. Matrix Metalloproteinases Promote Arterial Remodeling in Aging, Hypertension, and Atherosclerosis. Hypertension 2015, 65, 698–703. [Google Scholar] [CrossRef]
- Bai, B.; Yang, Y.; Wang, Q.; Li, M.; Tian, C.; Liu, Y.; Aung, L.H.H.; Li, P.; Yu, T.; Chu, X. NLRP3 Inflammasome in Endothelial Dysfunction. Cell Death Dis. 2020, 11, 1–18. [Google Scholar] [CrossRef]
- Tong, Y.; Wang, Z.; Cai, L.; Lin, L.; Liu, J.; Cheng, J. NLRP3 Inflammasome and Its Central Role in the Cardiovascular Diseases. Oxid. Med. Cell. Longev. 2020, 2020, 4293206. [Google Scholar] [CrossRef] [Green Version]
- Song, P.; Zhao, Q.; Zou, M.-H. Targeting Senescent Cells to Attenuate Cardiovascular Disease Progression. Ageing. Res. Rev. 2020, 60, 101072. [Google Scholar] [CrossRef]
- Xu, M.; Pirtskhalava, T.; Farr, J.N.; Weigand, B.M.; Palmer, A.K.; Weivoda, M.M.; Inman, C.L.; Ogrodnik, M.B.; Hachfeld, C.M.; Fraser, D.G.; et al. Senolytics Improve Physical Function and Increase Lifespan in Old Age. Nat. Med. 2018, 24, 1246–1256. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Tchkonia, T.; Fuhrmann-Stroissnigg, H.; Dai, H.M.; Ling, Y.Y.; Stout, M.B.; Pirtskhalava, T.; Giorgadze, N.; Johnson, K.O.; Giles, C.B.; et al. Identification of a Novel Senolytic Agent, Navitoclax, Targeting the Bcl-2 Family of Anti-Apoptotic Factors. Aging. Cell 2016, 15, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Doornebal, E.J.; Pirtskhalava, T.; Giorgadze, N.; Wentworth, M.; Fuhrmann-Stroissnigg, H.; Niedernhofer, L.J.; Robbins, P.D.; Tchkonia, T.; Kirkland, J.L. New Agents That Target Senescent Cells: The Flavone, Fisetin, and the BCL-XL Inhibitors, A1331852 and A1155463. Aging. Albany N. Y. 2017, 9, 955–963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Triana-Martínez, F.; Picallos-Rabina, P.; Da Silva-Álvarez, S.; Pietrocola, F.; Llanos, S.; Rodilla, V.; Soprano, E.; Pedrosa, P.; Ferreirós, A.; Barradas, M.; et al. Identification and Characterization of Cardiac Glycosides as Senolytic Compounds. Nat. Commun. 2019, 10, 4731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sweeney, M.; Cook, S.A.; Gil, J. Therapeutic Opportunities for Senolysis in Cardiovascular Disease. FEBS J. 2022. Online ahead of print. [Google Scholar] [CrossRef]
- Dookun, E.; Walaszczyk, A.; Redgrave, R.; Palmowski, P.; Tual-Chalot, S.; Suwana, A.; Chapman, J.; Jirkovsky, E.; Donastorg Sosa, L.; Gill, E.; et al. Clearance of Senescent Cells during Cardiac Ischemia-Reperfusion Injury Improves Recovery. Aging. Cell 2020, 19, e13249. [Google Scholar] [CrossRef]
- Walaszczyk, A.; Dookun, E.; Redgrave, R.; Tual-Chalot, S.; Victorelli, S.; Spyridopoulos, I.; Owens, A.; Arthur, H.M.; Passos, J.F.; Richardson, G.D. Pharmacological Clearance of Senescent Cells Improves Survival and Recovery in Aged Mice Following Acute Myocardial Infarction. Aging Cell 2019, 18, e12945. [Google Scholar] [CrossRef]
- Fuhrmann-Stroissnigg, H.; Ling, Y.Y.; Zhao, J.; McGowan, S.J.; Zhu, Y.; Brooks, R.W.; Grassi, D.; Gregg, S.Q.; Stripay, J.L.; Dorronsoro, A.; et al. Identification of HSP90 Inhibitors as a Novel Class of Senolytics. Nat. Commun. 2017, 8, 422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ungvari, Z.; Tarantini, S.; Sorond, F.; Merkely, B.; Csiszar, A. Mechanisms of Vascular Aging, A Geroscience Perspective: JACC Focus Seminar. J. Am. Coll. Cardiol. 2020, 75, 931–941. [Google Scholar] [CrossRef] [PubMed]
- Yamada, S.; Tatsumoto, N.; Tokumoto, M.; Noguchi, H.; Ooboshi, H.; Kitazono, T.; Tsuruya, K. Phosphate Binders Prevent Phosphate-Induced Cellular Senescence of Vascular Smooth Muscle Cells and Vascular Calcification in a Modified, Adenine-Based Uremic Rat Model. Calcif. Tissue Int. 2015, 96, 347–358. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.I.; Choi, S.; Roh, W.S.; Lee, J.H.; Kim, T.-G. Cellular Senescence and Inflammaging in the Skin Microenvironment. Int. J. Mol. Sci. 2021, 22, 3849. [Google Scholar] [CrossRef] [PubMed]
- Sung, J.Y.; Lee, K.Y.; Kim, J.-R.; Choi, H.C. Interaction between MTOR Pathway Inhibition and Autophagy Induction Attenuates Adriamycin-Induced Vascular Smooth Muscle Cell Senescence through Decreased Expressions of P53/P21/P16. Exp. Gerontol. 2018, 109, 51–58. [Google Scholar] [CrossRef]
- Hongo, A.; Okumura, N.; Nakahara, M.; Kay, E.P.; Koizumi, N. The Effect of a P38 Mitogen-Activated Protein Kinase Inhibitor on Cellular Senescence of Cultivated Human Corneal Endothelial Cells. Investig. Ophthalmol. Vis. Sci. 2017, 58, 3325–3334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennaceur, K.; Atwill, M.; Al Zhrany, N.; Hoffmann, J.; Keavney, B.; Breault, D.; Richardson, G.; von Zglinicki, T.; Saretzki, G.; Spyridopoulos, I. Atorvastatin Induces T Cell Proliferation by a Telomerase Reverse Transcriptase (TERT) Mediated Mechanism. Atherosclerosis 2014, 236, 312–320. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Uppal, H.; Demaria, M.; Desprez, P.-Y.; Campisi, J.; Kapahi, P. Simvastatin Suppresses Breast Cancer Cell Proliferation Induced by Senescent Cells. Sci. Rep. 2015, 5, 17895. [Google Scholar] [CrossRef] [Green Version]
- Efimova, E.V.; Ricco, N.; Labay, E.; Mauceri, H.J.; Flor, A.C.; Ramamurthy, A.; Sutton, H.G.; Weichselbaum, R.R.; Kron, S.J. HMG-CoA Reductase Inhibition Delays DNA Repair and Promotes Senescence After Tumor Irradiation. Mol. Cancer Ther. 2018, 17, 407–418. [Google Scholar] [CrossRef]
- Moon, S.-H.; Huang, C.-H.; Houlihan, S.L.; Regunath, K.; Freed-Pastor, W.A.; Morris, J.P.; Tschaharganeh, D.F.; Kastenhuber, E.R.; Barsotti, A.M.; Culp-Hill, R.; et al. P53 Represses the Mevalonate Pathway to Mediate Tumor Suppression. Cell 2019, 176, 564–580.e19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiley, C.D.; Schaum, N.; Alimirah, F.; Lopez-Dominguez, J.A.; Orjalo, A.V.; Scott, G.; Desprez, P.-Y.; Benz, C.; Davalos, A.R.; Campisi, J. Small-Molecule MDM2 Antagonists Attenuate the Senescence-Associated Secretory Phenotype. Sci. Rep. 2018, 8, 2410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, X.; Chen, H.; Gong, H.; Tang, X.; Huang, N.; Xu, W.; Tai, H.; Zhang, G.; Zhao, T.; Gong, C.; et al. Autolysosomal Degradation of Cytosolic Chromatin Fragments Antagonizes Oxidative Stress-Induced Senescence. J. Biol. Chem. 2020, 295, 4451–4463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bharath, L.P.; Agrawal, M.; McCambridge, G.; Nicholas, D.A.; Hasturk, H.; Liu, J.; Jiang, K.; Liu, R.; Guo, Z.; Deeney, J.; et al. Metformin Enhances Autophagy and Normalizes Mitochondrial Function to Alleviate Aging-Associated Inflammation. Cell Metab. 2020, 32, 44–55.e6. [Google Scholar] [CrossRef]
- Khavinson, V.; Linkova, N.; Kozhevnikova, E.; Dyatlova, A.; Petukhov, M. Transport of Biologically Active Ultrashort Peptides Using POT and LAT Carriers. Int. J. Mol. Sci. 2022, 23, 7733. [Google Scholar] [CrossRef]
- Fedoreyeva, L.I.; Smirnova, T.A.; Kolomijtseva, G.Y.; Khavinson, V.K.; Vanyushin, B.F. Interaction of Short Peptides with FITC-Labeled Wheat Histones and Their Complexes with Deoxyribooligonucleotides. Biochemistry 2013, 78, 166–175. [Google Scholar] [CrossRef]
- Anisimov, S.V.; Bokheler, K.R.; Khavinson, V.K.; Anisimov, V.N. Studies of the Effects of Vilon and Epithalon on Gene Expression in Mouse Heart Using DNA-Microarray Technology. Bull. Exp. Biol. Med. 2002, 133, 293–299. [Google Scholar] [CrossRef]
- Ashapkin, V.V.; Linkova, N.S.; Khavinson, V.K.; Vanyushin, B.F. Epigenetic Mechanisms of Peptidergic Regulation of Gene Expression during Aging of Human Cells. Biochemistry 2015, 80, 310–322. [Google Scholar] [CrossRef]
- Kozlov, K.L.; Bolotov, I.I.; Linkova, N.S.; Drobintseva, A.O.; Khavinson, V.K.; Dyakonov, M.M.; Kozina, L.S. Molecular aspects of vasoprotective peptide KED activity during atherosclerosis and restenosis. Adv. Gerontol. 2016, 29, 646–650. [Google Scholar]
- Kitachev, K.V.; Sazonov, A.B.; Kozlov, K.L.; Petrov, K.I.; Sliusarev, A.S.; Khavinson, V.K. [The efficacy of peptide bioregulators of vessels in lower limbs chronic arterial insufficiency treatment in old and elderly people]. Adv. Gerontol. 2014, 27, 156–159. [Google Scholar]
- Khavinson, V.K.; Ryzhak, G.A.; Grigoriev, E.I.; Ryadnova, I.Y. Peptide Substance Restoring Myocardium Function. U.S. Patent 7,662,789, 16 October 2010. [Google Scholar]
- Marso, S.P.; Bain, S.C.; Consoli, A.; Eliaschewitz, F.G.; Jódar, E.; Leiter, L.A.; Lingvay, I.; Rosenstock, J.; Seufert, J.; Warren, M.L.; et al. Semaglutide and Cardiovascular Outcomes in Patients with Type 2 Diabetes. N. Engl. J. Med. 2016, 375, 1834–1844. [Google Scholar] [CrossRef] [Green Version]
- Helmstädter, J.; Frenis, K.; Filippou, K.; Grill, A.; Dib, M.; Kalinovic, S.; Pawelke, F.; Kus, K.; Kröller-Schön, S.; Oelze, M.; et al. Endothelial GLP-1 (Glucagon-Like Peptide-1) Receptor Mediates Cardiovascular Protection by Liraglutide In Mice with Experimental Arterial Hypertension. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 145–158. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Yang, F.; Wang, W.; Li, X.; Liu, D.; Zhang, Y.; Yin, G.; Lv, F.; Guo, Z.; Mehta, J.L.; et al. Liraglutide Attenuates Myocardial Fibrosis via Inhibition of AT1R-Mediated ROS Production in Hypertensive Mice. J. Cardiovasc. Pharmacol. Ther. 2021, 26, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Savchenko, L.G.; Digtiar, N.I.; Selikhova, L.G.; Kaidasheva, E.I.; Shlykova, O.A.; Vesnina, L.E.; Kaidashev, I.P. Liraglutide Exerts an Anti-Inflammatory Action in Obese Patients with Type 2 Diabetes. Rom. J. Intern. Med. 2019, 57, 233–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiemer, A.K.; Fürst, R.; Vollmar, A.M. Vasoprotective Actions of the Atrial Natriuretic Peptide. Curr. Med. Chem. Cardiovasc. Hematol. Agents 2005, 3, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Krylatov, A.V.; Tsibulnikov, S.Y.; Mukhomedzyanov, A.V.; Boshchenko, A.A.; Goldberg, V.E.; Jaggi, A.S.; Erben, R.G.; Maslov, L.N. The Role of Natriuretic Peptides in the Regulation of Cardiac Tolerance to Ischemia/Reperfusion and Postinfarction Heart Remodeling. J. Cardiovasc. Pharmacol. Ther. 2021, 26, 131–148. [Google Scholar] [CrossRef] [PubMed]
- Curry, F.E.; Clark, J.F.; Jiang, Y.; Kim, M.; Adamson, R.H.; Simon, S.I. The Role of Atrial Natriuretic Peptide to Attenuate Inflammation in a Mouse Skin Wound and Individually Perfused Rat Mesenteric Microvessels. Physiol. Rep. 2016, 4, e12968. [Google Scholar] [CrossRef] [Green Version]
- Kikuchi, M.; Nakamura, M.; Suzuki, T.; Sato, M.; Takino, T.; Hiramori, K. Usefulness of Carperitide for the Treatment of Refractory Heart Failure Due to Severe Acute Myocardial Infarction. Jpn. Heart J. 2001, 42, 271–280. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, Y.; Ohtani, M.; Sawa, Y.; Hiraishi, T.; Akedo, H.; Kobayashi, Y.; Matsuda, H. Synthetic Human Alpha-Atrial Natriuretic Peptide Improves the Management of Postoperative Hypertension and Renal Dysfunction after the Repair of Abdominal Aortic Aneurysm. J. Cardiovasc. Pharmacol. 2003, 42, 636–641. [Google Scholar] [CrossRef]
- Nagai, T.; Iwakami, N.; Nakai, M.; Nishimura, K.; Sumita, Y.; Mizuno, A.; Tsutsui, H.; Ogawa, H.; Anzai, T. JROAD-DPC investigators Effect of Intravenous Carperitide versus Nitrates as First-Line Vasodilators on in-Hospital Outcomes in Hospitalized Patients with Acute Heart Failure: Insight from a Nationwide Claim-Based Database. Int. J. Cardiol. 2019, 280, 104–109. [Google Scholar] [CrossRef]
- Hasegawa, A.; Sato, K.; Shirai, R.; Watanabe, R.; Yamamoto, K.; Watanabe, K.; Nohtomi, K.; Hirano, T.; Watanabe, T. Vasoprotective Effects of Urocortin 1 against Atherosclerosis In Vitro and In Vivo. PLoS ONE 2014, 9, e110866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leo, C.H.; Ng, H.H.; Marshall, S.A.; Jelinic, M.; Rupasinghe, T.; Qin, C.; Roessner, U.; Ritchie, R.H.; Tare, M.; Parry, L.J. Relaxin Reduces Endothelium-Derived Vasoconstriction in Hypertension: Revealing New Therapeutic Insights. Br. J. Pharmacol. 2020, 177, 217–233. [Google Scholar] [CrossRef] [PubMed]
- Mallart, S.; Ingenito, R.; Bianchi, E.; Bresciani, A.; Esposito, S.; Gallo, M.; Magotti, P.; Monteagudo, E.; Orsatti, L.; Roversi, D.; et al. Identification of Potent and Long-Acting Single-Chain Peptide Mimetics of Human Relaxin-2 for Cardiovascular Diseases. J. Med. Chem. 2021, 64, 2139–2150. [Google Scholar] [CrossRef]
- Jasaszwili, M.; Billert, M.; Strowski, M.Z.; Nowak, K.W.; Skrzypski, M. Adropin as A Fat-Burning Hormone with Multiple Functions-Review of a Decade of Research. Molecules 2020, 25, 549. [Google Scholar] [CrossRef] [Green Version]
- Butler, A.A.; Zhang, J.; Price, C.A.; Stevens, J.R.; Graham, J.L.; Stanhope, K.L.; King, S.; Krauss, R.M.; Bremer, A.A.; Havel, P.J. Low Plasma Adropin Concentrations Increase Risks of Weight Gain and Metabolic Dysregulation in Response to a High-Sugar Diet in Male Nonhuman Primates. J. Biol. Chem. 2019, 294, 9706–9719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mushala, B.A.S.; Scott, I. Adropin: A Hepatokine Modulator of Vascular Function and Cardiac Fuel Metabolism. Am. J. Physiol. Heart Circ. Physiol. 2021, 320, H238–H244. [Google Scholar] [CrossRef]
- Zhao, L.-P.; You, T.; Chan, S.-P.; Chen, J.-C.; Xu, W.-T. Adropin Is Associated with Hyperhomocysteine and Coronary Atherosclerosis. Exp. Ther. Med. 2016, 11, 1065–1070. [Google Scholar] [CrossRef] [Green Version]
- Sato, K.; Yamashita, T.; Shirai, R.; Shibata, K.; Okano, T.; Yamaguchi, M.; Mori, Y.; Hirano, T.; Watanabe, T. Adropin Contributes to Anti-Atherosclerosis by Suppressing Monocyte-Endothelial Cell Adhesion and Smooth Muscle Cell Proliferation. Int. J. Mol. Sci. 2018, 19, 1293. [Google Scholar] [CrossRef] [Green Version]
- Park, Y.M.; Febbraio, M.; Silverstein, R.L. CD36 Modulates Migration of Mouse and Human Macrophages in Response to Oxidized LDL and May Contribute to Macrophage Trapping in the Arterial Intima. J. Clin. Investig. 2009, 119, 136–145. [Google Scholar] [CrossRef] [Green Version]
- Gao, S.; McMillan, R.P.; Zhu, Q.; Lopaschuk, G.D.; Hulver, M.W.; Butler, A.A. Therapeutic Effects of Adropin on Glucose Tolerance and Substrate Utilization in Diet-Induced Obese Mice with Insulin Resistance. Mol. Metab. 2015, 4, 310–324. [Google Scholar] [CrossRef]
- Zhang, S.; Chen, Q.; Lin, X.; Chen, M.; Liu, Q. A Review of Adropin as the Medium of Dialogue between Energy Regulation and Immune Regulation. Oxid. Med. Cell. Longev. 2020, 2020, 3947806. [Google Scholar] [CrossRef] [PubMed]
- Khavinson, V.K.; Grigoriev, E.I.; Malinin, V.V.; Ryzhak, G.A. Peptide Substance Enhancing Capillaries Resistance, Pharmaceutical Composition on Its Base and Method of Its Application. Israel Patent IL 194500, 23 May 2006. [Google Scholar]
- Khavinson, V.K.; Tarnovskaia, S.I.; Lin’kova, N.S.; Guton, E.O.; Elashkina, E.V. Epigenetic aspects of peptidergic regulation of vascular endothelial cell proliferation during aging. Adv. Gerontol. 2014, 27, 108–114. [Google Scholar] [PubMed]
- Khavinson, V.K.; Lin’kova, N.S.; Evlashkina, E.V.; Durnova, A.O.; Kozlov, K.L.; Gutop, O.E. Molecular Aspects of Anti-Atherosclerotic Effects of Short Peptides. Bull. Exp. Biol. Med. 2014, 158, 159–163. [Google Scholar] [CrossRef]
- Khavinson, V.K.; Lin’kova, N.S.; Polyakova, V.O.; Kvetnoy, I.M.; Benberin, V.V.; D’yakonov, M.M.; Titkov, Y.S. Tetrapeptide H-Ala-Glu-Asp-Arg-OH Stimulates Expression of Cytoskeletal and Nuclear Matrix Proteins. Bull. Exp. Biol. Med. 2012, 153, 559–562. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, L.V.; Flint, A.; Olsen, A.K.; Ingwersen, S.H. Liraglutide in Type 2 Diabetes Mellitus: Clinical Pharmacokinetics and Pharmacodynamics. Clin. Pharmacokinet. 2016, 55, 657–672. [Google Scholar] [CrossRef] [Green Version]
- Hasdemir, B.; Mhaske, P.; Paruthiyil, S.; Garnett, E.A.; Heyman, M.B.; Matloubian, M.; Bhargava, A. Sex- and Corticotropin-Releasing Factor Receptor 2- Dependent Actions of Urocortin 1 during Inflammation. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2016, 310, R1244–R1257. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Cell Type | Molecular Controller of SASP Development |
|---|---|
| Endothelial cells | eNOS, TIMP1, TSP1, ICAM1 |
| Vascular smooth muscle cells | IL-18, Sirt1, OPN, OPG, AngII, Prelamin A, Cyclin D |
| Cardiomyocytes | TGFβ, GDF15, pRb, p19, MCP1, Endothelin3, CCN1, CTP1, PDH, PDK4 |
| Peptide | Amino Acid Sequence | Vasoprotective and Cardioprotective Properties | References |
|---|---|---|---|
| Tetrapeptide | AEDR | Reduces acute myocardial infarction-related mortality in experimental animals; produces a protective effect on mitochondria; stimulates reparative processes in the myocardium in the experimental infarction; regulates the synthesis of cyto- and karyoskeleton proteins in fibroblasts. | [123,148] |
| Tripeptide | KED | Regulates endothelial synthesis of SIRT1, endothelin-1, connexin, Ki67, Cx43, VEGF, p53 in aging, atherosclerosis and restenosis; normalizes blood circulation in elderly atherosclerosis patients. | [7,121,122,145] |
| Liraglutide | HAEGTFTSDVSSYLEGQAAKEFIAWLVRGRG | Reduces blood pressure, inhibits fibrosis in in vivo CVD models, has anti-inflammatory properties in the blood mononuclear cells culture. | [126,127,149] |
| Atrial Natriuretic Peptide (ANP) | MSSFSTTTVSFLLLLAFQLLGQTRANPMYNAVSNADLMDFKNLLDHLEEKMPLEDEVVPPQVLSEPNEEAGAALSPLPEVPPWTGEVSPAQRDGGALGRGPWDSSDRSALLKSKLRALLTAPRSLRRSSCFGGRMDRIGAQSGLGCNSFRY | Induces vasodilation, natriuresis, diuresis and counteracts the renin-angiotensin-aldosterone system (RAAS). | [128,129,130,132] |
| Human urocortin 1 (Ucn1) | DNPSLSIDLTFHLLRTLLELARTQSQRERAEQNRIIFDSV | Inhibits the formation of foam cells; inhibits the synthesis of CD36, acyl-CoA, cholesterol acyltransferase 1 in macrophages, reduces the migration and proliferation of human GMSCs, increases the activity of MMP2 and MMP9 in GMSCs. | [134,150] |
| Mimetics of Relaxin | DSWMEEVIKLCGRELVRAQIAICGMSTWSLYSALANKCCHVGCTKRSLARFC (Serelaxin) | Enhances NO-dependent relaxation, leads to PGI2-IP-mediated vasorelaxation | [135,136] |
| Adropin | MGAAISQGALIAIVCNGLVGFLLLLLWVILCWACHSRSADVDSLSESSPNSSPGPCPEKAPPPQKPSHEGSYLLQP | Negatively correlates with homocysteine, highly sensitive C-reactive protein, CD36, TNF-a, IL-6; regulates the expression of PDK4 and PDH enzymes, which ensure the metabolism of fatty acids and are involved in the formation of SASP in cardiomyocytes. | [137,138,139,141,143,144] |
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.
|
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khavinson, V.; Linkova, N.; Dyatlova, A.; Kantemirova, R.; Kozlov, K. Senescence-Associated Secretory Phenotype of Cardiovascular System Cells and Inflammaging: Perspectives of Peptide Regulation. Cells 2023, 12, 106. https://doi.org/10.3390/cells12010106
Khavinson V, Linkova N, Dyatlova A, Kantemirova R, Kozlov K. Senescence-Associated Secretory Phenotype of Cardiovascular System Cells and Inflammaging: Perspectives of Peptide Regulation. Cells. 2023; 12(1):106. https://doi.org/10.3390/cells12010106
Chicago/Turabian StyleKhavinson, Vladimir, Natalia Linkova, Anastasiia Dyatlova, Raisa Kantemirova, and Kirill Kozlov. 2023. "Senescence-Associated Secretory Phenotype of Cardiovascular System Cells and Inflammaging: Perspectives of Peptide Regulation" Cells 12, no. 1: 106. https://doi.org/10.3390/cells12010106