





Abstract
Bovine viral diarrhea virus (BVDV) is one of the most important viruses affecting the health and well-being of bovine species throughout the world. Here, we used CRISPR-mediated homology-directed repair and somatic cell nuclear transfer to produce a live calf with a six amino acid substitution in the BVDV binding domain of bovine CD46. The result was a gene-edited calf with dramatically reduced susceptibility to infection as measured by reduced clinical signs and the lack of viral infection in white blood cells. The edited calf has no off-target edits and appears normal and healthy at 20 months of age without obvious adverse effects from the on-target edit. This precision bred, proof-of-concept animal provides the first evidence that intentional genome alterations in the CD46 gene may reduce the burden of BVDV-associated diseases in cattle and is consistent with our stepwise, in vitro and ex vivo experiments with cell lines and matched fetal clones.
Bovine viral diarrhea virus (BVDV) is one of the most burdensome viruses affecting the health and well-being of cattle throughout the world. The main host receptor mediating BVDV infection is CD46. This proof-of-concept study showed that substituting six amino acids in CD46 caused a dramatic reduction in BVDV susceptibility in a gene-edited calf without causing any obvious adverse effects in the first 20 months of life. This provides the first example of gene editing in cattle to reduce the impact of a major viral disease. This approach could significantly improve animal welfare, increase the long-term sustainability of cattle production, and provide an opportunity to reduce antibiotic use in agriculture, given that BVDV infection puts calves at risk for secondary bacterial diseases.
Introduction
Bovine viral diarrhea virus (BVDV, family Flaviviridae, genus Pestivirus) is a ubiquitous pathogen of cattle that is associated with gastrointestinal and respiratory diseases and reproductive failure in cattle worldwide (1, 2). Following exposure, BVDV replicates in the oronasal mucosa as well as the tonsil before spreading to the regional lymph nodes (3, 4). BVDV infection of lymphocyte and monocyte populations results in immune suppression and systemic dissemination of the virus to multiple organs, including the digestive and reproductive tracts (2). In pregnant cattle, BVDV can cross the placenta and infect the fetus, resulting in abortion, congenital malformations, or birth of immunotolerant and persistently infected (BVDV-PI) calves (5). While vaccines for BVDV have been available for more than 50 years, the extensive genetic and antigenic diversity in circulating field strains of BVDV poses a challenge to making these vaccines broadly protective (6). Thus, if available, cattle with reduced genetic susceptibility to BVDV would complement current strategies to control BVDV-associated diseases.
Modifying pathogen receptors via genome editing offers an innovative way to control viral infections in livestock (7–9). Bovine CD46 is the main cellular receptor for BVDV (10, 11), and CRISPR/Cas9-mediated knockout of the CD46 gene in Madin–Darby bovine kidney (MDBK) cells results in a significant reduction in BVDV susceptibility in vitro (12–14). Yet, it is unknown whether editing CD46 will reduce an animal's susceptibility to BVDV in vivo. Like many viruses grown in cell culture, BVDV has been reported to rapidly adapt in vitro to infect cells lacking CD46 by acquiring mutations that enhance heparan sulfate-mediated binding and entry (13). However, viral adaptations to host receptors occurring in vitro do not necessarily correlate with in vivo outcomes (15, 16). Thus, the success of a gene editing strategy for reducing viral infection can be difficult to accurately predict.
Deleting the entire CD46 gene in vivo would likely have severe deleterious consequences. In most mammals, CD46 is ubiquitously expressed on all nucleated cells and is critical in several diverse biological processes (17). The essential roles for CD46 function include complement inactivation, modulation of T-cell activation, and fertility (18, 19). For these reasons, minimal and precise genomic alterations are necessary if the normal cellular functions of CD46 are to be preserved in the host animal. Important work by Krey et al. mapped the BVDV receptor-binding sites of CD46 to two peptide domains, E66QIV69 and G82QVLAL87, which are located on antiparallel beta sheets in the amino terminus of the protein (10). Further, by expressing chimeric CD46 proteins in porcine cells, they showed that replacing the bovine CD46 G82QVLAL87 residues with ALPTFS prevented BVDV cellular entry and blocked infection in vitro (10).
Building on their discovery, our goal was to use gene editing via homology-directed repair to make a precise 18-nt (nucleotide) replacement in the endogenous CD46 gene to produce cells expressing homozygous CD46 protein with the A82LPTFS87 substitution in the BVDV binding domain. In stepwise experiments, we measured the impact of this edit on BVDV susceptibility in immortalized cells, primary cells from fetal tissues, cells from a live CD46-edited calf, and in a natural exposure challenge study with the same edited calf.
Materials and methods
Ethics statement
All protocols for reproductive cloning, fetal tissue collection, and birthing were reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) of Trans Ova Genetics (IACUC Project ID 68-000176-00). All protocols for the gene-edited calf and the BVDV challenge study were reviewed and approved by the IACUC of the University of Nebraska–Lincoln, an AAALAC International Accredited institution (IACUC Project ID 2111).
Protein folding prediction of CD46 extracellular domains
The predicted protein structure of the CD46 extracellular domain, minus the signal peptide, was compared with the wild-type G82QVLAL87 residues and the substituted A82LPTFS87 residues. These extracellular domains consisted of residues 43–313 of the National Center for Biotechnology Information reference sequence: NP_898903.2. An artificial general intelligence software that uses machine learning was used to fold the CD46 extracellular domain variants (AlphaFold v2.2.2) (20, 21). Each of the two CD46 variants was folded separately, and the .pdb structure file with the highest confidence was selected as the best representation of the biologically relevant structure. The two CD46 variant files were loaded into a molecular graphics system (PyMOL Molecular Graphics System v2.5.3; Schrödinger, LLC, New York, NY), aligned, and rotated to view the variant region and the BVDV binding platform.
CRISPR/Cas9 editing of CD46 in MDBK cells and Gir fibroblasts
BVDV-free MDBK cells (ATCC CCL-22) were obtained from the American Type Culture Collection (ATCC; Rockville, MD). Primary skin fibroblasts used for reproductive cloning were from a Gir breed (Bos indicus) female (TO470).
Gene editing and detection reagents
CRISPR/Cas9 technology was used to create altered alleles of CD46 in both MDBK and primary fibroblast cells. For the CD46 gene deletion (CD46Δ), the entire coding sequence of CD46 was removed, while for CD46 A82LPTFS87, 18-nt (six amino acid codons) were substituted. The combination of cell type, guide RNA, and homology-dependent repair templates for each edit is listed in Table 1 according to cell type and gene editing outcome desired. Individual gRNAs were designed using Cas-Designer (22) and selected based on proximity to the target sequence and fewest potential off-target sites. Editing efficiencies of gRNAs screened for this study are listed in Table S1.
Synthetic guide RNA (gRNA) and single-stranded oligonucleotide donor (ssODN) molecules used in gene editing.
| Intended outcome | gRNA(s) (5′−3′) | HDR template ssODN (5′−3′) |
|---|---|---|
| MDBK CD46 gene deletion | btCD46 5′ g4: GGTCCTCTCCTGGGAATTAC btCD46 3′ g3: GGCATTAGTAAAAATCCCCA |
btCD46 LD1: GTTATTTAATAATTTACCTTCCCAGGGTCCTCTCCTGGGAATAAGCTTGGATTTTTACTAATGCCTGGGTGCCCCCACCCCCTCCCCGTC |
| MDBK CD46 A82LPTFS87 substitution | btCD46 g2.5: ACGAGAGCCAGGACTTGACC | btCD46 AA sub: GTATGAATGTCGTCTGGGTTTCCAGCCAGTAACTCCGGCATTGCCTACATTCAGTGTTTGTCAGGATAATAATACATGGTCGTCTCTCCA |
| Gir skin fibroblast CD46 A82LPTFS87 substitution | btCD46 g2.5: ACGAGAGCCAGGACTTGACC | btCD46 ALPTFS Gir AS: C*C*TCCTGGAGAGACGACCATGTATTATTATCCTGACAAACACTGAAkGTAGGCAATGCCGGAGTTACTGGCTGGAAACCCAGATGACATTCATACACA*A*T |
| Intended outcome | gRNA(s) (5′−3′) | HDR template ssODN (5′−3′) |
|---|---|---|
| MDBK CD46 gene deletion | btCD46 5′ g4: GGTCCTCTCCTGGGAATTAC btCD46 3′ g3: GGCATTAGTAAAAATCCCCA |
btCD46 LD1: GTTATTTAATAATTTACCTTCCCAGGGTCCTCTCCTGGGAATAAGCTTGGATTTTTACTAATGCCTGGGTGCCCCCACCCCCTCCCCGTC |
| MDBK CD46 A82LPTFS87 substitution | btCD46 g2.5: ACGAGAGCCAGGACTTGACC | btCD46 AA sub: GTATGAATGTCGTCTGGGTTTCCAGCCAGTAACTCCGGCATTGCCTACATTCAGTGTTTGTCAGGATAATAATACATGGTCGTCTCTCCA |
| Gir skin fibroblast CD46 A82LPTFS87 substitution | btCD46 g2.5: ACGAGAGCCAGGACTTGACC | btCD46 ALPTFS Gir AS: C*C*TCCTGGAGAGACGACCATGTATTATTATCCTGACAAACACTGAAkGTAGGCAATGCCGGAGTTACTGGCTGGAAACCCAGATGACATTCATACACA*A*T |
*Denotes phosphorothioated base; k denotes G/T degenerate base; and underline denotes bases introducing the A82LPTFS87 edit.
Synthetic guide RNA (gRNA) and single-stranded oligonucleotide donor (ssODN) molecules used in gene editing.
| Intended outcome | gRNA(s) (5′−3′) | HDR template ssODN (5′−3′) |
|---|---|---|
| MDBK CD46 gene deletion | btCD46 5′ g4: GGTCCTCTCCTGGGAATTAC btCD46 3′ g3: GGCATTAGTAAAAATCCCCA |
btCD46 LD1: GTTATTTAATAATTTACCTTCCCAGGGTCCTCTCCTGGGAATAAGCTTGGATTTTTACTAATGCCTGGGTGCCCCCACCCCCTCCCCGTC |
| MDBK CD46 A82LPTFS87 substitution | btCD46 g2.5: ACGAGAGCCAGGACTTGACC | btCD46 AA sub: GTATGAATGTCGTCTGGGTTTCCAGCCAGTAACTCCGGCATTGCCTACATTCAGTGTTTGTCAGGATAATAATACATGGTCGTCTCTCCA |
| Gir skin fibroblast CD46 A82LPTFS87 substitution | btCD46 g2.5: ACGAGAGCCAGGACTTGACC | btCD46 ALPTFS Gir AS: C*C*TCCTGGAGAGACGACCATGTATTATTATCCTGACAAACACTGAAkGTAGGCAATGCCGGAGTTACTGGCTGGAAACCCAGATGACATTCATACACA*A*T |
| Intended outcome | gRNA(s) (5′−3′) | HDR template ssODN (5′−3′) |
|---|---|---|
| MDBK CD46 gene deletion | btCD46 5′ g4: GGTCCTCTCCTGGGAATTAC btCD46 3′ g3: GGCATTAGTAAAAATCCCCA |
btCD46 LD1: GTTATTTAATAATTTACCTTCCCAGGGTCCTCTCCTGGGAATAAGCTTGGATTTTTACTAATGCCTGGGTGCCCCCACCCCCTCCCCGTC |
| MDBK CD46 A82LPTFS87 substitution | btCD46 g2.5: ACGAGAGCCAGGACTTGACC | btCD46 AA sub: GTATGAATGTCGTCTGGGTTTCCAGCCAGTAACTCCGGCATTGCCTACATTCAGTGTTTGTCAGGATAATAATACATGGTCGTCTCTCCA |
| Gir skin fibroblast CD46 A82LPTFS87 substitution | btCD46 g2.5: ACGAGAGCCAGGACTTGACC | btCD46 ALPTFS Gir AS: C*C*TCCTGGAGAGACGACCATGTATTATTATCCTGACAAACACTGAAkGTAGGCAATGCCGGAGTTACTGGCTGGAAACCCAGATGACATTCATACACA*A*T |
*Denotes phosphorothioated base; k denotes G/T degenerate base; and underline denotes bases introducing the A82LPTFS87 edit.
For the CD46 gene deletion, homology-directed repair (HDR) templates were purchased from Integrated DNA Technologies (IDT; San Jose, CA). gRNA and Cas9 mRNA were synthesized by in vitro transcription. pDR274 transcription plasmids containing the gRNA sequences were linearized with DraI (NEB; Ipswich, MA) and amplified using Accustart Taq DNA Polymerase HiFi (Quanta Biosciences, Gaithersburg, MD) using the following primers and cycling program: pDR274 F (5′-TCCGCTCGCACCGCTAGCT-3′) and pDR274 R (5′-AGCACCGACTCGGTGCCAC-3′) and 1 cycle (95°C, 2 min), 35 cycles (95°C, 30 s; 48°C, 15 s; 68°C, 15 s), and 1 cycle (68°C, 30 s). Once completed, the amplification reactions were treated with RNAsecure (ThermoFisher Scientific; Waltham, MA) following the manufacturer's recommendations and purified using the QIAquick Gel Extraction Kit (Qiagen; Hilden, Germany). These amplicons were used as a template for transcription using the MEGAshortscript T7 Transcription Kit (ThermoFisher Scientific) and purified with the RNeasy Mini Kit (Qiagen), following the manufacturer's instructions. This RNA transcript constituted the gRNA.
For Cas9 mRNA synthesis, pT3Ts-nCas9n plasmid was linearized with XbaI (NEB), treated with RNAsecure (ThermoFisher Scientific), and purified using the QIAquick Gel Extraction Kit (QIAGEN). In vitro Cas9 mRNA transcription was performed using the mMESSAGE mMACHINE T3 Kit (ThermoFisher Scientific) followed by A-tailing using a Poly(A) Tailing Kit (ThermoFisher Scientific). These transcripts were purified with the RNeasy Mini Kit (Qiagen), following the manufacturer's instructions. The RNA transcript constituted the JECas9 mRNA.
For CD46 A82LPTFS87 substitutions, gRNAs (Alt-R CRISPR-Cas9 crRNA and Alt-R CRISPR-Cas9 tracrRNA), HDR templates [single-stranded oligo donor (ssODN)], and Cas9 protein (Cas9 Nuclease V3) were purchased from IDT. The relative position of the gRNA and the ssODN in relation to CD46 protein domains and amino acid sequence are shown in Fig. 1A and B. Ribonucleoprotein complexes were formed according to the manufacturers’ recommendations and incubated for 20 min immediately prior to transfection.
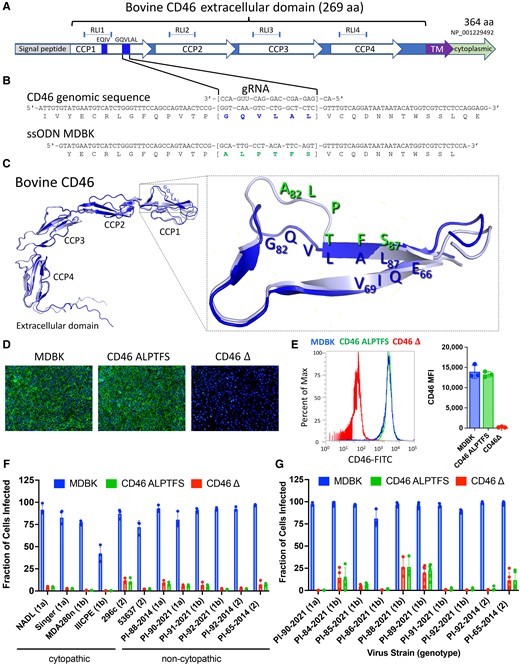
Comparing the CD46 A82LPTFS87 substitution with the CD46 gene deletion in MDBK cells. A) Physical map of CD46 protein domains. RLI, receptor–ligand interaction domain; CCP, complement control protein domain; TM, transmembrane domain. B) Genomic DNA sequence of CD46 in the region of the G82QVLAL87 motif showing the amino acid translation, the gRNA heteroduplex, and the alignment of the synthetic ssODN used for replacing the G82QVLAL87 residues with A82LPTFS87. gRNA, synthetic guide RNA; ssODN, single-stranded oligonucleotide donor template. C) Ribbon representations of AlphaFold2 predictions of the extracellular domain of wild-type bovine CD46 (G82QVLAL87 strand) aligned with that containing the A82LPTFS87 substitution. The inset shows a closeup of the BVDV binding platform rotated to visualize the predicted structure differences in residues 82–84. D) Immunofluorescence staining of CD46 (FITC) and nuclei (DAPI; 10× magnification). E) Flow cytometric quantification of CD46 protein expression levels (n = 3). MFI, median fluorescence intensity. F) CD46-edited cells were infected with cytopathic or non-cytopathic BVDV isolates at an MOI of 2, and infection efficiency was determined at 20 hpi by flow cytometry using a monoclonal anti-BVDV E2 antibody. G) Serum from BVDV-PI calves was inoculated on cells. Infection efficiency was quantified at 72 hpi by flow cytometry. In panels F and G, the results represent the mean±standard deviation of n ≥ 3 independent experiments.
MDBK cell culture and transfection
MDBK cells were maintained at 38.5°C, 5% CO2 in Dulbecco's modified Eagle's Medium (DMEM) supplemented with 10% irradiated fetal bovine serum, 100 I.U./mL penicillin and streptomycin, and 10 mm Hepes. Actively growing cells were prepared for transfection with the Neon Transfection System (Life Technologies; Carlsbad, CA). Briefly, 600 000 cells were resuspended in buffer “R.” For the CD46 gene deletion transfection, 1 μg of btCD46 5′ g4 RNA, 1 μg of btCD46 3′ g3 RNA, 2 μg of JECas9 mRNA, and 0.2 nmol of btCD46 LD1 ssODN were added to the cells. For the CD46 A82LPTFS87 substitution, 120 pMol gRNA:tracrRNA complex, 17.2 μg Alt-R S.p. HiFi Cas9 Nuclease V3, and 0.2 nmol of the ssODN were added to the cells. The cell suspension was electroporated using the 100 μL Neon Tip with the following parameters: input voltage, 1,600 V; pulse width, 20 ms; and pulse number, 1. Transfected cells were dispersed into one well of a six-well plate with 2 mL DMEM media and cultured for 4 days at 30°C.
Fibroblast culture and transfection
Primary Gir fibroblast cells were maintained at 37°C, 5% CO2 in DMEM supplemented with 20% irradiated fetal bovine serum and 100 I.U./mL penicillin and streptomycin. Actively growing cells were prepared for transfection with the Neon Transfection System (Life Technologies). Briefly, 600 000 cells were resuspended in buffer “R.” For the CD46 A82LPTFS87 substitution, 65 pmol of gRNA, 78 pmol of Alt-R S.p. HiFi Cas9 Nuclease V3, and 0.11 nmol of the ssODN were added to the cells. The cell suspension was electroporated using the 100 μL Neon Tip with the following parameters: input voltage, 1800 V; pulse width, 20 ms; and pulse number, 1. Transfected cells were dispersed into one well of a six-well plate with 2 mL DMEM media and cultured for 3 days at 37°C.
Single-cell derived clonal isolation and genotyping
After 4 days at 30°C for the MDBK cells or 3 days at 37°C for the fibroblasts, cells were seeded at low density, subcultured, and screened for homology-directed repair by PCR. Screening for homozygous CD46 A82LPTFS87 in MDBK and fibroblast cells was conducted using AccuStart II GelTrack PCR SuperMix (Quanta Biosciences) and the following primers and cycling program: btCD46 NJ F1 (5′-TTCTCCAACAGGCCAGAAGC-3′) and btCD46 NJ R1 (5′-AGGCAACCAATCGTGACGAA-3′) and 1 cycle (95°C, 2 min), 35 cycles (95°C, 20 s; 62°C, 20 s; 72°C, 45 s), and 1 cycle (72°C, 5 min). Amplicons were then digested with MspI (NEB) and visualized using agarose gel electrophoresis. Clones homozygous by restriction fragment length polymorphism were verified by Sanger sequencing (ACGT Inc.; Wheeling, Illinois or Eurofins; Louisville, Kentucky).
Screening for homozygous CD46 gene deletion in MDBK cells was conducted using AccuStart II GelTrack PCR SuperMix (Quanta Biosciences) and the following primers and cycling program: btCD46 5′ F2 (5′-CCAGCCCGCAATGTTTACAC-3′) and btCD46 5′ R2 (5′− TTTGGCCATTGCTCTCCCAA-3′) and 1 cycle (95°C, 2 min), 35 cycles (95°C, 20 s; 64°C, 20 s; 72°C, 45 s), and 1 cycle (72°C, 5 min) and btCD46 5′ F2 (5′-CCAGCCCGCAATGTTTACAC-3′) and btCD46 3′ NJ R1 (5′-TTCACATGCAGACGTGGACT-3′) and 1 cycle (95°C, 2 min), 35 cycles (95°C, 20 s; 64°C, 20 s; 72°C, 45 s), and 1 cycle (72°C, 5 min). Clones homozygous by junction PCR were verified by Sanger sequencing (ACGT).
Somatic cell nuclear transfer and reproductive cloning
Somatic cell nuclear transfer (SCNT) was performed by Trans Ova Genetics (Sioux Center, IA) as previously described (23, 24) on Gir fibroblast clones confirmed homozygous for the CD46 A82LPTFS87 substitution and wild-type unedited controls. Grade 1 embryos containing either the unedited wild-type CD46 (n = 8) or CD46 A82LPTFS87 substitution (n = 8) were implanted into each of 16 recipient cows. Pregnancies were confirmed by ultrasound at 30 and 90 days post-transplantation.
Fetal tissue collection and primary cell isolation
Fetal tissues were collected at 100 days of gestation from one CD46 A82LPTFS87-edited fetus and one unedited control fetus, and primary cells were isolated from the lungs, heart, small intestine, esophagus, kidney, and liver (25–30). Remaining fetuses were allowed to continue gestation with the goal of reaching full term.
Primary cells were isolated and maintained in various cell culture mediums. Alveolar cells were maintained in low-glucose DMEM (Corning, Corning, NY) supplemented with 20% irradiated fetal bovine serum and 1× antibiotic-antimycotic and cultured on plates coated with 2% gelatin (Sigma, St. Louis, MO). Renal epithelial cells were maintained in DMEM/Hams F-12 50/50 medium (Corning) supplemented with 10% irradiated fetal bovine serum, 1× antibiotic-antimycotic, 5 μg/mL transferrin (Sigma), 25 ng/mL rhEGF (Invitrogen), 0.1 μg/mL hydrocortisone (Sigma), and 10 μg/mL bovine insulin (Sigma). Renal epithelial cells were cultured on plates coated with collagen (Sigma). Small intestine epithelial cells were cultured in the same medium as listed for renal epithelial cells and cultured on plates coated with 0.1% gelatin. Esophageal fibroblasts were cultured in DMEM supplemented with 15% irradiated fetal bovine serum and 1× antibiotic-antimycotic. Epicardial cells were maintained in DMEM + M199 (ThermoFisher Scientific) mixed 1:1 and supplemented with 10% irradiated fetal bovine serum, 1× antibiotic-antimycotic, and 10 μM SB43152 (Sigma). Liver cells (mixed population) were cultured in the same medium as listed for the renal and intestinal epithelial cells and cultured on plates coated with 0.1% gelatin. All cells were tested for BVDV contamination prior to experimentation by RT-qPCR with a BVDV-specific primer/probe set (31) as described previously (32).
Birth of the CD46-edited calf
A CD46 A82LPTFS87-edited Gir calf was delivered by cesarean section at the calculated time of full gestation (∼285 days for Gir cattle). The calf was removed from the recipient cow without nursing and was fed a commercial bovine colostrum replacement solution (Calf's Choice Total Gold) by bottle. Thereafter, the calf was fed a commercial milk replacement solution formulated for calves. Simultaneously, an age- and sex-matched Holstein dairy calf was purchased from an Iowa dairy farm and fed the same commercial replacement solutions. At 1 week of age, both calves were moved to the BSL2 animal care facility at the University of Nebraska–Lincoln and housed in the same room.
Evaluating CD46 DNA sequences of cell lines and animals with 15x whole genome sequence
DNA was extracted from cell lines or tissues with standard procedures that used RNase/protease digestion, phenol/chloroform extraction, and ethanol precipitation. Purified DNAs were dissolved in a solution of 10 mm TrisCl, 1 mm EDTA (TE, pH 8.0), and stored at 4°C. For whole genome sequence (WGS), 2 μg of genomic DNA was fragmented and used to make indexed, 500 bp, paired-end libraries. Pooled, indexed libraries were sequenced with massively parallel sequencing machines (either NextSeq500 or NextSeq2000, Illumina Inc.) and the appropriate kits producing 2 × 150 bp paired-end reads. Samples were repeatedly sequenced to a threshold of 40 gigabases surpassing Q20 quality. This approach produced at least 12-fold mapped read coverage (15-fold average) and provided genotype scoring rates and accuracies that exceed 99% (33, 34).
FASTQ files were aggregated by animal and aligned individually to a bovine reference genome (ARS-UCD1.2) with the Burrows–Wheeler Alignment tool (BWA) aln algorithm version 0.7.1219. The files were merged and collated with the bwa sampe command, and the resulting sequence alignment map (SAM) files were converted to binary alignment map (BAM) files and sorted with SAMtools version 1.3.120. PCR duplicates were marked in the BAM files with the Genome Analysis Toolkit (GATK) version 3.621. The GATK module RealignerTargetCreator was used to identify regions with small indels, and those regions were realigned with the IndelRealigner module. The BAM files produced at each step were indexed with SAMtools and made available via object storage through a web service interface (Amazon Web Services, Inc. Seattle, WA).
Aligned genomes were viewed with the Integrative Genomics Viewer (IGV) version 2.12.2 by selecting the desired reference genome and loading a session file URL. For example, the TO470 cell line and its unedited and edited reproduction clones were viewed together in IGV by loading session URL: https://s3.us-west-2.amazonaws.com/usmarc.heaton.public/WGS/CellLines/ARS1.2/sessions/ARS1.2_CD46_Ginger4Tracks.xml. The genomic DNA sequences in a 150-kb region centered on the CD46 gene were visually inspected in IGV for differences at the nucleotide level. The edited site encoding CD46 residues 82 to 87 (ARS-UCD1.2; ch16:75,617,415 to ch16:75,617,432) was also inspected. Potential off-target sites were searched for in the ARS-UCD1.2 bovine reference genome with Cas-OFFinder (version 2.4 (35)) and the target gRNA sequence: 5′-ACGAGAGCCAGGACTTGACC-3′. Also, ad hoc GATK software analysis of WGS was used in an attempt to independently identify any significant differences in the genome sequences between the unedited Gir TO470 cell line, the unedited 100-day fetal calf, the CD46-edited 100-day fetal calf, and the born-live CD46-edited calf. Specifically, GATK-identified indels and structural variants were sorted by those that were unique to the live calf compared to the three other samples. Differences were ranked based on size and manually inspected in IGV.
Immunofluorescence staining and flow cytometric detection of bovine CD46
Cellular localization of CD46 protein was visualized by immunofluorescence (IF) staining and quantified by flow cytometry using a polyclonal anti-bovine CD46 antibody. Antibodies that detect bovine CD46 are not commercially available and were thus generated for use in this study. The sequence encoding, the extracellular domain of bovine CD46 (amino acids 43-310), was codon-optimized and synthesized with an artificial signal peptide, which caused the expressed protein to be secreted from cells. The synthetic gene was cloned into the mammalian expression vector pcDNA3.4 and transfected into mammalian cell cultures (Chinese hamster ovary cells, CHO-S). The secreted CD46 peptide was purified from the culture supernatant and injected into two rabbits as an immunogen for primary immunization and successive boosting. Rabbits were boosted three times before terminal blood collection. Antibodies from each rabbit were purified and quantified individually (Genscript BioTech; Piscataway, NJ).
For microscopy, cells were fixed with 4% paraformaldehyde (PFA) in phosphate-buffered saline (PBS) for 10 min at room temperature. Cells were blocked using 2% bovine serum albumin diluted in phosphate-buffered saline (BSA-PBS) for 1 h at room temperature. The CD46 polyclonal antibody was used at 5 μg/mL diluted in 2% BSA-PBS. Cells were incubated with the CD46 antibody for 1 h at room temperature. Following three washes with PBS, cells were incubated with a goat anti-rabbit IgG fluorescein isothiocyanate (FITC) conjugated secondary antibody (Abcam; Cambridge, United Kingdom; catalog no. ab6717) in 2% BSA-PBS for 1 h at room temperature in the dark. Following three PBS washes, the nuclei of the cells were stained with a 300 nm solution of 4′,6-diamidino-2-phenylindole (DAPI) in PBS for 10 min at room temperature. Cells were visualized using an EVOS FL Auto microscope at 10× magnification (Life Technologies).
For flow cytometry, cells were collected using TrypLE (Gibco; Waltham, MA) and centrifuged for 6 min at 300 × g. The supernatant was discarded, and 5 × 105 cells were resuspended in 100 μL of 2% BSA-PBS and incubated for 30 min on ice. An anti-bovine CD46 polyclonal antibody was added to the tubes at a concentration of 1 μg/mL and incubated for 30 min on ice. The cells were washed three times with PBS and resuspended with 2% BSA-PBS containing a goat anti-rabbit IgG FITC conjugated secondary antibody (Abcam catalog no. ab6717) and incubated for 30 min on ice in the dark. Cells were washed three times with PBS and analyzed using an Attune NxT Flow Cytometer (ThermoFisher Scientific) using the Attune cytometric software version 5.1.1.
Virus isolates and serum from calves persistently infected with BVDV
BVDV consists of two species, Pestivirus A (formerly BVDV-1) and Pestivirus B (formerly BVDV-2), according to the International Committee on Taxonomy of Viruses. Herein, for continuity with the literature on BVDV genotyping, these viruses will be termed BVDV-1 and BVDV-2. Information on the virus isolates used in this study can be found in Table S2.
Cytopathic isolates
BVDV-1a stain NADL (ATCC-VR1422) was purchased from ATCC. BVDV strains Singer (BVDV-1a), MDA280n (BVDV-1b), IIICPE (BVDV-1b), 53637 (BVDV-2), and 296c (BVDV-2) were obtained from the National Animal Disease Center collection in Ames, Iowa. Cytopathic BVDV strains were propagated in MDBK cells and quantitated in bovine turbinate cells (BT; ATCC CRL 1390). The infective titer was determined in two replicates using an endpoint dilution assay. Viral stocks were stored in 0.5 to 1 mL aliquots at −80°C.
Serum from BVDV-PI calves
To test non-cytopathic (NCP) field strains, whole blood was collected from cattle persistently infected with BVDV. Serum was separated at 1650 × g for 15 min at 4°C, aliquoted, and stored at −80°C. Virus from each sample was genotyped by PCR amplification and Sanger sequencing of the 5′ untranslated region as previously described (36).
Non-cytopathic isolates
NCP BVDV strains were isolated from serum (this study) or plasma (36) collected from BVDV-PI cattle. Virus was isolated on MDBK cells, and low-passage (passages 2–4) viral stocks were stored at −80°C. The infective titer of NCP BVDV isolates was estimated by RT-qPCR using log10 dilutions of the virus (32). A standard curve was made by plotting RT-qPCR Ct values against log10 dilutions of the NADL virus with a known infectious titer. Linear regression analysis was performed to create a standard for estimating the approximate titer of NCP stocks of virus.
IF staining and flow cytometric detection of BVDV antigen
For flow cytometric quantification of infected cells or IF visualization, cells were fixed with 4% PFA in PBS, washed, and processed similar to that described above for CD46 protein detection with the addition of 0.1% saponin (w/v) to all blocking, staining, and wash buffers for cell permeabilization. An anti-BVDV E2 monoclonal antibody (Creative Diagnostics; Shirley, NY, Catalog no. DMAB28412 or VMRD; Pullman, WA, Catalog no. 348) and an anti-mouse CruzFluor 488 (CFL 488) conjugated secondary antibody (Santa Cruz Biotechnology; Dallas, TX, catalog no. SC-533653) were used to detect BVDV antigen positive cells.
BVDV susceptibility testing in CD46-edited MDBK cells
Cells were seeded 1 day prior to infection in 24-well or 48-well plates in MEM supplemented with 7.5% horse serum (HS; ATCC). Prior to infection, cell culture medium was removed. Then, cells were inoculated with BVDV isolates or serum from BVDV-PI calves as described in the figure legends. Virus adsorption and entry were allowed to proceed for 2 h at 37°C, and unbound virus was removed by washing the cells four times with PBS. Cells were incubated with MEM supplemented with 5% HS for the desired time before infection efficiency was determined by IF staining or flow cytometry as described above.
Isolation and infection of primary skin fibroblasts
Ear punches were collected from the CD46 A82LPTFS87-edited calf and the unedited control Holstein calf using the Allflex tissue sampling unit for primary skin fibroblast isolation at Trans Ova Genetics. Primary skin fibroblasts were grown in DMEM supplemented with 15% irradiated fetal bovine serum and 1× antibiotic-antimycotic. For infection studies, primary fibroblasts were seeded in 24-well plates at a density of 5 × 104 cells per well in 200 μL DMEM supplemented with 1× antibiotic-antimycotic and 2 mM l-glutamine and 5% horse serum (HS; ATCC) and incubated 24 h at 37°C with 5% CO2. The following day, cells were infected with NCP BVDV from the serum of BVDV-PI calves for BVDV genotypes 1a and 1b or with low-passage cell culture isolates for BVDV genotype 2. Cell culture medium was removed, and cells were inoculated with serum from BVDV-PI calves diluted 1:1 with DMEM or BVDV-2 isolates at an multiplicity of infection (MOI) of 0.1 and incubated for 2 h at 37°C. Cells were then washed four times with PBS to remove unbound virus, and cells were incubated in DMEM supplemented with 5% HS at 37°C for 72 h. Infected cells were quantified by flow cytometry and visualized by IF as described above.
Peripheral blood mononuclear cell and monocyte isolation and ex vivo BVDV challenge
Blood was collected from the CD46 A82LPTFS87-edited Gir calf and the unedited control Holstein calf via jugular venipuncture into syringes containing EDTA as an anticoagulant. Peripheral blood mononuclear cell (PBMC) isolation was accomplished using SepMate tubes according to the manufacturer's instructions with a few modifications (Stemcell Technologies, Cambridge, MA). Each 9 mL of blood was diluted in 15 mL of room temperature PBS and gently mixed for 15 min on a nutator. Fifteen mL of Ficoll-Plaque Plus at a density of 1.077 g/mL (Cytiva, Marlborough, MA) was added to 50 mL SepMate tubes, and the diluted blood was overlaid followed by centrifugation at 1200 × g for 15 min at 21°C with the brake on. The PBMC layer was then collected by pouring the top layer into a fresh 50 mL tube. Approximately 30 mL of PBS was added to each tube, bringing the total volume to 50 mL, and cells were pelleted at 500 × g for 15 min at 21°C. Residual red blood cells were lysed using red blood cell lysis buffer (Sigma) according to the manufacturer's instructions. PBMCs were then washed three times by centrifugation (300 × g, 8 min, 21°C) with 50 mL PBS. On the fourth wash, cells were pelleted at 120 × g for 10 min at 21°C to remove platelets from the PBMC preparation. Cell pellets were then resuspended in RPMI plus 1× antibiotic-antimycotic (Gibco), and differential counts were obtained with the Heska Element HT5 (Heska; Loveland, CO).
For monocyte isolation, PBMCs were resuspended at 5 × 105 monocytes per mL and 700 μL of PBMC suspension containing 3.5 × 105 monocytes were added to each well in a 48-well plate. Plated cells were incubated at 37°C with 5% CO2 for 2 h. Non-adherent cells (mostly lymphocytes) were aspirated with a pipette, and wells were washed vigorously four times with RPMI plus 1× antibiotic-antimycotic to remove all non-adherent cells, leaving behind the monocytes. Two hundred fifty μL of RPMI supplemented with 1× antibiotic-antimycotic and 5% (v/v) heat-inactivated FBS was added to each well and incubated overnight at 37°C with 5% CO2.
Non-adherent cells collected from the 48-well plates following monocyte isolation were pelleted at 400 × g for 6 min at 4°C and resuspended in RPMI supplemented with 1× antibiotic-antimycotic and 10% (v/v) heat-inactivated FBS. Cell counts and viability were determined using the Heska Element HT5 and the automated cell counter, Countess II. Cells were then resuspended at 1.25 × 106 cells per mL, and 200 μL of the cell suspension containing 2.5 × 105 cells was plated in 96-well round bottom plates. Plated cells were incubated overnight at 37°C with 5% CO2.
For infection of monocytes, the medium was removed from the wells and replaced with 100 μL of RPMI with 10% (v/v) serum collected from persistently infected calves (genotypes 1a and 1b) or BVDV-2 isolate PI-92-2014 at an MOI of 0.01. Monocytes were inoculated for 2 h at 37°C with 5% CO2 with gentle agitation of the plates every 20 min. Two hundred fifty μL of RPMI supplemented with 1× antibiotic-antimycotic and 5% (v/v) heat-inactivated FBS was then added to each well, and cells were incubated for 48 h at 37°C with 5% CO2. Input (t = 0) samples were also collected and stored at −80°C. Duplicate plates were frozen at 48 hpi and processed for viral RNA detection. Following two freeze thaw cycles to release viral RNA from infected cells, RNA was extracted from clarified supernatants using the Qiagen viral RNA spin columns per the manufacturer's instructions. Viral RNA was quantified by RT-qPCR with a BVDV-specific primer/probe set (31) as previously described (32). Cycle threshold (Ct) values less than 38 were considered positive. Positive, negative, no template, and extraction controls were included on each run. The fold increase in viral RNA compared to the input concentration was determined with the delta Ct method (37) and graphed in Prism (v6, GraphPad Software; San Diego, CA).
For infection of lymphocytes, 80 μL of the medium was removed from each well and replaced with 80 μL of various virus isolates at varying MOI (average MOI = 4) so as to add the maximum amount of each virus isolate to the cells. Cells were incubated 24 h at 37°C with 5% CO2 then processed for flow cytometric quantification of BVDV-infected cells as described above.
Natural BVDV exposure challenge study
Serum titers of BVDV neutralizing antibodies were monitored in the CD46 A82LPTFS87-edited Gir calf and the unedited control Holstein calf, since the colostrum replacement solution contained anti-BVDV antibodies that were passively transferred during their first 24 h after birth. The natural exposure challenge was initiated only after the BVDV-1 and BVDV-2 serum neutralization antibody titers were negligible (≤1:2) in both calves.
For the challenge study, a calf persistently infected with BVDV was purchased from a dairy farm in Iowa. In routine testing, the calf tested positive for BVDV by RT-qPCR on pooled ear notches and by immunohistochemical detection on a fresh ear sample collected 10 days after the initial positive test. To ensure the animal was persistently and not transiently infected, infection status was confirmed after purchase by BVDV RT-qPCR on serum and nasal swab samples collected more than 2 weeks after the original positive sample. In addition, infectious virus was isolated on MDBK cells from both serum and nasal swab samples using standard protocols (Fig. S6). The complete viral sequence was determined by next-generation sequencing on the Illumina platform as previously described (36, 38). The virus genotype was determined by sequence alignment and phylogenetic analysis with known reference strains (36). The calf was ∼30 days of age and weighed ∼60 pounds at the time of the challenge study.
The CD46-edited Gir calf and the unedited control Holstein calf were challenged with BVDV by cohabitation with the BVDV-PI calf for 7 days in a BSL2 animal room. Samples collected throughout the study included blood, nasal swabs, and fecal swabs as outlined in Fig. 4A. Blood was collected by jugular venipuncture into syringes containing EDTA as an anticoagulant and Sarstedt Monovet tubes (Sarstedt, Inc; Newton, NC) for serum separation. Whole blood samples with EDTA were evaluated using an HT5 veterinary hematology instrument. A 1 mL aliquot of EDTA blood was stored at −80°C for BVDV RT-qPCR. PBMC was isolated from 20 mL EDTA blood using SepMate tubes similar to that described above but with minor modifications. Isolated PBMCs were washed once with PBS then counted and differentiated using the Heska instrument. Cells were diluted to 3 × 106 PBMC per 0.25 mL and stored in four replicate tubes at −80°C for detection of viral RNA by RT-qPCR. Serum was separated from clotted blood by centrifugation at 1660 × g for 15 min at 4°C and aliquoted into tubes and stored at −80°C. Nasal samples were collected by inserting two 6-inch nasal swabs into each nostril ∼4 inches and gently twisted back and forth for ∼3 to 5 s. One swab from each nostril was placed into duplicate cryovials containing 1 mL minimal essential medium (MEM; Gibco) and stored at −80°C. Fecal samples were collected by inserting one swab into the rectum ∼5–7 cm and gently swabbing in a circular motion. The fecal swab was placed into a cryovial containing 1 mL MEM and stored at −80°C. BVDV-1 and BVDV-2 virus neutralization assays were completed at the Nebraska Veterinary Diagnostic Center (University of Nebraska–Lincoln) on blinded serum samples.
In addition to sample collections by researchers, an animal health technician spent ∼30 min in the room daily to provide a clinical assessment of each calf's health. A score ranging from 0 to 16 was determined for each animal based on attitude, presence of cough, nasal discharge, eye discharge, fecal consistency, and rectal temperature according to a modified Wisconsin Health Scoring System (39) (Table S3).
Statistical analyses
Statistical analyses were performed using GraphPad Prism version 9.4.0 (GraphPad Software Inc.). Comparisons of means plus or minus the standard error (±SE) were analyzed using two-way ANOVA followed by Tukey's (three or more columns) or Sidak's (two columns) multiple comparisons test. For all comparisons, a P value less than 0.05 was considered significant.
Results
The effect of the CD46 A82LPTFS87 substitution in MDBK cells
CRISPR/Cas9 gene editing by homology-directed repair was used to generate MDBK cells with the A83LPTFS88 substitution in the BVDV binding domain of CD46 (Fig. 1A and B). Alignment of folded bovine CD46 protein structures containing the wild-type G82QVLAL87 or the A82LPTFS87 substitution showed a predicted structural change limited to the A82L83P84 site (Fig. 1C); however, CD46 surface expression remained normal in edited cells (Fig. 1D and E). For comparison, MDBK cells were also generated with a 42-kb deletion encompassing the entire CD46 gene (CD46Δ). WGS analysis showed the A82LPTFS87 and the CD46Δ edits were accurate (Fig. S1), and off-target site modifications were not found in 125 potential genome sites when allowing for up to 4-bp mismatches with the gRNA. Both the CD46Δ and CD46 A82LPTFS87 cells showed a dramatic reduction in susceptibility to BVDV compared to the parent MDBK cell line, regardless of the BVDV strain type (Figs. 1f and g and S2). For example, infection efficiency was reduced by 95% on average (range 87–99%) across all virus isolates in the CD46-edited cells compared to MDBK at 20 hpi (Fig. 1F) and by 92% (range 73–99%) at 72 hpi (Fig. 1G). Conversely, infection efficiency was equivalent between CD46Δ and CD46 A82LPTFS87-edited cells for all but one BVDV strain tested (PI-85-2022, P = 0.03, Fig. 1G). Together, these data suggest that bovine CD46 with the A82LPTFS87 substitution may reduce BVDV infection similar to that of a CD46 gene deletion while allowing CD46 protein to be expressed normally at the cell surface.
Production of gene-edited CD46 A82LPTFS87 cloned calves
To test the impact of the CD46 A82LPTFS87 edit in vivo, the same strategy was used to make the edit in a primary Gir fibroblast cell line (TO470) for reproductive cloning (Fig. 2A). Three of eight embryos of each type (CD46-edited and unedited control) had established successful pregnancies 30 days after implantation. However, at 90 days, two of the unedited fetuses had been lost and resorbed. At 100 days gestation, one unedited and one CD46 A82LPTFS87-edited fetus were collected for primary cell isolation from lung, heart, small intestine, esophagus, kidney, and liver tissues. The two remaining CD46 A82LPTFS87-edited fetuses continued gestating to test for normal calf development. One edited CD46 A82LPTFS87 fetus was spontaneously aborted at 212 days gestation but had no obvious physical defects (Fig. S3). The last remaining CD46 A82LPTFS87-edited Gir calf was delivered by cesarean section at full term (285 days) and was born healthy on 2021 July 19 (Fig. S4). This calf has continued to thrive since and provided the first evidence that a CD46 A82LPTFS87 substitution may not negatively impact pre- or postnatal development through at least 20 months of age.
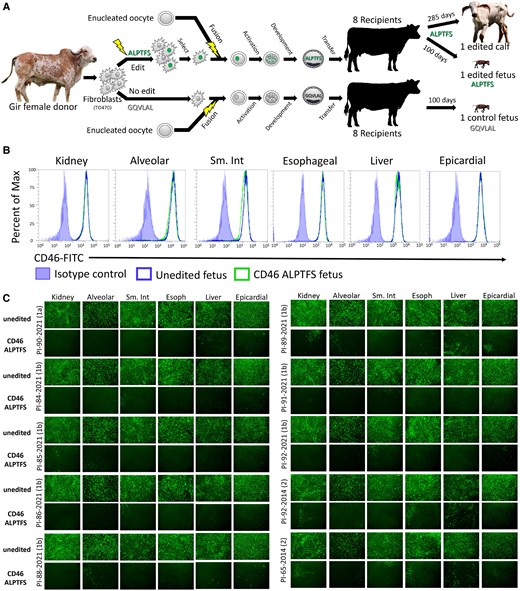
Reproductive cloning and BVDV susceptibility testing of primary cells from 100-day fetal tissues. A) Schematic representation of reproductive cloning. Primary skin fibroblasts were edited and subsequently fused to enucleated oocytes (somatic cell nuclear transfer) and the resultant embryos implanted into synchronized recipient cows. B) Flow cytometric quantification of CD46 surface expression. C) Cells were inoculated with serum (genotypes 1a and 1b) or low-passage virus isolates (genotype 2) from BVDV-PI calves. Infection efficiency was determined at 48 hpi using an anti-BVDV monoclonal antibody and FITC labeled secondary antibody. Nuclei were stained with DAPI to ensure images were taken in regions with complete cell monolayers (not shown). Cells imaged at 10× magnification. Sm. Int, small intestine; Esoph, esophageal.
Off-target editing not detected in CD46-edited calves
Analysis of WGS did not reveal any off-target edits in the edited CD46 A83LPTFS88 100-day fetus or live-born calf. Except for the intended A83LPTFS88 substitutions, the 42-kb region spanning CD46 was homozygous and identical in the donor Gir fibroblast line, the unedited fetus, the edited fetus, and the live-born calf (Fig. S5). The first heterozygous SNPs upstream and downstream of CD46 are at 42 and 48-kb, respectively (chr16:75,663,583 and chr16:75,572,857), and are also identical in all four samples. No off-target sites were found matching the gRNA in the bovine reference genome (ARS-UCD1.2) when zero, one, or two mismatches with zero bulge size was allowed. When 3 or 4 mismatches were allowed, 7 and 118 potential genomic sites were identified, respectively. Manual inspection of these 125 sites with IGV showed no genomic sequence differences between the parental Gir fibroblast, the unedited fetus, the CD46-edited fetus, and the live-born calf. Also, systematic GATK software analysis of WGS data did not detect any significant off-target insertions or deletions in the genome sequences that were unique to the live-born edited calf. Thus, phenotypic differences observed in BVDV susceptibility between unedited and edited calves were not readily attributable to off-target site modifications.
Reduced BVDV susceptibility in CD46-edited fetal tissues
The CD46 A83LPTFS88 substitution had a significant impact on reducing BVDV susceptibility in primary cells from all tissues from the 100-day fetus. The CD46 A83LPTFS88 substitution did not appreciably alter the surface expression levels of CD46 when measured in primary cultures by flow cytometry (Fig. 2B). Yet, the primary cells from all tissue types with the A83LPTFS88 substitution had a significant reduction in BVDV-infected cells (as visualized by IF) compared to cells from the unedited clone, regardless of BVDV strain type (Fig. 2C). Based on these results, multiple tissues/organs in a CD46 A83LPTFS88-edited calf were expected to have reduced BVDV susceptibility.
Ex vivo challenge of cells from the CD46-edited Gir calf
Three available and relevant cell types were collected from the live-born CD46-edited calf for ex vivo challenge: skin fibroblasts, peripheral blood lymphocytes, and monocytes. For comparison, a healthy 1-day old Holstein calf was purchased and housed with the CD46-edited Gir calf to serve as an unedited sex- and age-matched control, since no unedited Gir calves survived to term (Fig. 3A). CD46 protein localization (Fig. 3B) and expression (Fig. 3C) were unaltered in primary skin fibroblasts from the CD46-edited calf, yet infection efficiency was reduced by 96% on average (range 88–99%) as quantified by flow cytometry (Fig. 3D) and visualized by IF (Fig. 3E). Peripheral blood cells can also be infected by BVDV, and these cells are thought to disseminate virus throughout the host. Lymphocytes from the CD46-edited calf had on average a 96% reduction (range 84–100%) in BVDV susceptibility as quantified by flow cytometry (Fig. 3F). For all 13 BVDV isolates tested in unedited (wild-type) lymphocytes, the fraction of infected cells was significantly greater than the uninfected controls (P < 0.05). However, for the same isolates tested in CD46-edited lymphocytes, the fraction of infected cells was not statistically different from the uninfected controls (P > 0.05; Fig. 3F). Similarly, monocytes from the CD46-edited calf had a dramatic reduction in susceptibility to BVDV as quantified by RT-qPCR, with an average 163-fold reduction (range 7 to 446-fold) in viral RNA accumulation (Fig. 3G). Thus, each of the three cell types tested from the CD46-edited calf displayed a significant reduction in BVDV susceptibility ex vivo and suggested that the edited calf might resist infection in a natural exposure challenge experiment.
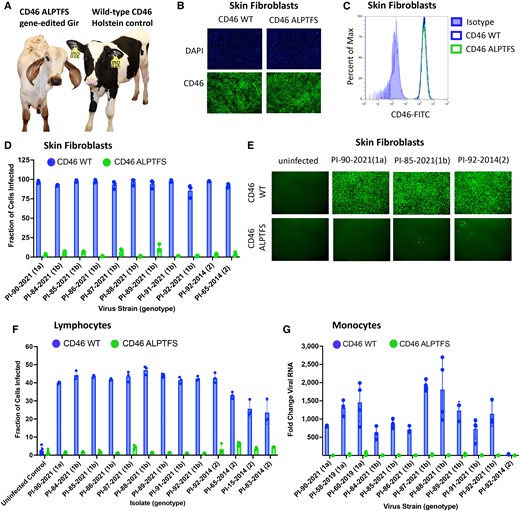
Ex vivo challenge of fibroblasts, monocytes, and lymphocytes from CD46 A82LPTFS87-edited calf. A) Picture of study participants. B) Immunofluorescence staining of CD46 (FITC) and nuclei were stained with DAPI . C) Flow cytometric quantification of CD46 protein expression levels. D) Cells were inoculated with serum from BVDV-PI calves, and infection efficiency was quantified at 72 hpi by flow cytometry using an anti-BVDV E2 antibody. Results represent the mean±standard deviation of n = 3 independent experiments. E) BVDV infection was visualized for a representative set of samples from panel D by IF using an anti-BVDV antibody and FITC labeled secondary antibody (10× magnification). F) Flow cytometric quantification of BVDV-infected cells at 24 hpi. Results represent the mean±SD of n = 3 independent experiments. G) Monocytes were inoculated with serum from BVDV-PI calves (genotypes 1a and 1b) or a low-passage BVDV-2 isolate at an MOI of 0.01 (PI-92-2014). Viral RNA was detected by RT-qPCR at 48 hpi, and fold change in viral RNA relative to the input sample (0 hpi) was calculated. Results represent the mean±SD of n ≥ 3 independent experiments. WT, wild-type.
Natural challenge of a CD46-edited Gir calf by exposure to a BVDV-PI calf
At 10 months of age, the CD46 A83LPTFS88-edited Gir calf and the wild-type CD46 control Holstein were co-housed with a week-old Holstein calf naturally and persistently infected with BVDV (genotype 1b, Fig. S6). Both calves received significant BVDV exposure from the BVDV-PI calf during the 7 days of cohabitation as measured by virus detection in nasal and fecal swabs (Fig. 4A and B). Importantly, swabs of the nasal passages on challenge days 2 through 7 revealed viral RNA loads were equivalent or higher in the CD46-edited calf compared to those in the control calf, suggesting similar exposure levels. Both calves also developed a fever (Fig. 4C) and had a reduction in circulating white blood cells (Table S5), but only the wild-type CD46 calf displayed other signs of infection, including a cough, rhinitis, and redness and chafing around the nostrils (Table S4). Concurrent with clinical signs of infection, BVDV viremia, as measured by RT-qPCR of whole blood, was detected in the wild-type calf and lasted 28 days. Conversely, BVDV RNA was only detected in the whole blood of the CD46-edited calf for 3 days, and the peak viral RNA load was reduced ∼2-log (Fig. 4B). Furthermore, PBMCs isolated from the CD46-edited calf remained uninfected throughout the challenge, whereas PBMCs from the wild-type CD46 calf were BVDV RNA-positive for 12 days. While low levels of cell-free BVDV RNA could be detected in the serum of both calves (Fig. 4B), infectious virus could not be isolated from the serum when inoculated on MDBK cells and blindly passaged three times. In contrast, replication-competent virus was easily isolated from the serum of the BVDV-PI calf (Fig. S6). Both calves generated neutralizing antibodies to BVDV, with a peak antibody titer of 1:256 and 1:64 for the wild-type and CD46-edited calf, respectively (Fig. 4D). This result indicates that the CD46 A83LPTFS88-edited Gir calf's immune system was competent to respond to the viral challenge. Together, these results suggest that in the face of overwhelming natural BVDV exposure, the CD46 A83LPTFS88-edited Gir calf displayed reduced susceptibility to BVDV, which resulted in no observable adverse health effects on the animal.
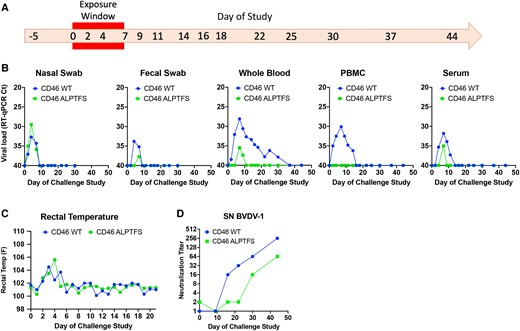
Natural exposure challenge study of CD46 A82LPTFS87-edited calf. A) Timeline of challenge study. The CD46-edited Gir calf and the unedited wild-type (WT) control Holstein calf were challenged with BVDV by cohabitation with a BVDV-PI calf for 7 days (exposure window). Nasal and fecal samples were collected on days −5 to 30. Blood was collected on days −5 to 44. B) BVDV RT-qPCR. Samples were extracted in duplicate and run on the same PCR plate. Samples that tested negative for BVDV RNA are plotted having a cycle threshold (Ct) equal to 40. C) Rectal temperatures taken daily through day 21. D) BVDV-1 serum neutralization (SN) titers.
Discussion
The present report describes the use of CRISPR/Cas9 technology to make an A82LPTFS87 substitution in the BVDV binding domain of CD46. In stepwise experiments, we showed this edit dramatically reduced BVDV susceptibility in MDBK cells, primary cells from multiple organs obtained from a CD46-edited fetus, fibroblast and immune cell populations from a live CD46-edited calf, and in a natural exposure challenge study with the same edited calf. Together, these results provide proof-of-concept for using intentional genome alterations in CD46 to reduce the burden of BVDV-associated diseases in cattle. This represents the first demonstration of editing a bovine gene to reduce susceptibility to a viral pathogen.
The substitution of six amino acids in CD46 is unique compared to previous reports of gene-edited cattle with reduced susceptibility to bacterial pathogens. For example, exogenous whole gene-knockin strategies have been used in cattle to reduce mastitis (Staphylococcus aureus) (40, 41) and bovine tuberculosis (Mycoplasma bovis) (42–44). In another example, a single amino acid was changed in bovine CD18 (ITGB2), causing the normally retained signal peptide to be cleaved (45). Although leukocytes isolated from a ITGB2-edited fetus were resistant to Manheimia haemolytica leukotoxin when challenged ex vivo, live calves were not reported. Here, homology-directed repair was used to make a precise 18-nt replacement in the endogenous CD46 gene. Subsequently, somatic cell nuclear transfer was used to produce an edited calf in a single generation without introducing any off-target modifications. Importantly, the edit did not disrupt the predicted tertiary structure of the protein or its expression levels, and the live CD46-edited calf has no obvious adverse effects from the on-target edit in the first 20 months after birth.
This study had four important limitations. First, only one live CD46-edited animal could be tested for altered BVDV susceptibility. Second, a live unedited wild-type CD46 clone was not available to serve as a matched control. Third, a breed-matched control was not available since Gir are uncommon in the United States. Although differences in breed-associated BVDV susceptibility have not been reported, ideally, comparisons would be made between clones that were identical in genomic sequence except for the 18-nt substitution in CD46. Fourth, our in vivo challenge study design did not allow direct determination of viral entry and replication in various tissues of the edited calf, since it would have required sacrificing the only available CD46-edited animal. Although nasal and fecal swab samples were collected to detect potential amplification of virus in the upper respiratory and gastrointestinal tracts, samples from both the CD46-edited and control calves were only positive for BVDV on the days they were co-housed with the BVDV-PI calf. Thus, these positive samples could be from environmental exposure and not active viral replication. As such, questions remain about the susceptibility of various tissues in vivo. Future studies should include additional animals from the most common beef and dairy breeds to help address these limitations.
Despite the limitations, the concordance of results between the in vitro and ex vivo BVDV susceptibility studies with the in vivo challenge study suggests that this edit could be useful for reducing BVDV susceptibility in cattle. Fetal kidney, lung, small intestine, esophagus, liver, and heart cells all had significantly reduced susceptibility to BVDV when infected ex vivo. Consistent with this result, primary skin fibroblasts, lymphocytes, and monocytes from the live CD46-edited calf had dramatically reduced BVDV susceptibility. Of note, the edit reduced susceptibility to all BVDV isolates tested, including cytopathic and non-cytopathic isolates belonging to the two genotypes of BVDV that infect cattle globally. This component of our study was the first to measure CD46 dependence in diverse cell types and provides new insights into CD46 receptor usage. Although the in vivo challenge study design did not directly determine viral replication in individual organs and tissues of the edited calf, the strong reduction in both the duration and peak viral RNA load in the blood of the CD46-edited calf was consistent with reduced viral replication in tissues in vivo. This coincides with PBMC results from the CD46-edited calf remaining uninfected during the entire challenge study. Thus, although questions remain about viral replication in vivo, our data suggest a profound decrease in viral susceptibility that limits the amount of virus in the blood. Although untested, we hypothesize that this reduced viremia would limit the dissemination of virus to other target organs.
Reducing viremia in acute infections may significantly impact BVDV control efforts. The most devastating outcome of transient infection in pregnant animals is viremia leading to transplacental infection of the developing fetus. Fetal infections can result in abortion, congenital malformations, or worse, the birth of BVDV-PI calves. These calves have lifelong viremia and continuously shed virus in all bodily secretions, making them the most important source of virus spread in the population and the target of national eradication programs (46). In addition, the immunosuppressive effects of BVDV during an acute infection increase the incidence of secondary infection and disease (47). For example, BVDV is an important cofactor of bovine respiratory disease complex (48, 49), one of the most economically important infectious diseases affecting the cattle industry. Therefore, reducing transplacental BVDV infections could significantly improve animal health and welfare, reduce the burden of BVDV infections on the industry, and provide a significant opportunity to reduce antibiotic use in agriculture. Accordingly, it will be important for future studies to determine whether CD46-edited dams are able to protect their developing fetuses from transplacental BVDV infection.
Conclusion
Stepwise experiments showed that substituting A82LPTFS87 in CD46 dramatically reduced BVDV susceptibility in vitro, ex vivo, and in a natural challenge study with one edited calf. The results provide proof-of-concept for using intentional genome alterations in CD46 to reduce the burden of BVDV-associated diseases in cattle. However, determining the ability of CD46-edited animals to withstand BVDV viral challenges in vivo will require experimental replication in other breeds and with more animals.
Acknowledgments
We thank Susan Hauver and the USMARC Core Facility for technical support, the University of Nebraska animal care staff, and Janel Nierman for secretarial and administrative support. Mention of trade names or commercial products in this publication is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the US Department of Agriculture. The USDA is an equal opportunity provider and employer. This manuscript was posted on a preprint server: https://doi.org/10.1101/2022.12.08.519336.
Supplementary material
Supplementary material is available at PNAS Nexus online.
Funding
Funding for this research was provided by the USDA, ARS appropriated project 3040-32000-034-00D (A.W. and M.H.), Recombinetics, Inc., and the University of Nebraska–Lincoln School of Veterinary Medicine and Biomedical Sciences/Great Plains Veterinary Education Center (B.V.L.) and the Nebraska Beef Industry Endowment (B.V.L.). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author contributions
Conceptualization, A.W., M. H., and T.S.; data curation, A.W., M.H., and T.K.; formal analysis, A.W., D.W., D.C., L.S., M.H., and T.K.; funding acquisition, A.W., M.H, and T.S.; investigation, A.W., B.V.L., D.W., E.J., J.B., L.S., and M.H.; methodology, A.W., B.V.L., D.W., D.C., G.H., J.B., L.S., M.H., and T.K.; project administration, A.W., D.C., M.H., S.L., and T.S.; resources, B.V.L., E.J., G.H., and T.K.; software, G.H. and T.K; supervision, A.W., B.V.L., D.C., M.H., and T.S.; validation, A.W., D.C., and M.H.; visualization, A.W. and M.H.; writing—original draft preparation, A.W. and M.H; and writing—review and editing, all authors reviewed and approved the final draft.
Data availability
Raw WGS files (fastq) for the MDBK and TO470 cell lines and their edited clones are available in the NCBI SRA under accession number SRR21834845-50. The sequence data have also been deposited with links to BioProject accession number PRJNA887820 (BioSamples SAMN31186839-44) in the NCBI BioProject database.
In addition, IGV access to the aligned sequences (bam files and IGV session files) is available:
MDBK CD46 gene deletion: https://s3.us-west-2.amazonaws.com/usmarc.heaton.public/WGS/CellLines/ARS1.2/bams/LIB102516_BovMDBK_CD46KO3.Bt_ARS-UCD1.2.realigned.bam
MDBK CD46 ALPTFS substitution: https://s3.us-west-2.amazonaws.com/usmarc.heaton.public/WGS/CellLines/ARS1.2/bams/LIB109073_BovMDBK_CD46Por6AA_296.Bt_ARS-UCD1.2.realigned.bam
MDBK IGV Session URL: https://s3.us-west-2.amazonaws.com/usmarc.heaton.public/WGS/CellLines/ARS1.2/sessions/ARS12._MDBK_CD46_3tracks.xml
Gir cell line (TO470): https://s3.us-west-2.amazonaws.com/usmarc.heaton.public/WGS/CellLines/ARS1.2/bams/LIB108537_TO470CellLine.Bt_ARS-UCD1.2.realigned.bam
Unedited fetus: https://s3.us-west-2.amazonaws.com/usmarc.heaton.public/WGS/CellLines/ARS1.2/bams/LIB113017_TO470FetalCalfSkin.Bt_ARS-UCD1.2.realigned.bam
CD46 ALPTFS edited fetus: https://s3.us-west-2.amazonaws.com/usmarc.heaton.public/WGS/CellLines/ARS1.2/bams/LIB113018_TO470_CD46_6AAFetalCalfSkin.Bt_ARS-UCD1.2.realigned.bam
CD46 ALPTFS edited live calf: https://s3.us-west-2.amazonaws.com/usmarc.heaton.public/WGS/CellLines/ARS1.2/bams/LIB110781_Ginger.Bt_ARS-UCD1.2.realigned.bam
Gir clone IGV Session URL: https://s3.us-west-2.amazonaws.com/usmarc.heaton.public/WGS/CellLines/ARS1.2/sessions/ARS1.2_CD46_Ginger4Tracks.xml
CD46-edited MDBK cells will be made available upon request (Contact: Dan Carlson at dan@recombinetics.com). Primary cells from fetal tissues will not be made available due to very limited quantities.
References
Author notes
Competing interest: D.W., J.B., and D.C. are full-time employees of Recombinetics, Inc.; S.L. and T.S. are employees of Acceligen, a wholly owned subsidiary of Recombinetics, Inc. Recombinetics, Inc., is a company that commercializes animal gene editing and associated applied technologies for biomedical research, regenerative medicine, and animal agriculture. There are no patents to declare, and the interests do not alter the authors’ adherence to all the journal's policies on sharing data and materials published herein.

{kind=link}
{kind=link}
{kind=link}
{kind=link}