





Abstract
Children ≤36 months with diffuse intrinsic pontine glioma (DIPG) have increased long-term survival (LTS, overall survival (OS) ≥24 months). Understanding distinguishing characteristics in this population is critical to improving outcomes.
Patients ≤36 months at diagnosis enrolled on the International DIPG Registry (IDIPGR) with central imaging confirmation were included. Presentation, clinical course, imaging, pathology and molecular findings were analyzed.
Among 1183 patients in IDIPGR, 40 were eligible (median age: 29 months). Median OS was 15 months. Twelve patients (30%) were LTS, 3 (7.5%) very long-term survivors ≥5 years. Among 8 untreated patients, median OS was 2 months. Patients enrolled in the registry but excluded from our study by central radiology review or tissue diagnosis had median OS of 7 months. All but 1 LTS received radiation. Among 32 treated patients, 1-, 2-, 3-, and 5-year OS rates were 68.8%, 31.2%, 15.6% and 12.5%, respectively. LTS had longer duration of presenting symptoms (P = .018). No imaging features were predictive of outcome. Tissue and genomic data were available in 18 (45%) and 10 patients, respectively. Among 9 with known H3K27M status, 6 had a mutation.
Children ≤36 months demonstrated significantly more LTS, with an improved median OS of 15 months; 92% of LTS received radiation. Median OS in untreated children was 2 months, compared to 17 months for treated children. LTS had longer duration of symptoms. Excluded patients demonstrated a lower OS, contradicting the hypothesis that children ≤36 months with DIPG show improved outcomes due to misdiagnosis.
Children ≤36 months with DIPG have improved OS of 15 months; 2-year OS of 26%
Treatment in this population further improves OS; untreated patient OS was 2 months
Longer symptom duration in LTS; H3K27M status was known for 9 patients, present in 6
Children ≤36 months with DIPG have reported improved overall survival and increased long-term survival, though the reasons for this are poorly understood. Herein we examine the largest cohort of children ≤36 months of age at diagnosis to date, 40 children with central radiological confirmation of DIPG, evaluating clinical course, imaging characteristics and genomic information. Improved median overall survival of 15 months is confirmed, with 30% LTS, as well as improved outcomes at 2-, 3- and 5-years. It has been hypothesized that younger children with DIPG show improved outcomes secondary to misdiagnosis. However, the 14 children excluded from our study upon radiology review or tissue diagnosis actually showed a shorter median OS of 7 months, in contrast to the previously reported idea that this population has improved outcomes secondary to misdiagnosis. Longer duration of symptoms was a good prognosticator of OS. H3K27M status was known in nine patients, present in only six.
Diffuse intrinsic pontine glioma (DIPG) is a devastating pediatric brainstem malignancy, responsible for more deaths among children than any other brain tumor.1 In spite of more than 250 clinical trials aimed at improving outcomes over the past forty years, median survival remains <1 year.2–4 However, children with DIPG who are ≤36 months at diagnosis have a higher rate of long-term survival (LTS), traditionally defined as an overall survival (OS) ≥24 months.2,5–8 While characteristics of LTS have been reported, including longer duration of symptoms prior to diagnosis, the absence of cranial nerve palsies, and a lack of ring enhancement on magnetic resonance imaging (MRI) at diagnosis,2,5,7,8 these factors have not been analyzed specifically within this younger population.
In light of reported improved overall survival, a prevailing hypothesis is that patients diagnosed with DIPG at ≤36 months of age represent a distinct subtype of disease, or perhaps an entirely different diagnosis.8 Analyzing patients enrolled on the IDIPGR, an international collaborative effort initiated in 2012 to share information and advance understanding of DIPG across 115 institutions from 15 countries worldwide, we aimed to gain a greater understanding of this critical cohort. We centrally reviewed diagnostic imaging of younger patients to assess any radiologic characteristics that define this population and examined their clinical courses. The aim of this study was to define the clinical, radiologic, histologic and molecular characteristics of this population, and examine their significance in predicting outcomes for these patients.
Materials & Methods
Patient Population
This study was reviewed and approved by the Institutional Review Board at Cincinnati Children’s Hospital Medical Center. Our study population was extracted from the IDIPGR. These patients were referred to the IDIPGR as previously described.9 From 1990–2018, 59 patients ≤36 months of age at diagnosis with radiographic diagnosis of DIPG were reviewed. DIPG was defined as a tumor centered in the brainstem occupying ≥50% of the pons with features consistent with DIPG,10 as previously described. No patients with neurofibromatosis type 1 (NF1) were included in the study. Further exclusion criteria are outlined in Figure 1. Demographics of patients who met inclusion criteria for our analyzed cohort are outlined in Table 1.
Univariate analysis of clinical characteristics seen in LTS and STS patients with DIPG ≤36 months. All available data is presented, with some pieces of clinical information unavailable for some patients. Symptom duration is the only variable of statistical significance, with STS more likely to present with shorter symptom duration
| Clinical variables, n(%) | LTS (n = 12) | STS (n = 28) | P-value |
|---|---|---|---|
| Sex | 0.74 | ||
| Male | 5 (42%) | 10 (36%) | |
| Female | 7 (58%) | 18 (64%) | |
| Age (mo.) | 0.81 | ||
| Median (IQR) | 29 (23–34) | 29 (19–35) | |
| Race | 0.22 | ||
| African | 0 (0%) | 5 (25%) | |
| Asian | 0 (0%) | 1 (5%) | |
| Caucasian | 7 (78%) | 13 (65%) | |
| Other | 2 (22%) | 1 (5%) | |
| Symptom duration | 0.018 | ||
| <6 weeks | 4 (33%) | 20 (77%) | |
| 6–12 weeks | 4 (33%) | 3 (12%) | |
| 12–24 weeks | 2 (17%) | 3 (12%) | |
| >24 weeks | 2 (17%) | 0 (0%) | |
| Palsy | 0.42 | ||
| Y | 5 (56%) | 13 (72%) | |
| N | 4 (44%) | 5 (28%) | |
| Pyramidal sign | 0.41 | ||
| Y | 2 (25%) | 9 (47%) | |
| N | 6 (75%) | 10 (53%) | |
| Cerebellar sign | 1.00 | ||
| Y | 4 (50%) | 11 (58%) | |
| N | 4 (50%) | 8 (42%) | |
| Spinal metastasis | 1.00 | ||
| Y | 0 (0%) | 8 (89%) | |
| N | 5 (100%) | 1 (11%) | |
| Shunt placement | 0.24 | ||
| Y | 2 (22%) | 13 (50%) | |
| N | 7 (78%) | 13 (50%) |
| Clinical variables, n(%) | LTS (n = 12) | STS (n = 28) | P-value |
|---|---|---|---|
| Sex | 0.74 | ||
| Male | 5 (42%) | 10 (36%) | |
| Female | 7 (58%) | 18 (64%) | |
| Age (mo.) | 0.81 | ||
| Median (IQR) | 29 (23–34) | 29 (19–35) | |
| Race | 0.22 | ||
| African | 0 (0%) | 5 (25%) | |
| Asian | 0 (0%) | 1 (5%) | |
| Caucasian | 7 (78%) | 13 (65%) | |
| Other | 2 (22%) | 1 (5%) | |
| Symptom duration | 0.018 | ||
| <6 weeks | 4 (33%) | 20 (77%) | |
| 6–12 weeks | 4 (33%) | 3 (12%) | |
| 12–24 weeks | 2 (17%) | 3 (12%) | |
| >24 weeks | 2 (17%) | 0 (0%) | |
| Palsy | 0.42 | ||
| Y | 5 (56%) | 13 (72%) | |
| N | 4 (44%) | 5 (28%) | |
| Pyramidal sign | 0.41 | ||
| Y | 2 (25%) | 9 (47%) | |
| N | 6 (75%) | 10 (53%) | |
| Cerebellar sign | 1.00 | ||
| Y | 4 (50%) | 11 (58%) | |
| N | 4 (50%) | 8 (42%) | |
| Spinal metastasis | 1.00 | ||
| Y | 0 (0%) | 8 (89%) | |
| N | 5 (100%) | 1 (11%) | |
| Shunt placement | 0.24 | ||
| Y | 2 (22%) | 13 (50%) | |
| N | 7 (78%) | 13 (50%) |
Univariate analysis of clinical characteristics seen in LTS and STS patients with DIPG ≤36 months. All available data is presented, with some pieces of clinical information unavailable for some patients. Symptom duration is the only variable of statistical significance, with STS more likely to present with shorter symptom duration
| Clinical variables, n(%) | LTS (n = 12) | STS (n = 28) | P-value |
|---|---|---|---|
| Sex | 0.74 | ||
| Male | 5 (42%) | 10 (36%) | |
| Female | 7 (58%) | 18 (64%) | |
| Age (mo.) | 0.81 | ||
| Median (IQR) | 29 (23–34) | 29 (19–35) | |
| Race | 0.22 | ||
| African | 0 (0%) | 5 (25%) | |
| Asian | 0 (0%) | 1 (5%) | |
| Caucasian | 7 (78%) | 13 (65%) | |
| Other | 2 (22%) | 1 (5%) | |
| Symptom duration | 0.018 | ||
| <6 weeks | 4 (33%) | 20 (77%) | |
| 6–12 weeks | 4 (33%) | 3 (12%) | |
| 12–24 weeks | 2 (17%) | 3 (12%) | |
| >24 weeks | 2 (17%) | 0 (0%) | |
| Palsy | 0.42 | ||
| Y | 5 (56%) | 13 (72%) | |
| N | 4 (44%) | 5 (28%) | |
| Pyramidal sign | 0.41 | ||
| Y | 2 (25%) | 9 (47%) | |
| N | 6 (75%) | 10 (53%) | |
| Cerebellar sign | 1.00 | ||
| Y | 4 (50%) | 11 (58%) | |
| N | 4 (50%) | 8 (42%) | |
| Spinal metastasis | 1.00 | ||
| Y | 0 (0%) | 8 (89%) | |
| N | 5 (100%) | 1 (11%) | |
| Shunt placement | 0.24 | ||
| Y | 2 (22%) | 13 (50%) | |
| N | 7 (78%) | 13 (50%) |
| Clinical variables, n(%) | LTS (n = 12) | STS (n = 28) | P-value |
|---|---|---|---|
| Sex | 0.74 | ||
| Male | 5 (42%) | 10 (36%) | |
| Female | 7 (58%) | 18 (64%) | |
| Age (mo.) | 0.81 | ||
| Median (IQR) | 29 (23–34) | 29 (19–35) | |
| Race | 0.22 | ||
| African | 0 (0%) | 5 (25%) | |
| Asian | 0 (0%) | 1 (5%) | |
| Caucasian | 7 (78%) | 13 (65%) | |
| Other | 2 (22%) | 1 (5%) | |
| Symptom duration | 0.018 | ||
| <6 weeks | 4 (33%) | 20 (77%) | |
| 6–12 weeks | 4 (33%) | 3 (12%) | |
| 12–24 weeks | 2 (17%) | 3 (12%) | |
| >24 weeks | 2 (17%) | 0 (0%) | |
| Palsy | 0.42 | ||
| Y | 5 (56%) | 13 (72%) | |
| N | 4 (44%) | 5 (28%) | |
| Pyramidal sign | 0.41 | ||
| Y | 2 (25%) | 9 (47%) | |
| N | 6 (75%) | 10 (53%) | |
| Cerebellar sign | 1.00 | ||
| Y | 4 (50%) | 11 (58%) | |
| N | 4 (50%) | 8 (42%) | |
| Spinal metastasis | 1.00 | ||
| Y | 0 (0%) | 8 (89%) | |
| N | 5 (100%) | 1 (11%) | |
| Shunt placement | 0.24 | ||
| Y | 2 (22%) | 13 (50%) | |
| N | 7 (78%) | 13 (50%) |
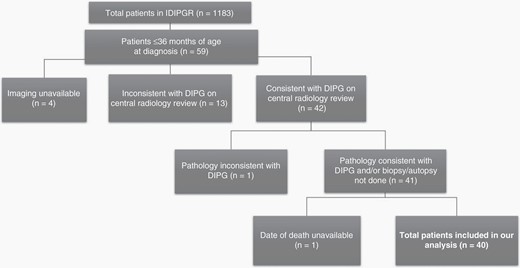
Description of included/excluded patients on the basis of our study criteria, demonstrating a total of 40 patients in our analysis.
Clinical Parameters
Clinical data were abstracted from patient medical records utilizing standardized case report forms.9 Each patient chart was reviewed for the presence of cranial nerve palsies, cerebellar signs, and pyramidal signs at presentation. Cerebellar signs included dysmetria, ataxia, dysarthria and nystagmus. Pyramidal signs included any evidence of paresis, hyperreflexia or a positive Babinski sign. Overall survival (OS) was defined as the time from diagnosis to death, or last available follow-up. Patients were further classified as short-term survivors (STSs), long-term survivors (LTSs) or very long-term survivors (VLTSs), with OS times of <24 months, ≥24 months, and ≥60 months, respectively.
Radiology Review
Diagnostic magnetic resonance images (MRI) were anonymized and centrally reviewed as recently described.10 Imaging was then classified into one of three categories based on appearance: typical DIPG, atypical features but likely DIPG, or DIPG diagnosis in question. Those in question were excluded from the study. Primary exclusionary criteria on review included involvement of <50% of the pons, focally exophytic morphology, marked diffusion restriction, or secondary brainstem involvement by a tumor centered elsewhere in the brain or spine.10
Histologic and Molecular Variables
Available tumor specimens were centrally reviewed. Histology was defined according to 2016 WHO criteria, with the exception of cases where tissue was obtained prior to implementation of these guidelines and not available for central review.11,12 Genomic mutations were assessed by whole-exome sequencing, whole-genome sequencing, Sanger sequencing, limited genomics panel or mutant-specific immunohistochemistry (H3 K27M). Mutations in H3F3A (H3.3 K27M) or HIST1H3B (H3.1 K27M) were considered mutually exclusive, with the presence of either mutation eliminating the possibility of the other mutation, even if both were not evaluated.
Statistical Analysis
Patient characteristics were analyzed and summarized using medians and ranges or frequencies and percentages. Univariable analyses were performed using the Fisher exact test or Wilcoxon rank sum test. Survival was estimated using the Kaplan-Meier method. Statistical evaluation was performed using R (Version 3.1.3). P < .05 was considered significant.
Results
Clinical Presentation
Among 1183 patients enrolled on the IDIPGR, 40 patients met inclusion criteria, as outlined in Figure 1. Presentation varied among 27 patients with available clinical data, 67% had one or more cranial nerve (CN) palsy, 56% showed cerebellar signs and 41% demonstrated pyramidal tract deficiencies. The heterogeneity observed in presentation did not prove statistically significant in predicting LTS within this population. The majority of patients, 24 of 40 (60%), were diagnosed <6 weeks from symptom onset, while some demonstrated longer symptom duration of 6–12 weeks (18%), 12–24 weeks (13%) and >24 weeks (5%). On univariate analysis, LTS were more likely to demonstrate a longer duration of symptoms (p = .018). Additional analyses comparing clinical presentation between LTSs and STSs did not reveal statistically significant parameters predictive of survival (Table 1).
Survival
For 40 patients who met inclusion criteria, median survival time was 15 months (IQR: 7 to 32 months). Median survival time amongst those who received treatment (n = 32) was 17 months (Range: 2–135 months, IQR: 11–33 months). Within this cohort of treated patients, 31% were LTSs, and 1-, 2-, 3-, and 5-year OS rates were 68.8%, 31.2%, 15.6% and 12.5%, respectively. Of the 8 patients (20%) who did not receive any radiation therapy or chemotherapy, median survival time was only 2 months (Range: 1–45 months, IQR 1–11 months), including two LTSs. Characteristics of LTSs from each group are outlined in Figure 2. Kaplan-Meier survival analyses for treated patients, untreated patients and excluded patients are shown in Figure 3.
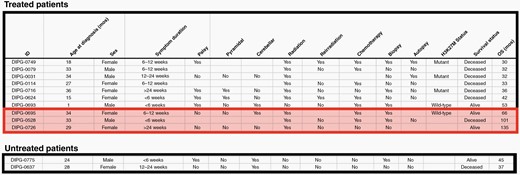
Characteristics of the 12 long-term survivors in our cohort, divided by those who received treatment, and untreated long-term survivors. Very long-term survivors highlighted.
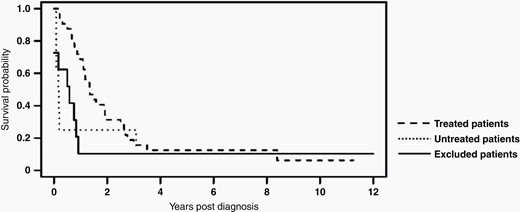
Kaplan-Meier survival analysis is shown for the 32 patients in our cohort who received treatment (Median OS: 17 mos, IQR: 11-33 mos), the 8 patients in our cohort who did not receive any radiation or systemic chemotherapy (Median OS: 2 mos, IQR: 1-11 mos), and the 14 patients excluded on central radiology and/or pathology review (Median OS: 7 mos, IQR: 1-10 mos).
Therapy
For the 31 treated patients with all available therapy data, 21 (54%) received radiation therapy (RT) and systemic therapy, 8 patients (21%) were treated with RT alone, and 2 patients (5%) with systemic therapy only. Distribution of types of therapy administered, comparing LTSs and STSs, did not reveal significant differences nor yield a survival advantage on univariate analysis (Figure 4). All irradiated patients received focal, photon beam radiation, both upfront and at progression. Radiation dose ranged from 4–60 Gray (Gy), with the majority of irradiated patients receiving 54 Gy as their initial treatment; four patients were re-irradiated at progression. All but one treated LTS received RT. The type of systemic therapy used was reported for 19 of 23 patients and can be found in Figure 5. Twelve patients received systemic therapy at progression; 11 of these patients were previously irradiated. Eight patients, including 3 LTSs, did not pursue any additional therapy at progression.
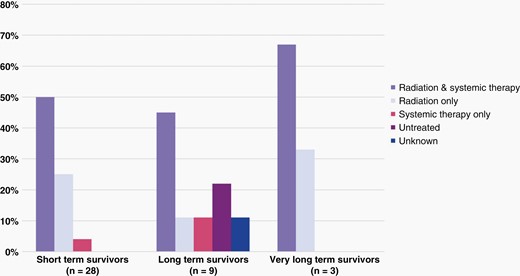
Distribution of various treatment strategies is shown for (a) short term survivors (b) long-term survivors and (c) very long-term survivors.
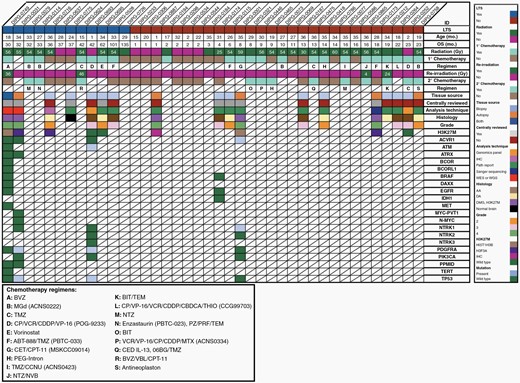
Clinical, molecular, and genomic data in DIPG patients ≤ 36 months of age at diagnosis with available tissue from biopsy and/or autopsy. Radiation dosing and chemotherapy regimens provided as clinically available. Abbreviations: 06BG: MGMT inhibitor; ABT-888: veliparib; BIT: bevacizumab/irinotecan/temozolomide; BVZ: bevacizumab; CBDCA: carboplatin; CCNU: lomustine; CDDP: cisplatin; CED: convection enhanced delivery: CET: cetuximab; CP: cyclophosphamide; CPT-11: irinotecan; MGd: motexafin-gadolinium; MTX: methotrexate; NTZ: nimotuzumab; NVB: vinorelbine; PRF: perifosine; PZ: pazopanib; TEM: temsirolimus; THIO: thiotepa; TMZ: temozolomide; VBL: vinblastine; VCR: vincristine; VP-16: etoposide.
Eight patients of 40 (20%) did not receive any therapy following diagnosis. These untreated patients were analyzed separately from those who received medical intervention. Median OS was 2 months (range: 1–45 months), with two LTSs.
Imaging
All diagnostic imaging was centrally reviewed for confirmation of radiologic diagnosis. Images were characterized as typical DIPG (n = 26, 65%) or DIPG with atypical features (n = 14, 35%). Atypical features were described individually and included eccentric location, low positioning in the pons, portions that appeared exophytic, well-defined margin along the entire tumor and diffusion restriction. Most patients with typical features on diagnostic imaging were STS (73%), while 7 of these patients were LTS (27%). The same was true for patients with atypical features on imaging, 9 patients (64%) with atypical features were STS, while five patients (36%) were LTS.
MRIs were subsequently analyzed for characteristics associated with DIPG, some of which have been previously associated with improved outcomes.10 Each parameter was compared between LTSs and STSs, evaluating for clinical significance. The median measurements of the tumor in anteroposterior (AP), transverse and craniocaudal (CC) dimensions were comparable between LTSs and STSs (P = .17, P = .57, P = .28, respectively). The percentage of the pons involved did not vary between the two groups, with most patients showing 67–100% involvement (P = .22). Few tumors had well-defined margins (n = 4, 11%), and hemorrhage was very uncommon, with only one patient showing more than minimal hemorrhage at diagnosis. Similarly, necrosis at diagnosis was rare, with only 2 patients (6%) with 34–66% necrosis on their initial scan, both STSs. No patients demonstrated >66% necrosis at diagnosis, and the extent of necrosis at this timepoint was not predictive of survival (P = 1.0). Extent of enhancement, presence of diffusion restriction, intensity of T1 and T2 FLAIR signal proved similar between LTSs and STSs. While we observed some atypical features on central review, none of the parameters analyzed proved to be statistically significant in predicting improved outcome. Individual analyses of each diagnostic imaging characteristic are listed in Supplemental Table 1.
Histology and Molecular
Of the 40 patients who met inclusion criteria, 13 patients (33%) underwent biopsy, and 6 (17%) autopsy. Of these patients with tissue samples collected (45%), one had both biopsy and autopsy performed. Nine tissue samples (47%) of the nineteen collected in this cohort, were available for central pathology review. Amongst the 13 patients with biopsies, specimens included anaplastic astrocytoma (AA; grade III, n = 6), diffuse astrocytoma (DA; grade II, n = 4; grade unknown, n = 1), diffuse midline glioma, H3K27M (DMG; grade IV, n = 1), and one with only normal brain tissue to analyze. Histology from autopsy tissue of patients included in our analysis revealed DMG, H3K27M (grade IV, n = 5) and DA (grade II, n = 1). The eligible patient with both specimens had DMG, H3K27M at both biopsy and autopsy.
Of 18 patients who had biopsy, autopsy or both, tissue and/or genomic data was available for 56% (n = 10) of patients, representing 25% of the analyzed cohort and including 5 LTSs. Whole-genome or whole-exome sequencing was performed for four patients, limited genomic sequencing was completed for two patients, and immunohistochemical analysis alone was conducted for five patients. Mutations in this group were observed in ACVR1 (n = 1), ATM (n = 1), NTRK1 (n = 1), PDGFRA (n = 2) and TP53 (n = 2). H3K27M status was determined for 9 patients, including all 5 LTSs with available tissue. Six patients, including 3 LTSs, had H3K27M mutations (2 H3.1 K27M; 2 H3.3 K27M; 2 unspecified as based on IHC only), with a median OS of 24 months (range: 1–36 months). The 3 LTSs demonstrated 1 H3.1 K27M, 1 H3.3 K27M and 1 IHC only. These small numbers preclude conclusions for the population at large, however remaining biologic data explored is outlined in Figure 5.
Patients excluded from our analysis were subsequently reviewed for available tissue. Of 19 patients who did not meet inclusion criteria, 5 of the 13 patients who were excluded based on central radiology review had tissue available. Biopsy specimens from these five patients included astrocytoma (grade unknown, n = 1), pilocytic astrocytoma (PA; grade I, n = 1), low grade glioma (LGG; grade unknown, n = 1), primitive neuroectodermal tumor (PNET; grade unknown, n = 1) and glioblastoma (GBM; grade IV, n = 1). The patient with GBM at biopsy also had an autopsy, which was also a GBM. Imaging for this patient revealed a tumor which was not centered in the pons. One final patient had imaging which met criteria for DIPG but was excluded when autopsy tissue was consistent with PNET.
Discussion
Our study of 40 centrally-confirmed DIPG patients ≤36 months of age at diagnosis is, to our knowledge, the largest cohort of such patients reported in the literature, confirms the improved OS reported in smaller and mixed patient cohorts,5,7,8 and provides additional insight into this subgroup of patients. Median OS of the 40 patients who met inclusion criteria was 15 months, further improved to 17 months when considering only patients who received therapy. The OS of these cohorts surpasses the previously reported median OS of 8–11 months2,4,5,13,14 for all patients with DIPG. Included in the treated patient group were 10 LTSs (31%), with a median survival time of 39 months (range: 30 to 135 months), as well as 2 untreated LTS with survival of 37 and 45 months. Previously reported frequency of LTS (defined as patients with OS of ≥24 months) amongst all patients with DIPG is 10%,5 which we demonstrate has tripled in this younger patient population. Four of these LTS remain alive at the time of publication, with a median follow up of 60 months (range: 45 – 135 months).
In addition to a higher percentage of LTS, younger patients are also more likely to be very long-term survivors (VLTS), defined as an OS ≥60 months. Less than 50 patients diagnosed with DIPG have been described as VLTS in the literature.2,5 Of our 12 LTS, three were VLTS at 66 months, 101 months and 135 months, representing 7.5% of our analyzed patients, a significant increase from 1.6% VLTS reported by Hoffman et al5 for patients of all ages with DIPG.
Similarly, these younger children show improved rates of survival frequency years after diagnosis. Previously reported 1-year OS rate in two large pediatric DIPG patient cohorts with >1000 patients each was 41–42%,5,15 while the 1 year OS in our study was 50.1%. Improved survival frequency in our population continued at the 2-, 3-, and 5-year time points at 26%, 16%, and 11.7%, respectively. This is particularly notable, as the survival frequency in this population at 5 years is markedly improved from <3% in children of all ages previously reported by Jackson et al2 and Hoffman et al.5
In addition to age, previously observed clinical predictors of LTS in patients with DIPG include prolonged duration of symptoms at presentation, the absence of cranial nerve palsies, and receipt of systemic therapy at diagnosis.1,2,5–8,14,16,17 Analysis of our cohort reinforced a survival advantage for patients with longer duration of symptoms at presentation, with the majority of STS and few LTS having symptoms <6 weeks at diagnosis. Similarly, most STS presented with CN palsies, however this was not a statistically significant predictor of outcome. Other aspects of clinical presentation evaluated included the presence of pyramidal or cerebellar symptoms, as well as hydrocephalus and spinal metastasis. Of the patients evaluated for spinal metastasis, no LTS had this finding. No additional clinical factors were found to be predictive of improved OS.
Administration of systemic treatment has previously been correlated with LTS.5 In our cohort, most patients received both radiation and systemic therapy. A wide variety of both targeted and cytotoxic agents were employed, with rare repetition, precluding analysis for a superior regimen. Of the 31 patients with comprehensive treatment histories available, median OS of patients who received radiation therapy alone was 14 months (range: 4–136 months), comparable to the median OS of 16 months (range: 2–101 months) for patients who received both radiation and systemic therapy. Only two patients were treated with systemic therapy alone, with survival of 23 and 42 months. Perhaps most notably, all but one treated LTS received radiation, who was 1 month old at the time of diagnosis.
Treatment decisions made at progression likewise varied significantly. While improved OS has previously been reported by Janssens et al.18 in patients with DIPG who received reirradiation at progression, only one of our younger patients pursued RT alone at progression. Given the small numbers and the wide variability in treatment, no conclusions can be drawn regarding which therapeutic choices might confer OS advantage at progression in this younger population.
The eight patients (20%) in our cohort who did not receive any therapy had a median OS of only 2 months, with six of these patients surviving ≤2 months. Notably two LTSs, one who lived 37 months and one who remains alive 45 months after diagnosis were untreated. The majority had a short duration of symptoms, and most of these children had cranial nerve palsies on presentation. Central imaging review of scans performed at diagnosis confirmed typical appearance of DIPG tumors for five patients, while three scans showed some atypical features, but were considered likely DIPG. One of these eight patients had an autopsy which showed GBM. Another of these patients had a biopsy showing only normal brain tissue, however progression on imaging was consistent with that of a typical DIPG, raising concern for possible inadequate tissue sampling. Comparison of this untreated group to those who received therapy in our cohort demonstrates the clear survival advantage of treating DIPG in this age group, as median OS was 2 months and 17 months, respectively.
Utilizing the radiographic criteria we developed based on Barkovich et al.19 for our central imaging review, 13 patients were excluded on the basis of diagnostic MRI appearance. Median OS of these excluded patients was 7 months (range: 0–144 months). This notably opposes prior reports5,17 that excluded patients may have improved OS, and counters the hypothesis that younger patients diagnosed with DIPG show improved outcomes as a result of being misdiagnosed. On the contrary, the excluded patients ≤36 months of age at diagnosis in our cohort with imaging available for radiology review had a lower median OS compared to those considered radiographically typical DIPGs.
Additional radiographic parameters analyzed included tumor dimensions, tumor extension, margin characteristics, presence of hemorrhage, diffusion restriction, enhancement morphology, necrosis and hydrocephalus. Previously described radiologic factors influencing OS include tumor size, tumor necrosis and ring enhancement5,20; none of these were found to be predictive of long-term survival in this younger population. No other significant predictors of outcome emerged from our radiographic analysis.
Analysis of biologic and molecular data is paramount in DIPG, especially in patient populations like ours, which consistently show improved OS. Histone mutations were of particular interest, with previous studies reporting a prevalence of 70–78% for histone mutations amongst DIPG patients of all ages.21–23 Their presence has previously been considered a prognostic indicator, with H3.1 K27M associated with LTS and H3.3 K27M associated with STS in DIPG.5,22,24 Histone mutation status was known for half of the patients with available tissue, including five LTS. With a prevalence of 67% (n = 6) for histone mutations across nine available tissue samples, numbers were too small to draw conclusions for the whole population.
We were particularly interested in the 10 patients in this cohort with tissue and/or genomic information available. With data for only a few patients for each mutation, tissue amount, and availability were significant limitations. Half of these were LTS, with a median OS of 26.5 months and a median age at diagnosis of 29.5 months. Treatment upfront varied, with two patients receiving only radiation, two patients receiving only chemotherapy, four patients were given a combination of radiation and chemotherapy, one was untreated and 1 was unknown. At progression, only two patients received additional therapy, one was re-irradiated and the other received chemotherapy. The only chemotherapy regimen used more than once in this group was POG-9322, used in two patients ≤2 months of age. Univariate analysis did not reveal any statistically significant predictors of survival within this group.
This study, in accordance with those that have come before it, highlights our increasing understanding of the complexity, heterogeneity and spectrum of behavior of DIPG from a biologic, radiographic, histologic, and clinical perspective.25–31 Focusing on this contingent of younger patients with DIPG, a few unique considerations emerge. Compared to adult tumors of the CNS, pediatric brain tumors often harbor fewer aberrations, which typically increase with age.32,33 Younger DIPG patients follow this trend, with low mutational burden.34,35 Our cohort reinforced this finding, with rare patients demonstrating more than one mutation of the 23 investigated. While improved prognosis cannot be attributed to lower mutational burden alone, it has been hypothesized that key biologic drivers of the disease require subsequent genetic aberrations or events for disease progression,25 which would occur more rarely in this younger population.
As outlined in our previous review of predictors of survival in patients abstracted from the IDIPGR,5 the use of disease-specific registry data leaves our study population susceptible to enrollment bias, which remains a limitation of our current investigation. Additional confounding factors include the absence of standards of care for DIPG, resulting in a varied treatment approach, which may have influenced our findings. While the importance of anonymity of these subjects for research purposes cannot be overstated, it allows for the possibility of redundant findings in the literature, as some patients reported herein may have been a part of previous cohorts, as well. However, this de-identification also serves as a significant strength of our study, as it allows for blinded internal radiology and pathology review by highly experienced physicians to confirm the diagnosis for each patient.
This study represents the largest cohort of this special subset of patients with DIPG who are ≤36 months of age at diagnosis, confirming their improved OS and increased proportion of LTS and VLTS. While a longer duration of symptoms at presentation is associated with LTS in this subset, additional previously reported predictors of LTS did not predict improved OS. Treated patients in this younger group have a markedly improved OS compared to those who do not receive radiation or chemotherapy. Analysis of available biologic data in this younger cohort was fascinating, but largely descriptive. As biopsies and autopsies are being performed more commonly, in conjunction with the ready pursuit of genetic and molecular testing, we anticipate a host of biologic data will soon be available to supplement these often-small sample sizes and bridge the gaps in our understanding of this devastating disease.
Funding
For financial support of the IDIPGR, we thank The Cure Starts Now Foundation, The Cure Starts Now Australia, Brooke Healey Foundation, Wayland Villars Foundation, Aidan’s Avengers, Aubreigh’s Army, Austin Strong, Cure Brain Cancer, Jeffrey Thomas Hayden Foundation, Laurie’s Love Foundation, Love Chloe Foundation, Musella Foundation, Pray Hope Believe, Reflections Of Grace, Storm the Heavens Fund, Whitley’s Wishes, Gabriella’s Smile Foundation, The Gold Hope Project, The Isabella and Marcus Foundation, Lauren’s Fight for Cure, Robert Connor Dawes Foundation, Ryan’s Hope, Benny’s World, Lily Larue Foundation, Marlee’s Mission, RUN DIPG, American Childhood Cancer Organization, The DIPG Collaborative, and Snapgrant.com as well as the Kyler Strong Foundation and Keris Kares.
Acknowledgments
We are indebted to the children and families who have suffered from DIPG for their invaluable contribution to this research. We acknowledge the outstanding regulatory support of Dr. Renee Doughman and Marianne Torontali.
Conflict of interest statement. Authors have no conflicts of interest to disclose.
Authorship statement. Experimental Design—Allison Bartlett, Adam Lane, Maryam Fouladi. Experimental implementation(By providing cases, data, and/or clinical annotation) – All authors.Analysis and interpretation of the data—Allison Bartlett, Adam Lane, Mariko DeWire-Schottmiller, James Leach, Blaise Jones, Christine Fuller, Anne Cochrane, Rachid Drissi, and Maryam Fouladi. All authors were involved in the writing of the manuscript at draft and any revision stages and have read and approved the final version. The DIPG Registry’s mission is to ensure that there is collaborative research in this rare disease, so that sites band together and feel that their contributions are being recognized. Our constitution states that when patients, data, imaging and genomics data that the sites have submitted to us are part of any of our papers, PIs from sites who contribute, who are instrumental in ensuring we are provided with the data the registry requires, are recognized as authors. All of the authors listed contributed data, all reviewed the manuscript and approved and suggested modifications.
References



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}