Laboratory Tests
The serum titer of the acetyl-choline receptor antibodies does not correlate with disease severity. Their value is mainly in the initial diagnosis of myasthenia gravis (MG), or in the case of modulating antibodies as a potential marker for thymoma. Unlike anti-AChR-abs, there appears to be a correlation between anti-MuSK titers, disease severity, and the application of immunomodulatory therapy. [42]
Anti–acetylcholine receptor antibody
The anti-acetylcholine receptor (AChR) antibody (Ab) test is reliable for diagnosing autoimmune myasthenia gravis (MG). It is highly specific (as high as 100%, according to Padua et al). [4] Results are positive in as many as 90% of patients who have generalized MG but in only 50-70% of those who have only ocular MG; thus false negatives are common in cases of purely ocular MG. However, these may be present at the NMJ, the site of disease pathology, and are causal, but may not be detectable in the serum at an early stage of disease.
Anti-AChR antibodies are predominantly IgG1 and IgG3. They effectively activate complement, leading to the formation of the membrane attack complex resulting in damage to the NMJ in the form of simplification of postsynaptic junctional folds, removal of AChR from the membrane, and widening of the synaptic cleft.
Anti-AChR ab (binding)
Sensitivity 88%–93% for generalized MG, and 50%–71% for ocular MG. False positives are rare and may be seen in thymoma without MG, Lambert-Eaton Mysthenic Syndrome (LEMS), graft-versus-host-disease (GVHD), autoimmune liver disease, small cell cancer, rheumatoid arthritis treated with d-penicillamine, and motor neuron disease. There is essentially an identical phenotype in patient with and without anti-AChR-ab (binding). [43]
Anti-AChR ab (modulating)
2%–4% of MG cases with negative AChR-ab (binding) will have the modulating antibody. It is implicated with an increased risk of thymoma. 73% of patients with thymoma and MG will have modulating antibodies. [44]
Anti-AChR ab (blocking)
These are present in approximately half of the patients with generalized MG but only 30% of patients with ocular disease. Less than 1% of MG patients have anti-AChR-ab (blocking) without detectable binding or modulating antibodies, thus, this test not clinically useful. [45]
One should be aware that laboratory testing for autoantibodies using different methods yields different sensitivity and specificty. The diagnostic accuracy varies between cell-based assay (CBA) and radioimmunoprecipitation assay (RIPA) and enzyme-linked immunosorbent assay (ELISA) and type of ELISA used. [73, 74]
Anti–striated muscle antibody
The anti–striated muscle (anti-SM) Ab refers to a class of antibodies against components of skeletal muscle including titin, the ryanodine receptor, myosin, and alpha-actin. Anti-SM Ab is present in about 70–80%of patients with thymoma and MG who are younger than 40 years, and 30% of adult patients with MG without thymoma and 24% of patients with thymoma without MG. [46] Thus, a positive test result should prompt a search for thymoma in patients younger than 40 years. In individuals older than 40 years, anti-SM Ab can be present without thymoma. Unlike AChR antibodies, striational antibodies can be of value in monitoring the disease course. Persistence or recurrence of high titers of these antibodies may indicate incomplete resection or recurrence of thymoma, respectively.
Anti-MuSK antibody
About half of the patients with negative results for anti-AChR Ab (seronegative MG) may have positive test results for antibody to muscle-specific kinase (MuSK), a receptor tyrosine kinase that is essential for neuromuscular junction development. [47] These patients may represent a distinct group of autoimmune MG, in that they show some collective characteristics that are different from those of anti-AChR–positive patients. [17]
Anti-MuSK–positive individuals tend to have more pronounced bulbar weakness and may have tongue and facial atrophy. They may have neck, shoulder and respiratory involvement without ocular weakness. They are also less likely to respond to acetylcholine esterase (AChE) inhibitors, and their symptoms may actually worsen with these medications. [48, 36]
Anti-lipoprotein-related protein 4 (LRP4) antibody
Lipoprotein-related protein 4 is present on the postsynaptic membrane and is a coreceptor for agrin and is essential for for agrin-induced activation of MuSK in concert with Dok-7. Full activation of MuSK results in activation of rapsyn, which then induces clustering of AChRs by binding them to the post-synaptic scaffold. Antibody to this protein is present in 9.2% of double seronegative myasthenics (absence of AChR and Anti-MuSK antobodies). However, 2 recent studies showed prevalence of anti-LRP4 antibodies varied from 2% to 50% of patients with double-seronegative MG from different geographic locations. [49, 50, 51] LRP4 autoantibodies are predominantly IgG1, and studies suggest that they are pathogenic.
Anti-agrin antibody
About 80–90% of MG patients have detectable serum antibodies against AChRs with 40–70% of the remaining patients being positive for anti-MuSK antibodies and 2–50% for anti-LRP4 antibodies. This would leave approximately 2–5% of the MG patients triple seronegative, i.e., without detectable antibodies against any known autoantigen (AChR, MuSK or LRP4) at the NMJ.The presence of agrin antibodies in ‘triple seronegative’ patients with MG suggests that agrin may be a novel antigen in some triple seronegative MG patients. [52]
Other autoantibodies in MG
Besides LRP4 and agrin, collagen Q, cortactin, and voltage-gated potassium channel Kv1.4 are detected.
Other laboratory studies
Testing for rheumatoid factor and antinuclear antibodies (ANAs) is indicated to rule out systemic lupus erythematosus (SLE) and rheumatoid arthritis (RA).
Thyroid function tests are indicated to rule out associated Graves disease or hyperthyroidism. This is essential, especially in patients with ocular MG where the concomitant hyperthyroidism is most frequent.
Radiography, CT, and MRI
Imaging of the chest in myasthenia gravis (MG) is used to rule out tumors. On plain anteroposterior and lateral views, radiography may identify a thymoma as an anterior mediastinal mass. A negative chest radiograph does not rule out a smaller thymoma, in which case a chest computed tomography (CT) scan is required. Chest CT scan should be obtained to identify or rule out thymoma or thymic enlargement in all cases of MG (see the images below). This is especially true in older individuals.
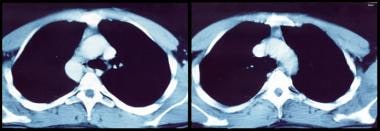 CT scan of chest and mediastinum showing thymoma in patient with myasthenia gravis.
CT scan of chest and mediastinum showing thymoma in patient with myasthenia gravis.
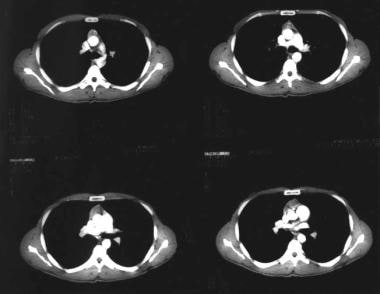 CT scan of chest showing an anterior mediastinal mass (thymoma) in a patient with myasthenia gravis.
CT scan of chest showing an anterior mediastinal mass (thymoma) in a patient with myasthenia gravis.
It is essential to rule out mass lesions compressing the cranial nerves in strictly ocular MG. CT or preferably magnetic resonance imaging (MRI) of the brain and orbit is indicated. It is helpful when the diagnosis of MG is not established and to rule out other causes of cranial nerve deficits. MRI can evaluate for intraorbital or intracranial lesions, basal meningeal pathology, or multiple sclerosis.
Electrodiagnostic Studies
Routine motor and sensory nerve conduction studies (NCS) in myasthenia gravis (MG) must not be omitted before embarking on electrodiagnostic studies that demonstrate a defect of neuromuscular transmission. Routine NCS is done to ensure the integrity of any nerve that subsequently will be used in RNS. A decrement in RNS can be seen in other conditions (neuropathies, motor neuron disease, inflammatory myopathies) and myotonic disorders. At least one motor and sensory conduction study should be performed in an upper and lower extremity. Compound mucle action potentials (CMAP) amplitudes should be normal with only 3% to 15% showing CMAPs that are diffusely low. If CMAP amplitudes are low or borderline, repeat distal stimulation after 10 seconds of exercise to exclude a presynaptic NMJ transmission disorder such as LEMS. The routine needle EMG of distal and proximal muscles, especially weak muscles may reveal unstable or small amplitude, short duration, polyphasic motor unit potentials, with or without early recruitment. Unstable motor units represent muscle fibers that are blocked or come into action potential at varying intervals, resulting in MUAPs that change in configuration from impulse to impulse. Fibrillation potentials and other abnormal spontaneous activity are not seen in NMJ disorder, except in the case of botulism.
The following 2 studies are commonly performed:
-
Repetitive stimulation of a muscle at 2-3 Hz, also known as repetitive nerve stimulation (RNS)
-
Single-fiber electromyography (SFEMG), aimed at evaluating neuromuscular block, jitter, and fiber density
SFEMG is more sensitive than RNS in assessing MG. However, SFEMG is technically more difficult and much more dependent on the experience and skill of the testing physician. Consequently, RNS is the most frequently performed neurophysiologic test of neuromuscular transmission.
Repetitive nerve stimulation
RNS is abnormal in more than 50% to 70% of patients with generalized MG but are often normal in patients with purely ocular form of MG. During low-frequency (1-5 Hz) RNS, the locally available acetylcholine (ACh) becomes depleted at all neuromuscular junctions (NMJs), and less is therefore available for immediate release. This results in smaller excitatory postsynaptic potentials (EPSPs).
In patients without MG, all EPSPs exceed the threshold to generate an action potential (ie, there is a safety factor). No change in the summated compound muscle action potential (CMAP) is noted. In patients with MG, the number of AChRs is reduced, lowering the safety factor. During RNS, some EPSPs may not reach threshold, which means that no action potential is generated. This results in the decrement in the amplitude of the CMAP.
In patients with myasthenia gravis, this decremental response usually has a maximum decrement at the fourth or fifth response, followed by a tendency toward repair (see the image below). A stimulation rate of 1-5 per second should result in a 10% or more decrease in amplitude by the fourth or fifth action potential; any decrement over 10% is considered abnormal. The most common employed stimulation rate is 3 Hz.
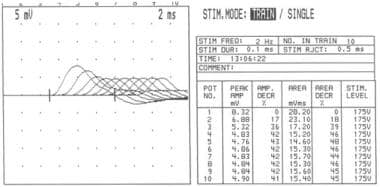 Repetitive nerve stimulation at frequency of 2 Hz showing increasing decrement in amplitude of compound muscle action potential up to fourth response (42% amplitude loss), after which it stabilizes.
Repetitive nerve stimulation at frequency of 2 Hz showing increasing decrement in amplitude of compound muscle action potential up to fourth response (42% amplitude loss), after which it stabilizes.
Patients with MG rarely have a decreased response in a clinically normal muscle. Thus, testing a proximal weak muscle gives a better yield than testing a unaffected distal muscle, even if the latter is technically easier. Testing a facial muscle (eg, the orbicularis oculi) is useful because most patients suffer from eyelid weakness or ptosis. RNS results are less likely to be positive in patients with ocular MG. Facial RNS is especially important to perform in suspected anti-MuSK MG due to much higher facial and bulbar involvement than a limb muscle.
Factors affecting results
Several factors can affect RNS results. Lower temperatures increase the amplitude of the CMAPs. Patients with MG may report clinically significant improvement in cold temperatures, and they typically report worsening of ptosis in bright sunlight or on a warm day. Therefore, maintaining a constant and perhaps higher-than-ambient temperature during RNS testing is important to bring out abnormalities of NMJ function. The temperature of the skin overlying the tested muscle should be at least 34°C.
Administration of AChE inhibitors before testing may mask the abnormality and consequently should be avoided for at least 1 day beforehand (even longer for long-acting agents).
Factors related to tetanic contraction may also affect RNS findings. A tetanic contraction of muscle is followed by 2 distinct phases:
-
Post-exercise facilitation or repair of decrement, occurring for the first 2 minutes after tetanic contraction
-
Post-exercise exhaustion, lasting an additional 15 minutes after posttetanic potentiation
During post-exercise facilitation, accumulation of calcium inside the terminal axon causes enhanced mobilization and release of ACh, which overcomes the reduced number of AChRs at the NMJ and thus leads to larger EPSPs with additional recruitment of muscle fibers, resulting in a larger CMAP. In MG, this potentiation may normalize RNS results.
In the post-exercise exhaustion phase, the NMJ is less excitable, and even fewer EPSPs reach threshold. Thus, some patients with an equivocal abnormality on RNS during the resting phase may show clear-cut abnormalities during the post-exercise exhaustion phase.
Tetanic contraction of the muscle can be achieved by applying electrical stimulation to the nerve at a rate of 50 per second for 20-30 seconds. However, this is painful. Voluntary contraction of the muscle for 10 seconds at the maximum force can achieve the same goal without discomfort and is preferred. This principle is utilized in RNS studies. If on slow RNS (3 Hz) there is no significant decrement (< 10%) on RNS at baseline, the patient should peform maximual voluntary contraction of the muscle being tested for 1 minute. This is followed by RNS immediately and at 1-minute intervals for the next 4 minutes, looking for a >10% CMAP decrement that results from post-exercise exhaustion. If at any time, either at baseline or following exercise, a significant decrement (>10%) develops, the patient should perform a brief 10 seconds of maximum voluntary contraction of the muscle being tested. Immediately following this maneuver, slow RNS is performed looking for an increment in the CMAP that suggests post-exercise facilitation or “repair” of decrement. This finding should be demonstrated in at least two nerves for the definite diagnosis of a neuromuscular transmission defect.
It is useful to get a baseline RNS on patients who are clinically and have serological positive status for MG. Typically, these patients who are initially weak will on slow RNS (3Hz) demonstrate significant decrement (>10%). When such patients who are on treatment return later at some point with worsening of symptoms, a normal RNS (showing no decrement) may be helpful in ruling out worsening of weakness due to myasthenia.
Single-fiber electromyography
SFEMG provides the most sensitive measure of myasthenia gravis. A normal SFEMG of a clinically weak muscle effectively rules out the diagnosis of MG.
A concentric needle electrode and other monopolar and bipolar needle electrodes can record single motor unit potentials, but they cannot discriminate individual muscle fibers within the motor unit. The single-fiber needle used in SFEMG, which has a small recording surface, allows recording from individual muscle fibers.
SFEMG is capable of determining jitter (ie, variability of the time interval between the action potentials of 2 single adjacent muscle fibers in the same motor unit) and fiber density (ie, number of single-fiber action potentials within recording radius of the needle). Increased jitter (with or without impulse blocking) and normal fiber density are suggestive of a neuromuscular fiber transmission defect (see the image below).
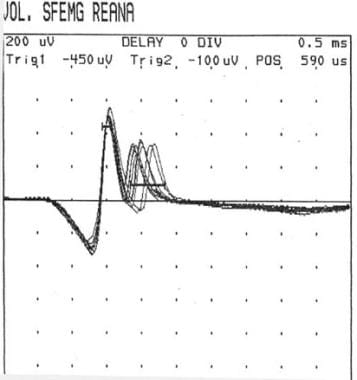 Single-fiber electromyography showing so-called jitter phenomenon (second action potential wave group).
Single-fiber electromyography showing so-called jitter phenomenon (second action potential wave group).
Examination of a weak muscle with SFEMG is more useful than examination with RNS in demonstrating abnormal neuromuscular transmission. SFEMG of the extensor digiti communis (EDC) yields abnormal results in 87% of patients with generalized MG. Examination of a second muscle raises the sensitivity to 99%. In ocular MG, examination of the frontalis is more useful than examination of the EDC. Frontalis findings are abnormal in almost 100% of patients, but only about 60% of EDC findings are abnormal.
Treatment with AChR inhibitors does not normalize SFEMG results. SFEMG findings are abnormal in almost 100% of patients, whereas RNS findings are abnormal in only 44-65%. SFEMG is a good substitute for RNS in patients with ocular MG; a study by Padua et al on 86 patients with ocular MG showed 100% sensitivity. [4] However, SFEMG is technically demanding and highly operator-dependent. In addition, it has a lower specificity, and it can give positive results in other neuromuscular disorders.
Pharmacological Testing
In patients with myasthenia gravis (MG), the number of AChRs at the NMJ is low, which results in a decreased number of interactions between ACh and its receptor. ACh released from motor nerve terminals is metabolized by AChE. As a result, pharmacologic inhibition of AChE increases ACh concentration at the NMJ, improving the chance for interactions between ACh and its receptor. Edrophonium (tensilon) is a short-acting AChE inhibitor that improves muscle weakness in patients with MG.
This test evaluates weakness (eg, ptosis, partial or complete ophthalmoplegia, and forced hand grip) in an involved group of muscles before and after intravenous (IV) administration of edrophonium. Blinding of both the examiner and the patient increases the validity of the test.
To perform the test, a butterfly needle is placed in an accessible vein. A 2 mg (0.2 mL) test dose is administered initially, as some patients are extremely sensitive to low doses. If no response and no untoward effects are noted after 30 seconds, remainder of the drug 8 mg (0.8 mL) is injected in 2 mg increments every 10-15 seconds. If the patient has objective improvement (improvement in ptosis or ophthalmoparesis) or a severe side effect, the test is aborted. The test is not considered positive if the patient reports feeling stronger. Sinus bradycardia due to excessive cholinergic stimulation of the heart is a serious complication; consequently, an ampule of atropine should be available at the bedside or in the clinic room while the test is performed.
This test may give both false-negative results and false-positive results. It has a low sensitivity in ocular MG; 50% of patients presenting with eye symptoms will be missed. On the other hand, diseases other than MG, such as amyotrophic lateral sclerosis (ALS) and cavernous sinus lesions, LEMS, botulism, congenital myasthenic syndromes, and GBS can score positive on the test. [39] This test has been combined with electromyography (EMG) and ocular tonography to increase its sensitivity in ocular MG; however, it still produces false-negative and false-positive results. Thus, a positive edrophonium test indicates abnormal neuromuscular transmission and does not specify a disease condition.
Edrophonium is being used in combination with the Lancaster red-green screen testing for diplopia. Most patients would show an improvement in some fields of gaze and a worsening in other directions of gaze. Other patients would not appreciate any change.
The combination of edrophonium with electronystagmographic analysis of optokinetic nystagmus, seems promising for the diagnosis of ocular MG. [40, 41]
Many patients receiving edrophonium besides sinus bradycardia may complain of fasciculations, borborygmi, flatus, eructations, nausea, vomiting, increased lacrimation, and in extreme cases syncope as a result of bradycardia and transient heart block.
Ice Pack Test
The ice pack test (ie, placing ice over the lid) has gained interest among ophthalmologists for assessing improvement in ptosis and diplopia in ocular myasthenia gravis (MG). The rationale behind this test is that cooling might improve neuromuscular transmission.
Movaghar and Slavin questioned the validity of such a test by demonstrating that patients with ocular MG actually improve on the ice, heat, and modified sleep tests. [42] Hence, rest might be the cause of the improvement in ocular signs. Both the ice test and the rest test are sensitive and specific in ocular MG. [43]
Histologic Findings
Routine histopathology is not part of the evaluation of myasthenia gravis (MG).
Routine light microscopy reveal mild, nonspecific abnormalities on muscle biopsy including type 1 fiber predominance, mild fiber type grouping, or type 2 fiber atrophy. [53] Studies of muscle biopsy specimens showed that the NMJs of patients with MG had only one third as many AChRs as average normal individuals. Morphologic changes, such as simplification of the pattern of postsynaptic membrane folding and an increase in the gap between the nerve terminal and the postsynaptic muscle membrane, also are present.
Lymphofollicular hyperplasia of thymic medulla occurs in 65% of patients with MG and thymoma occurs in 15%.
-
Normal neuromuscular junction showing a presynaptic terminal with a motor nerve ending in an enlargement (bouton terminale): Synaptic cleft and postsynaptic membrane with multiple folds and embedded with several acetylcholine receptors.
-
Acetylcholine receptor. Note 5 subunits, each with 4 membrane-spanning domains forming a rosette with a central opening. The central opening acts as an ion channel.
-
CT scan of chest showing an anterior mediastinal mass (thymoma) in a patient with myasthenia gravis.
-
Increasing left ptosis developing upon sustained upward gaze in patient with myasthenia gravis (A through F). Note limited elevation of left eye, denoting superior rectus palsy (A). A initially, C after around 20 seconds, F after 1 minute.
-
Cogan sign. Patient changes gaze from downward position (A) to primary position (B). Both lids are seen to overshoot in twitch (B) before gaining their initial ptotic position (D). In this case, Cogan sign is seen more obviously on right, whereas left lid is more ptotic.
-
CT scan of chest and mediastinum showing thymoma in patient with myasthenia gravis.
-
Repetitive nerve stimulation at frequency of 2 Hz showing increasing decrement in amplitude of compound muscle action potential up to fourth response (42% amplitude loss), after which it stabilizes.
-
Single-fiber electromyography showing so-called jitter phenomenon (second action potential wave group).
-
What is myasthenia gravis? Myasthenia gravis is an autoimmune disease that's categorized as a type II hypersensitivity that involves autoantibodies binding acetylcholine receptors on skeletal muscle cells. Courtesy of Osmosis.org (https://www.osmosis.org/).
-
Motor end plate and innervation. Courtesy of Wikimedia Commons.
Tables
| Distribution of Weakness | Percentage of Patients |
|---|---|
| Localized ocular | 17% |
| Ocular and generalized | 50% |
| Ocular and bulbar | 13% |
| Ocular and limb | 20% |