

Novel Thiazolidine-2,4-dione-trimethoxybenzene-thiazole Hybrids as Human Topoisomerases Inhibitors

 ,
,
,
,
,
,
,
,
 ,
,
Abstract
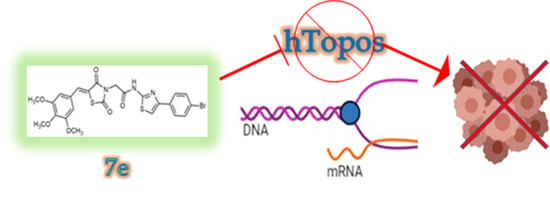
:
1. Introduction
2. Results
2.1. Chemistry
2.2. Docking Studies
2.3. Biology
2.3.1. Effects on Cell Viability
2.3.2. Inhibition of Human Topoisomerases I and II
2.3.3. Compound 7e Induced Apoptosis via Cytochrome c Release in MCF-7 Cells
3. Discussion
4. Materials and Methods
4.1. Chemistry
4.1.1. General Methods
4.1.2. Preparation of 2-amino-4-aryl-1,3-thiazoles 4b–e
4.1.3. Chloroacetylation of 2-Aminothiazoles; Preparation of 2-chloroacetamides 5a–f
4.1.4. Preparation of 2-Iodoacetamides 6a–f
4.1.5. General Procedure for Synthesis of the Target Compounds 7a–f
4.2. Docking Studies
4.3. Cell Cultures
4.4. Biology
4.4.1. MTT Assay
4.4.2. hTopo I Relaxation Assay and hTopo II Decatenation Assay
4.4.3. TUNEL Assay
4.4.4. Immunofluorescence Assays
4.4.5. Caspase Assay
4.4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mohan, M.; Murugan, V.; Geetha, K.; Kataria, M.K. Bis-[2-(Chloroethyl) Amino] Acetamido-4-Substituted Phenyl Thiazole Derivatives as Possible Antioxidant and Alky-lating Anticancer Agents. Int. J. Pharm. Drug Anal. 2018, 6, 352–360. [Google Scholar]
- Asati, V.; Thakur, S.S.; Upmanyu, N.; Bharti, S.K. Virtual Screening, Molecular Docking, and DFT Studies of Some Thiazolidine-2,4-diones as Potential PIM-1 Kinase Inhibitors. ChemistrySelect 2018, 3, 127–135. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: A Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Edwards, B.K.; Brown, M.L.; Wingo, P.A.; Howe, H.L.; Ward, E.; Ries, L.A.; Schrag, D.; Jamison, P.M.; Jemal, A.; Wu, X.C. Annual report to the nation on the status of cancer, 1975–2002, featuring population-based trends in cancer treatment. J. Natl. Cancer Inst. 2005, 97, 1407–1427. [Google Scholar] [CrossRef] [Green Version]
- WHO. Breast Cancer. Available online: https://www.who.int/news-room/fact-sheets/detail/breast-cancer (accessed on 2 August 2022).
- Liu, J.; Ming, B.; Gong, G.-H.; Wang, D.; Bao, G.-L.; Yu, L.-J. Current research on anti-breast cancer synthetic compounds. RSC Adv. 2018, 8, 4386–4416. [Google Scholar] [CrossRef] [Green Version]
- Naim, M.J.; Alam, M.J.; Ahmad, S.; Nawaz, F.; Shrivastava, N.; Sahu, M.; Alam, O. Therapeutic journey of 2,4-thiazolidinediones as a versatile scaffold: An insight into structure activity relationship. Eur. J. Med. Chem. 2017, 129, 218–250. [Google Scholar] [CrossRef]
- Day, C. Thiazolidinediones: A new class of antidiabetic drugs. Diabet. Med. 1999, 16, 179–192. [Google Scholar] [CrossRef]
- Tuncbilek, M.; Altanlar, N. Synthesis of New 3-(Substituted Phenacyl)-5-[3′-(4H-4-oxo-1-benzopyran-2-yl)-benzylidene]-2, 4-thiazolidinediones and their Antimicrobial Activity. Arch. Der Pharm. Int. J. Pharm. Med. Chem. 2006, 339, 213–216. [Google Scholar] [CrossRef]
- Bahare, R.S.; Ganguly, S.; Choowongkomon, K.; Seetaha, S. Synthesis, HIV-1 RT inhibitory, antibacterial, antifungal and binding mode studies of some novel N-substituted 5-benzylidine-2,4-thiazolidinediones. DARU J. Pharm. Sci. 2015, 23, 6. [Google Scholar] [CrossRef] [Green Version]
- Reddy, K.A.; Lohray, B.; Bhushan, V.; Reddy, A.S.; Kishore, P.H.; Rao, V.V.; Saibaba, V.; Bajji, A.; Rajesh, B.; Reddy, K.V. Novel euglycemic and hypolipidemic agents: Part-2 antioxidant moiety as structural motif. Bioorganic Med. Chem. Lett. 1998, 8, 999–1002. [Google Scholar] [CrossRef]
- Prabhakar, C.; Madhusudhan, G.; Sahadev, K.; Reddy, C.M.; Sarma, M.; Reddy, G.O.; Chakrabarti, R.; Rao, C.S.; Kumar, T.D.; Rajagopalan, R. Synthesis and biological activity of novel thiazolidinediones. Bioorganic Med. Chem. Lett. 1998, 8, 2725–2730. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Landreth, G.E. PPARs in the brain. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2007, 1771, 1031–1045. [Google Scholar] [CrossRef]
- Patil, V.; Tilekar, K.; Mehendale-Munj, S.; Mohan, R.; Ramaa, C. Synthesis and primary cytotoxicity evaluation of new 5-benzylidene-2, 4-thiazolidinedione derivatives. Eur. J. Med. Chem. 2010, 45, 4539–4544. [Google Scholar] [CrossRef]
- Asati, V.; Mahapatra, D.K.; Bharti, S.K. Thiazolidine-2, 4-diones as multi-targeted scaffold in medicinal chemistry: Potential anticancer agents. Eur. J. Med. Chem. 2014, 87, 814–833. [Google Scholar] [CrossRef] [PubMed]
- Kosurkar, U.B.; Mamilla, J.; Dadmal, T.L.; Choudante, P.C.; Mali, S.N.; Misra, S.; Kumbhare, R.M. Synthesis of Novel Thiazolidine-4-One Derivatives, Their Cytotoxicity, Antifungal Properties, Molecular Docking and Molecular Dynamics. Russ. J. Bioorganic Chem. 2023, 49, 314–323. [Google Scholar] [CrossRef]
- Liu, K.; Rao, W.; Parikh, H.; Li, Q.; Guo, T.L.; Grant, S.; Kellogg, G.E.; Zhang, S. 3, 5-Disubstituted-thiazolidine-2, 4-dione analogs as anticancer agents: Design, synthesis and biological characterization. Eur. J. Med. Chem. 2012, 47, 125–137. [Google Scholar] [CrossRef] [PubMed]
- Pomel, V.; Klicic, J.; Covini, D.; Church, D.D.; Shaw, J.P.; Roulin, K.; Burgat-Charvillon, F.; Valognes, D.; Camps, M.; Chabert, C. Furan-2-ylmethylene thiazolidinediones as novel, potent, and selective inhibitors of phosphoinositide 3-kinase γ. J. Med. Chem. 2006, 49, 3857–3871. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.G.; Beaumont, K.A.; Smit, D.J.; Thurber, A.E.; Cook, A.L.; Boyle, G.M.; Parsons, P.G.; Sturm, R.A.; Muscat, G.E. PPARγ agonists attenuate proliferation and modulate Wnt/β-catenin signalling in melanoma cells. Int. J. Biochem. Cell Biol. 2009, 41, 844–852. [Google Scholar] [CrossRef]
- Xia, Z.; Knaak, C.; Ma, J.; Beharry, Z.M.; McInnes, C.; Wang, W.; Kraft, A.S.; Smith, C.D. Synthesis and evaluation of novel inhibitors of Pim-1 and Pim-2 protein kinases. J. Med. Chem. 2009, 52, 74–86. [Google Scholar] [CrossRef] [Green Version]
- Trotsko, N.; Przekora, A.; Zalewska, J.; Ginalska, G.; Paneth, A.; Wujec, M. Synthesis and in vitro antiproliferative and antibacterial activity of new thiazolidine-2, 4-dione derivatives. J. Enzym. Inhib. Med. Chem. 2018, 33, 17–24. [Google Scholar] [CrossRef] [Green Version]
- Elancheran, R.; Saravanan, K.; Divakar, S.; Kumari, S.; Maruthanila, V.L.; Kabilan, S.; Ramanathan, M.; Devi, R.; Kotoky, J. Design, synthesis and biological evaluation of novel 1, 3-thiazolidine-2, 4-diones as anti-prostate cancer agents. Anti-Cancer Agents Med. Chem. (Former. Curr. Med. Chem. Anti-Cancer Agents) 2017, 17, 1756–1768. [Google Scholar] [CrossRef] [PubMed]
- Yoshizumi, T.; Ohta, T.; Ninomiya, I.; Terada, I.; Fushida, S.; Fujimura, T.; Nishimura, G.-I.; Shimizu, K.; Yi, S.; Miwa, K. Thiazolidinedione, a peroxisome proliferator-activated receptor-γ ligand, inhibits growth and metastasis of HT-29 human colon cancer cells through differentiation-promoting effects. Int. J. Oncol. 2004, 25, 631–639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LOZYNSKYI, A.; Zimenkovsky, B.; Lesyk, R. Synthesis and anticancer activity of new thiopyrano [2, 3-d] thiazoles based on cinnamic acid amides. Sci. Pharm. 2014, 82, 723–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bharti, S.; Nath, G.; Tilak, R.; Singh, S. Synthesis, anti-bacterial and anti-fungal activities of some novel Schiff bases containing 2,4-disubstituted thiazole ring. Eur. J. Med. Chem. 2010, 45, 651–660. [Google Scholar] [CrossRef]
- Yang, B.V.; Weinstein, D.S.; Doweyko, L.M.; Gong, H.; Vaccaro, W.; Huynh, T.; Xiao, H.-Y.; Doweyko, A.M.; McKay, L.; Holloway, D.A. Dimethyl-diphenyl-propanamide derivatives as nonsteroidal dissociated glucocorticoid receptor agonists. J. Med. Chem. 2010, 53, 8241–8251. [Google Scholar] [CrossRef] [PubMed]
- Andreani, A.; Granaiola, M.; Leoni, A.; Locatelli, A.; Morigi, R.; Rambaldi, M. Synthesis and antitubercular activity of imidazo [2, 1-b] thiazoles. Eur. J. Med. Chem. 2001, 36, 743–746. [Google Scholar] [CrossRef]
- Aoyama, T.; Murata, S.; Arai, I.; Araki, N.; Takido, T.; Suzuki, Y.; Kodomari, M. One pot synthesis using supported reagents system KSCN/SiO2–RNH3OAc/Al2O3: Synthesis of 2-aminothiazoles and N-allylthioureas. Tetrahedron 2006, 62, 3201–3213. [Google Scholar] [CrossRef]
- Bell, F.W.; Cantrell, A.S.; Hoegberg, M.; Jaskunas, S.R.; Johansson, N.G.; Jordan, C.L.; Kinnick, M.D.; Lind, P.; Morin, J.M., Jr. Phenethylthiazolethiourea (PETT) compounds, a new class of HIV-1 reverse transcriptase inhibitors. 1. Synthesis and basic structure-activity relationship studies of PETT analogs. J. Med. Chem. 1995, 38, 4929–4936. [Google Scholar] [CrossRef]
- Bozdağ-Dündar, O.; Ceylan-Uenluesoy, M.; Eugen, V.J.; Ertan, R. Synthesis and antidiabetic activity of novel 2,4-thiazolidinedione derivatives containing a thiazole ring. Arzneimittelforschung 2006, 56, 621–625. [Google Scholar] [CrossRef]
- González Cabrera, D.; Douelle, F.; Feng, T.-S.; Nchinda, A.T.; Younis, Y.; White, K.L.; Wu, Q.; Ryan, E.; Burrows, J.N.; Waterson, D. Novel orally active antimalarial thiazoles. J. Med. Chem. 2011, 54, 7713–7719. [Google Scholar] [CrossRef]
- Kachroo, M.; Rao, G.; Rajasekaran, S.; Pai, S.; Hemalatha, Y. Synthesis, antibacterial and antioxidant activity of N-[(4E)-arylidene-5-oxo-2-phenyl-4, 5-dihydro-1H-imidazol-1-yl]-2-(2-methyl-1, 3-thiazol-4-yl) acetamide. Der Pharma Chem. 2011, 3, 241–245. [Google Scholar]
- Kouatly, O.; Geronikaki, A.; Kamoutsis, C.; Hadjipavlou-Litina, D.; Eleftheriou, P. Adamantane derivatives of thiazolyl-N-substituted amide, as possible non-steroidal anti-inflammatory agents. Eur. J. Med. Chem. 2009, 44, 1198–1204. [Google Scholar] [CrossRef] [PubMed]
- Omarx, A.-M.M.; Eshba, N.H. Synthesis and biological evaluation of new 2, 3-dihydrothiazole derivatives for antimicrobial, antihypertensive, and anticonvulsant activities. J. Pharm. Sci. 1984, 73, 1166–1168. [Google Scholar] [CrossRef] [PubMed]
- Spector, F.; Liang, L.; Giordano, H.; Sivaraja, M.; Peterson, M. Inhibition of herpes simplex virus replication by a 2-amino thiazole via interactions with the helicase component of the UL5-UL8-UL52 complex. J. Virol. 1998, 72, 6979–6987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ucar, H.; Van derpoorten, K.; Cacciaguerra, S.; Spampinato, S.; Stables, J.P.; Depovere, P.; Isa, M.; Masereel, B.; Delarge, J.; Poupaert, J.H. Synthesis and anticonvulsant activity of 2 (3 H)-benzoxazolone and 2 (3 H)-benzothiazolone derivatives. J. Med. Chem. 1998, 41, 1138–1145. [Google Scholar] [CrossRef]
- Weikert, R.J.; Bingham, S., Jr.; Emanuel, M.A.; Fraser-Smith, E.B.; Loughhead, D.G.; Nelson, P.H.; Poulton, A.L. Synthesis and anthelmintic activity of 3′-benzoylurea derivatives of 6-phenyl-2,3,5,6-tetrahydroimidazo [2, 1-b] thiazole. J. Med. Chem. 1991, 34, 1630–1633. [Google Scholar] [CrossRef]
- Franchetti, P.; Cappellacci, L.; Grifantini, M.; Barzi, A.; Nocentini, G.; Yang, H.; O’Connor, A.; Jayaram, H.N.; Carrell, C.; Goldstein, B.M. Furanfurin and thiophenfurin: Two novel tiazofurin analogs. Synthesis, structure, antitumor activity, and interactions with inosine monophosphate dehydrogenase. J. Med. Chem. 1995, 38, 3829–3837. [Google Scholar] [CrossRef]
- Li, X.; He, Y.; Ruiz, C.H.; Koenig, M.; Cameron, M.D. Characterization of dasatinib and its structural analogs as CYP3A4 mechanism-based inactivators and the proposed bioactivation pathways. Drug Metab. Dispos. 2009, 37, 1242–1250. [Google Scholar] [CrossRef] [Green Version]
- Hu-Lieskovan, S.; Mok, S.; Homet Moreno, B.; Tsoi, J.; Robert, L.; Goedert, L.; Pinheiro, E.M.; Koya, R.C.; Graeber, T.G.; Comin-Anduix, B. Improved antitumor activity of immunotherapy with BRAF and MEK inhibitors in BRAF V600E melanoma. Sci. Transl. Med. 2015, 7, 279ra241. [Google Scholar] [CrossRef] [Green Version]
- Mishra, S.; Singh, P. Hybrid molecules: The privileged scaffolds for various pharmaceuticals. Eur. J. Med. Chem. 2016, 124, 500–536. [Google Scholar]
- Tilekar, K.; Shelke, O.; Upadhyay, N.; Lavecchia, A.; Ramaa, C. Current status and future prospects of molecular hybrids with thiazolidinedione (TZD) scaffold in anticancer drug discovery. J. Mol. Struct. 2022, 1250, 131767. [Google Scholar] [CrossRef]
- El-Kashef, H.; Badr, G.; El-Maali, N.A.; Sayed, D.; Melnyk, P.; Lebegue, N.; Abd El-Khalek, R. Synthesis of a novel series of (Z)-3, 5-disubstituted thiazolidine-2, 4-diones as promising anti-breast cancer agents. Bioorganic Chem. 2020, 96, 103569. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Li, W.; Yang, X.; Chen, G.; Jin, X.; Chen, W.; Ye, L. Design, Synthesis, anticancer evaluation and in silico studies of 2, 4, 6-trimethoxychalcone derivatives. Saudi Pharm. J. 2023, 31, 65–84. [Google Scholar] [CrossRef] [PubMed]
- Aziz, H.A.; El-Saghier, A.M.; Badr, M.; Abuo-Rahma, G.E.-D.A.; Shoman, M.E. Thiazolidine-2, 4-dione-linked ciprofloxacin derivatives with broad-spectrum antibacterial, MRSA and topoisomerase inhibitory activities. Mol. Divers. 2022, 26, 1743–1759. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.-C.; Chen, B.; Yang, J.-L.; Weng, J.-Q.; Yu, Q.; Hu, D.-X. Design, Synthesis and Cytotoxicity of Thiazole-Based Stilbene Analogs as Novel DNA Topoisomerase IB Inhibitors. Molecules 2022, 27, 1009. [Google Scholar] [CrossRef]
- Mastrangelo, S.; Attina, G.; Triarico, S.; Romano, A.; Maurizi, P.; Ruggiero, A. The DNA-topoisomerase Inhibitors in Cancer Therapy. Biomed. Pharmacol. J. 2022, 15, 553–562. [Google Scholar] [CrossRef]
- Liang, X.; Wu, Q.; Luan, S.; Yin, Z.; He, C.; Yin, L.; Zou, Y.; Yuan, Z.; Li, L.; Song, X. A comprehensive review of topoisomerase inhibitors as anticancer agents in the past decade. Eur. J. Med. Chem. 2019, 171, 129–168. [Google Scholar] [CrossRef]
- Mague, J.; Mohamed, S.; Akkurt, M.; El-Kashef, H.; Lebegue, N.; Albayati, M. (5Z)-3-(2-Oxopropyl)-5-(3,4,5-trimethoxybenzylidene)-1, 3-thiazolidine-2,4-dione. IUCrData 2016, 1, x161959. [Google Scholar] [CrossRef] [Green Version]
- Dodson, R.; King, L.C. The reaction of ketones with halogens and thiourea1. J. Am. Chem. Soc. 1945, 67, 2242–2243. [Google Scholar] [CrossRef]
- Sinicropi, M.S.; Iacopetta, D.; Rosano, C.; Randino, R.; Caruso, A.; Saturnino, C.; Muià, N.; Ceramella, J.; Puoci, F.; Rodriquez, M. N-thioalkylcarbazoles derivatives as new anti-proliferative agents: Synthesis, characterisation and molecular mechanism evaluation. J. Enzym. Inhib. Med. Chem. 2018, 33, 434–444. [Google Scholar] [CrossRef] [Green Version]
- Viale, M.; Cordazzo, C.; De Totero, D.; Budriesi, R.; Rosano, C.; Leoni, A.; Ioan, P.; Aiello, C.; Croce, M.; Andreani, A. Inhibition of MDR1 activity and induction of apoptosis by analogues of nifedipine and diltiazem: An in vitro analysis. Investig. New Drugs 2011, 29, 98–109. [Google Scholar] [CrossRef]
- Wallace, A.C.; Laskowski, R.A.; Thornton, J.M. LIGPLOT: A program to generate schematic diagrams of protein-ligand interactions. Protein Eng. Des. Sel. 1995, 8, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Cascioferro, S.; Parrino, B.; Carbone, D.; Schillaci, D.; Giovannetti, E.; Cirrincione, G.; Diana, P. Thiazoles, their benzofused systems, and thiazolidinone derivatives: Versatile and promising tools to combat antibiotic resistance. J. Med. Chem. 2020, 63, 7923–7956. [Google Scholar] [CrossRef] [PubMed]
- Bergant Loboda, K.; Janežič, M.; Štampar, M.; Žegura, B.; Filipič, M.; Perdih, A. Substituted 4,5′-Bithiazoles as catalytic inhibitors of human DNA topoisomerase IIα. J. Chem. Inf. Model. 2020, 60, 3662–3678. [Google Scholar] [CrossRef]
- Zelelew, D.; Endale, M.; Melaku, Y.; Kedir, F.; Demissie, T.B.; Ombito, J.O.; Eswaramoorthy, R. Synthesis, Antibacterial, and Antioxidant Activities of Thiazolyl-Pyrazoline Schiff Base Hybrids: A Combined Experimental and Computational Study. J. Chem. 2022, 2022, 3717826. [Google Scholar] [CrossRef]
- Aki-Yalcin, E.; Ertan-Bolelli, T.; Taskin-Tok, T.; Ozturk, O.; Ataei, S.; Ozen, C.; Yildiz, I.; Yalcin, I. Evaluation of inhibitory effects of benzothiazole and 3-amino-benzothiazolium derivatives on DNA topoisomerase II by molecular modeling studies. SAR QSAR Environ. Res. 2014, 25, 637–649. [Google Scholar] [CrossRef]
- Rizza, P.; Pellegrino, M.; Caruso, A.; Iacopetta, D.; Sinicropi, M.S.; Rault, S.; Lancelot, J.C.; El-Kashef, H.; Lesnard, A.; Rochais, C.; et al. 3-(Dipropylamino)-5-hydroxybenzofuro[2,3-f]quinazolin-1(2H)-one (DPA-HBFQ-1) plays an inhibitory role on breast cancer cell growth and progression. Eur. J. Med. Chem. 2016, 107, 275–287. [Google Scholar] [CrossRef]
- Sordet, O.; Khan, Q.A.; Kohn, K.W.; Pommier, Y. Apoptosis induced by topoisomerase inhibitors. Curr. Med. Chem. Anti-Cancer Agents 2003, 3, 271–290. [Google Scholar] [CrossRef]
- Ceramella, J.; Mariconda, A.; Sirignano, M.; Iacopetta, D.; Rosano, C.; Catalano, A.; Saturnino, C.; Sinicropi, M.S.; Longo, P. Novel Au carbene complexes as promising multi-target agents in breast cancer treatment. Pharmaceuticals 2022, 15, 507. [Google Scholar] [CrossRef]
- Mariconda, A.; Iacopetta, D.; Sirignano, M.; Ceramella, J.; Costabile, C.; Pellegrino, M.; Rosano, C.; Catalano, A.; Saturnino, C.; El-Kashef, H. N-Heterocyclic Carbene (NHC) Silver Complexes as Versatile Chemotherapeutic Agents Targeting Human Topoisomerases and Actin. ChemMedChem 2022, 17, e202200345. [Google Scholar] [CrossRef]
- Bousbaa, H. Novel Anticancer Strategies. Pharmaceutics 2021, 13, 275. [Google Scholar] [CrossRef]
- Alkhzem, A.H.; Woodman, T.J.; Blagbrough, I.S. Design and synthesis of hybrid compounds as novel drugs and medicines. RSC Adv. 2022, 12, 19470–19484. [Google Scholar] [CrossRef] [PubMed]
- Romagnoli, R.; Baraldi, P.G.; Salvador, M.K.; Prencipe, F.; Lopez-Cara, C.; Schiaffino Ortega, S.; Brancale, A.; Hamel, E.; Castagliuolo, I.; Mitola, S.; et al. Design, Synthesis, in Vitro, and in Vivo Anticancer and Antiangiogenic Activity of Novel 3-Arylaminobenzofuran Derivatives Targeting the Colchicine Site on Tubulin. J. Med. Chem. 2015, 58, 3209–3222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, S.; Pattnaik, S.; Pathak, K.; Kumar, S.; Pathak, D.; Jain, S.; Vaidya, A. Anticancer Potential of Thiazole Derivatives: A Retrospective Review. Mini-Rev. Med. Chem. 2018, 18, 640–655. [Google Scholar] [CrossRef]
- Buzun, K.; Bielawska, A.; Bielawski, K.; Gornowicz, A. DNA topoisomerases as molecular targets for anticancer drugs. J. Enzym. Inhib. Med. Chem. 2020, 35, 1781–1799. [Google Scholar] [CrossRef]
- Skok, Ž.; Zidar, N.; Kikelj, D.; Ilaš, J. Dual Inhibitors of Human DNA Topoisomerase II and Other Cancer-Related Targets. J. Med. Chem. 2019, 63, 884–904. [Google Scholar] [CrossRef] [Green Version]
- Banothu, J.; Vaarla, K.; Bavantula, R.; Crooks, P.A. Sodium fluoride as an efficient catalyst for the synthesis of 2, 4-disubstituted-1, 3-thiazoles and selenazoles at ambient temperature. Chin. Chem. Lett. 2014, 25, 172–175. [Google Scholar] [CrossRef]
- Wolf, L.; Quoos, N.; Mayer, J.C.; de Souza, D.; Sauer, A.C.; Meichtry, L.; Bortolotto, V.; Prigol, M.; Rodrigues, O.E.; Dornelles, L. Synthesis and free radical scavenging activity of 2-alkyl/arylchalcogenyl-N-(4-aryl-1,3-thiazol-2-yl) acetamide compounds. Tetrahedron Lett. 2016, 57, 1031–1034. [Google Scholar] [CrossRef]
- Geronikaki, A.; Theophilidis, G. Synthesis of 2-(aminoacetylamino) thiazole derivatives and comparison of their local anaesthetic activity by the method of action potential. Eur. J. Med. Chem. 1992, 27, 709–716. [Google Scholar] [CrossRef]
- Liu, H.-L.; Li, Z.; Anthonsen, T. Synthesis and fungicidal activity of 2-imino-3-(4-arylthiazol-2-yl)-thiazolidin-4-ones and their 5-arylidene derivatives. Molecules 2000, 5, 1055–1061. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, S.; Sen, J. Synthesis and biological evaluation of 2- aminobenzothiazole derivatives. Indian J. Chem. 2008, 47B, 1583–1586. [Google Scholar]
- Iacopetta, D.; Mariconda, A.; Saturnino, C.; Caruso, A.; Palma, G.; Ceramella, J.; Muià, N.; Perri, M.; Sinicropi, M.S.; Caroleo, M.C. Novel gold and silver carbene complexes exert antitumor effects triggering the reactive oxygen species dependent intrinsic apoptotic pathway. ChemMedChem 2017, 12, 2054–2065. [Google Scholar] [CrossRef] [PubMed]
- Redinbo, M.R.; Stewart, L.; Kuhn, P.; Champoux, J.J.; Hol, W.G. Crystal structures of human topoisomerase I in covalent and noncovalent complexes with DNA. Science 1998, 279, 1504–1513. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-R.; Chen, S.-F.; Wu, C.-C.; Liao, Y.-W.; Lin, T.-S.; Liu, K.-T.; Chen, Y.-S.; Li, T.-K.; Chien, T.-C.; Chan, N.-L. Producing irreversible topoisomerase II-mediated DNA breaks by site-specific Pt (II)-methionine coordination chemistry. Nucleic Acids Res. 2017, 45, 10861–10871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanner, M.F.; Duncan, B.S.; Carrillo, C.J.; Olson, A.J. Integrating computation and visualization for biomolecular analysis: An example using python and AVS. In Biocomputing’99; World Scientific: Singapore, 1999; pp. 401–412. [Google Scholar]
- Rosano, C.; Lappano, R.; F Santolla, M.; Ponassi, M.; Donadini, A.; Maggiolini, M. Recent advances in the rationale design of GPER ligands. Curr. Med. Chem. 2012, 19, 6199–6206. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Iacopetta, D.; Rosano, C.; Puoci, F.; Parisi, O.I.; Saturnino, C.; Caruso, A.; Longo, P.; Ceramella, J.; Malzert-Fréon, A.; Dallemagne, P. Multifaceted properties of 1, 4-dimethylcarbazoles: Focus on trimethoxybenzamide and trimethoxyphenylurea derivatives as novel human topoisomerase II inhibitors. Eur. J. Pharm. Sci. 2017, 96, 263–272. [Google Scholar] [CrossRef]
- Sirignano, E.; Saturnino, C.; Botta, A.; Sinicropi, M.S.; Caruso, A.; Pisano, A.; Lappano, R.; Maggiolini, M.; Longo, P. Synthesis, characterization and cytotoxic activity on breast cancer cells of new half-titanocene derivatives. Bioorganic Med. Chem. Lett. 2013, 23, 3458–3462. [Google Scholar] [CrossRef]
- Iacopetta, D.; Rosano, C.; Sirignano, M.; Mariconda, A.; Ceramella, J.; Ponassi, M.; Saturnino, C.; Sinicropi, M.S.; Longo, P. Is the Way to Fight Cancer Paved with Gold? Metal-Based Carbene Complexes with Multiple and Fascinating Biological Features. Pharmaceuticals 2020, 13, 91. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IC50 (µM) | |||||
|---|---|---|---|---|---|
| Compounds | MDA-MB-231 | MCF-7 | A2058 | 3T3 BALB | MCF-10A |
| 7a | 27.5 ± 0.9 | >100 | >100 | 89.2 ± 1.1 | >100 |
| 7b | 17.9 ± 1.1 | 8.5 ± 1.2 | 3.5 ± 1.1 | >100 | >100 |
| 7c | 32.8 ± 0.6 | 19.5 ± 0.6 | 4.8 ± 1.0 | >100 | 46.4 ± 1.1 |
| 7d | 33.4 ± 1.0 | 34.1 ± 0.7 | 6.4 ± 0.9 | 36.1 ± 1.0 | >100 |
| 7e | 41.2 ± 0.9 | 3.1 ± 0.6 | 8.4 ± 0.9 | 35.0 ± 0.6 | >100 |
| 7f | 50.5 ± 0.8 | 17.4 ± 1.1 | 24.6 ± 1.1 | >100 | >100 |
| colchicine | 8.1 (± 1.0) × 10−2 | 1.6 (± 0.8) × 10−2 | 3.4 (± 0.7) × 10−2 | 5.2 (± 0.9) × 10−1 | 8.9 (± 1.2) × 10−3 |
| ellipticine | 1.73 ± 0.4 | 1.15 ± 0.5 | 1.33 ± 0.9 | 0.97 ± 0.1 | 1.09 ± 0.1 |
| Topoisomerase Inhibition | |||
|---|---|---|---|
| Compounds | [µM] | hTopo I | hTopo II |
| 7b | 10 | − | − |
| 50 | − | ||
| 7c | 10 | − | − |
| 50 | + | ||
| 7d | 10 | − | − |
| 50 | + | ||
| 7e | 10 | − | − |
| 50 | + | ||
| 7f | 10 | + | − |
| 50 | + | ||
| ellipticine | 10 | + | +/− |
| 50 | + | + | |
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.
|
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sinicropi, M.S.; Ceramella, J.; Vanelle, P.; Iacopetta, D.; Rosano, C.; Khoumeri, O.; Abdelmohsen, S.; Abdelhady, W.; El-Kashef, H. Novel Thiazolidine-2,4-dione-trimethoxybenzene-thiazole Hybrids as Human Topoisomerases Inhibitors. Pharmaceuticals 2023, 16, 946. https://doi.org/10.3390/ph16070946
Sinicropi MS, Ceramella J, Vanelle P, Iacopetta D, Rosano C, Khoumeri O, Abdelmohsen S, Abdelhady W, El-Kashef H. Novel Thiazolidine-2,4-dione-trimethoxybenzene-thiazole Hybrids as Human Topoisomerases Inhibitors. Pharmaceuticals. 2023; 16(7):946. https://doi.org/10.3390/ph16070946
Chicago/Turabian StyleSinicropi, Maria Stefania, Jessica Ceramella, Patrice Vanelle, Domenico Iacopetta, Camillo Rosano, Omar Khoumeri, Shawkat Abdelmohsen, Wafaa Abdelhady, and Hussein El-Kashef. 2023. "Novel Thiazolidine-2,4-dione-trimethoxybenzene-thiazole Hybrids as Human Topoisomerases Inhibitors" Pharmaceuticals 16, no. 7: 946. https://doi.org/10.3390/ph16070946