
Abstract
Its excellent spatial resolution makes scanning probe microscopy a capable method to investigate chemical reactions at the single-molecule level and obtain fascinating and unprecedented insights into the mechanisms of chemical transformations. Particularly exciting are recent advances in atomic force microscopy that allow bond-resolved imaging and thus make the chemical identification of organic molecular reaction intermediates and products possible. In this chapter we will give an overview about recent fundamental research on reaction mechanisms and kinetics of surface-supported reactions by scanning probe microscopy. Particular emphasis will be placed on the stabilization and statistical analysis of intermediates, which provides fundamental understanding of the microscopic driving forces of complex chemical transformations of organic molecules.
You have full access to this open access chapter, Download conference paper PDF
Similar content being viewed by others

1 Introduction
One of the greatest challenges in fundamental as well as technological research in chemistry is the determination of mechanistic aspects of reactions of organic molecules. Design of novel synthesis protocols and thus controlled fabrication of new materials critically rely on the understanding of reaction mechanisms. Information about the reaction mechanisms can be obtained from measurements of the time-dependence of the concentration of reactants and products of a chemical reaction. But such analysis of chemical kinetics cannot uniquely determine the precise sequence of transformation steps, because certain details of the mechanisms, such as the occurrence of intermediate steps, might be overlooked [1, 2].
Therefore—particularly for complex organic reactions—it is crucial to identify reaction intermediates and their respective structures. In chemistry, structure identification is commonly based on spectroscopic techniques, such as nuclear magnetic resonance, infrared spectroscopy, and mass spectrometry. These techniques provide a plethora of information about the chemical (and electronic) structure and in most cases yield unique fingerprints for different species. However, chemical reactions of organic molecules commonly yield reaction mixtures containing different chemical species, which hampers the use of such ensemble-averaging techniques. The respective reaction intermediates are difficult to separate from the reaction mixture and often are not available in sufficiently high concentrations. Also, for unknown intermediate species (which in many cases are highly reactive), reference spectra are not available and thus structure determination can be difficult.
Recently, progress has been made in the visualization of intermolecular reactions by aberration corrected transmission electron microscopy in real space [3, 5,6,7,8,9]. Reactions can be induced via the electron beam and followed in situ. The rather high energy of the electron beam imposes requirements on the stability of the chemical species to be investigated, as well as on the support. In a recent study, hetero-atoms (such as Cl and S) have been introduced for enhanced contrast for single-molecule imaging and higher stability under the electron beam [3] (Fig. 1a, b).

a, b were included with permission from Ref. [3]. Copyright 2017 American Chemical Society; original figure is available under the terms of the ACS AuthorChoice license
Real-space imaging of chemical reactions at the single-molecule level by TEM, STM, and AFM. a Aberration-corrected high-resolution transmission electron microscopy (AC-HRTEM) images showing the transition of perchlorocoronene from a face-on orientation to an edge-on orientation on graphene. This reaction is triggered by the 80 keV electron beam and has been recorded in situ [3]. b Reaction scheme derived from the experimental data and additional DFT calculations shows cleavage of C–Cl bonds by the electron beam and subsequent Diels-Alder cycloaddition of the formed aryne to the graphene substrate [3]. c STM and d AFM images along with e the derived chemical structure models show the thermally induced cyclization of 1,2-bis(2-ethynylphenyl)ethyne on Au(111) towards an (E)-1,1′-bi(indenylidene) diradical, which then reacts towards poly-(E)-1,1′-bi-(indenylidene) chains via radical step growth [4].
Scanning probe techniques, scanning tunneling microcopy (STM) and atomic force microscopy (AFM), provide superior spatial resolution for single-molecule investigations of chemical transformations [10,11,12,13,14,15,16] (Fig. 1c–e). Recent advances in AFM with functionalized tips allow to image the chemical structure of organic molecules [17,18,19,20] making this technique particularly suitable for investigations of on-surface chemistry. One of the main drawbacks of this technique, however, is the relatively long acquisition time that is needed to record images with chemical-structure resolution. In principle it is possible to control the reaction rate of a chemical transformation by variation of the reaction temperature, which opens up the possibility to investigate reactions in situ. In practice, however, it is substantially more difficult to overcome experimental difficulties associated with tip functionalization and sample drift when scanning at non-cryogenic temperatures. Another possibility to follow reactions in situ and even observe intermediates is to trigger reactions with light [21,22,23,24,25], by a change of the chemical potential of the surface by gating [26, 27], or via the scanning probe tip [11, 28,29,30,31,32,33] (different mechanisms have been reported, such as the creation of electronically and/or vibrationally excited states in the molecules, as well as force-induced chemical changes [28, 33]). However, so far this has only been demonstrated for relatively simple chemical transformations. Thus, a major research focus is put on thermally induced chemical transformations that allow exploration of a wide variety of chemical synthesis routes.
To investigate reaction mechanisms of thermally induced chemical transformations, it is necessary to stabilize potential reaction intermediates. As will be discussed below, whether and which intermediates are stabilized depends on a variety of parameters: the potential-energy landscape, energy flow at the microscopic scale, as well as entropic effects. One of the main parameters that can be controlled is the reaction temperature, the adjustment of which can be used to slow down reaction rates and in certain cases lead to “freezing” of the reaction at different steps (see for example [34,35,36,37,38,39,40,41,42,43,44,45]). Furthermore, it is possible to chemically quench a reaction at a certain step, for instance by use of additional reactants that are dosed to passivate (reversibly or irreversibly) specific reactive centers of intermediates along the reaction pathway.
In the following, a few examples will be shown demonstrating what level of understanding can be achieved based on scanning probe investigation of surface-supported reactions. In most of these examples, the experimental studies are corroborated by theoretical calculations [46]. Based on data gained through experiments, calculations can unveil chemical and physical details of molecular behavior at length and time scales that are not accessible experimentally. In the context of the investigation of reaction mechanisms and intermediate species it is particularly important to provide compelling evidence that the theoretical simulations are indeed resulting in (or based on) a relevant reaction path, i.e. a reaction path that is observed in experiment. Such crucial connections between theory and experiment can be established indirectly (for instance by comparing simulated and experimentally observed reaction kinetics) and/or, preferably, directly by identifying reaction intermediates.
2 Imaging and Counting Intermediates
In a recent study the reaction pathway of Ullmann-type coupling between bromotriphenylene molecules on a Cu(111) surface was elucidated using AFM measurements supported by theoretical calculations [37]. The exact reaction mechanism of the Ullmann reaction, i.e. coupling of aryl halides, is still under debate [10, 47,48,49,50,51]. Of particular interest is the occurrence and nature of radicals and organometallic intermediates (the molecules can form carbon-metal-carbon moieties with surface atoms), which can shed light on the catalytic role of substrate.
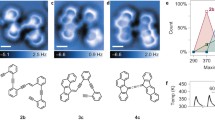
Intermediates of the transformation and coupling of bromotriphenylene molecules can be observed when subjecting the sample to a series of annealing steps to temperatures between 100 and 660 K (Fig. 2). After each annealing step, the sample was cooled down to cryogenic temperatures for AFM experiments (high-resolution AFM measurements are commonly performed at 4–6 K with CO modified tips [17, 18, 37, 52]). When the sample temperature is kept below 100 K, the precursor molecule bromotriphenylene can be detected (labeled “Br–TP” in Fig. 2). Increase of the sample temperature triggers cleavage of the bromine atom to form triphenylene molecules (labeled “TP” in Fig. 2) that—as the asymmetric distortions and local reduction of the brightness in the AFM images suggest—exhibit radical character. In the next step, at temperatures closer to room temperature, where TP molecules can more freely diffuse across the surface, intermolecular coupling leads to organometallic intermediates (labeled “TP–Cu–TP” in Fig. 2). These exist in two configurations: cis and trans. In both cases, the experimental data in conjunction with theoretical calculations shows that the molecular units are connected by Cu adatoms (as opposed to surface atoms). Finally, when the sample is heated above 500 K, bistriphenylene (“TP–TP”) are formed via transformation of the C–Cu–C bond into a covalent C–C bond.

Adapted with permission from Ref. [37]. Copyright 2017 American Chemical Society
Ullmann-type coupling reaction of Br–TP via TP and TP–Cu–TP towards TP–TP. a Chemical structure models and b corresponding AFM images show the stepwise transformation that includes an intermediate with radical character (“TP”) and an organometallic intermediate (“TP–Cu–TP”). The trans configurations of TP–TP and TP–Cu–TP are shown, cis configurations were observed as well. c The relative counts of the respective chemical species were determined as a function of annealing temperature.
Statistical analysis of the observed chemical species as a function of annealing temperature is shown in Fig. 2c. The precursor molecule “Br–TP” can only be observed when the sample temperature is kept below 300 K. The intermediates “TP” and “TP–Cu–TP” start to form at sample temperatures of 100 and 200 K, respectively. The reaction product “TP–TP” starts to form at 500 K. At 600 K the majority of the molecules have been transformed into “TP–TP”.
A recent scanning tunneling microscopy study investigated the kinetics of the transformation of 2,3,7,8,12,13,17,18-octaethylporphyrin Fe(III) chloride (FeOEP-Cl) towards iron-II-tetra-benzo-porphyrin (FeTBP) on a Cu(111) surface [40, 53]. The reaction proceeds via a dechlorination step and dehydrogenation of the molecule’s eight terminal ethyl groups. Dehydrogenation of an ethyl group leads to the formation of a vinyl group. When neighboring ethyl group are dehydrogenated, benzene rings are formed. Even though only these few types of reaction steps occur, 22 intermediate states are found depending on the sequence of these steps (Fig. 3). These transformations give rise to characteristic changes of molecular features, based on which the different species can be identified by STM measurements (with support of theoretical simulations).

Adapted from Ref. [40] with permission of The Royal Society of Chemistry
Transformation of FeOEP-Cl towards FeTBP. a STM images after heating FeOEP-Cl on Cu(111) to 430 K for 60 min. (T = 80 K, VS = 0.2 V, I = 50 pA). b STM image of FeOEP-Cl, c corresponding simulated STM image based on the DFT calculations. d STM image of FeTBP, e corresponding simulated STM image. Structural models of f FeOEP-Cl and g FeTBP. The graph shows the relative population of the reactant, intermediate species and the product after the heating step.
Using STM, the authors were able to identify and count more than 10,000 molecular species that were present after one annealing step. This gives a snapshot of the reaction kinetics, which is fitted to a model that is based on a few reasonable assumptions about the reaction sequence and its energetics (these assumptions are needed to reduce the number of free fitting parameters). Based on this fit, differences in activation energies of the respective types of reaction steps (vinyl group formation of chlorinated molecules, benzene ring formation of chlorinated molecules, dechlorination, vinyl group formation of dechlorinated molecules, benzene ring formation of dechlorinated molecules) can be estimated.
The prowess of single molecule bond-resolved AFM measurements is shown in recent studies on enediyne cyclization reactions [4, 29, 35, 36, 54]. These reactions are more complex than the reactions presented above, as multiple chemical bonds are affected in such transformations. Furthermore, different reaction pathways can occur leading to a complex reaction mixture that needs to be analyzed molecule by molecule [4, 36, 54, 55].
On a Ag(100) surface, 1,2-bis(2-ethynyl phenyl)ethyne (1) can undergo intermolecular coupling and different types of intramolecular cyclization reactions upon thermal annealing. These transformations yield a plethora of different chemical species that were analyzed using bond-resolved AFM using CO-functionalized tips [36]. The major focus of this study was put on the dimeric species, which are formed through monomer coupling by formation of covalent bonds between the terminal alkyne groups of two precursor molecules (1). Different chemical coupling of the precursors (1) and different types of cyclization reactions can occur giving rise to a complex reaction mixture consisting of a wide variety of chemically distinct dimers. However, the dimers can be grouped based on their cyclization grade: uncyclized dimers (dimers that consist of two uncyclized subunits), half-cyclized dimers (dimers that consist of one cyclized subunit and one uncyclized monomer subunit) and fully cyclized dimers (dimers that consist of two cyclized monomer subunits). With each annealing step the relative ratio of these different forms of dimers gradually shifts towards increasing cyclization grades.
Figure 4 shows molecular species representing one of many competitive reaction pathways, identified by high-resolution AFM. An intermolecular C–C bond is formed between two precursor molecules (1) leading to the eneyne intermediate 2b. Via C1–C6 and C1–C5 cyclizations and hydrogen transfers within 2b, the benzo[a]fluoren intermediate 3c is formed. Another cyclization and hydrogen transfer sequence leads to formation of the cumulene 4c, the final product of this reaction sequence.

Adapted by permission from Macmillan Publishers Ltd: Nat. Chem. 8, 678 (2016), copyright 2016
Thermally induced intermolecular coupling and intramolecular cyclization of enediyne molecules. a–d AFM images show the reaction pathway from the precursor molecules 1 via the intermediate species 2b and 3c towards the product 4c. e The relative abundance of the experimentally observed species after each annealing step was determined by single-molecule counting. f Sample temperature measured for each annealing cycle.
The relative abundance of the species associated with this reaction pathway was determined by identification of individual species in the complex reaction mixture via AFM and single-molecule counting. As can be seen in Fig. 4e, sequential annealing of the sample initially causes an increase of the relative abundance of the intermediate species (2b and 3c) and eventually leads to the prevalence of the reaction product 4c (Fig. 4e, f).
Theoretical calculations reveal the reaction barriers associated with every transformation step in the reaction pathway from 1 to 4c (Fig. 5). Surprisingly, the experimentally observed intermediates 2b and 3c exhibit the lowest transformation enthalpy barriers: ΔH‡ = 0.7 eV for 2b and ΔH‡ = 0.5 eV for 3c. This can at first seem like a contradiction to the experiment. However, the next section will give an in-depth look at why intermediates are stabilized and which other factors need to be considered to assess reaction kinetics of surface-supported reactions.

Reprinted by permission from Macmillan Publishers Ltd: Nat. Chem. 8, 678 (2016), copyright 2016
Calculated energy landscape of the transformation from 1 to 4c. Strikingly, the two experimentally observed intermediates exhibit comparably low activation enthalpy barriers (ΔH‡ = 0.7 eV for 2b and ΔH‡ = 0.5 eV for 3c).
3 Why Are Intermediates Stabilized?
The concentration \(c_{I}\) of a particular reaction intermediate \(I\) is increased by reaction steps leading towards intermediate \(I\), and at the same time decreased by any possible reaction steps, in which the intermediate \(I\) is transformed into other intermediates or a reaction product. For simplicity we will restrict our considerations to reactions consisting of a sequence of unimolecular transformations of first order without side-reactions (the main insights obtained for this simple case are also relevant for more complex reaction types). In this case the change of the concentration of the reaction intermediate \(I\) with time (\(t\)) can be written as:
where \(c_{I}\) and \(c_{I - 1}\) are the concentrations of the intermediate \(I\), as well as the preceding intermediate \(I - 1\), while \(k_{I}\) and \(k_{I - 1}\) are the respective rate constants [2]. The temperature-dependence of the rate constants can be expressed by the Arrhenius equation [56,57,58,59]:
Here, \(A\) is the so-called preexponantial factor, \(\Delta E_{A}\) is the activation energy for the respective transformation step, \(k_{B}\) is the Boltzmann constant and \(T\) is the temperature. Using the Eqs. (1) and (2), one can establish a system of coupled differential equations that—even for the simple case of a sequence of unimolecular transformations of first order without side-reactions—can give rise to complex reaction kinetics, where stabilization of potential intermediates is strongly influenced by the applied reaction temperature.
However, this apparently simple description of reaction kinetics hides certain intricacies. In particular, the local temperature T can deviate from the “macroscopic” sample temperature due to energy release associated with chemical transformations in previous reactions steps. Particularly for surface-supported reactions, this can lead to complex dynamics demanding further refinements of the kinetic modelling. Energy dissipation to the surface can occur at the same timescale as subsequent chemical transformations. The efficiency of the energy exchange between substrate and adsorbed molecular species can strongly vary dependent on the chemical structure and adsorption configuration of the respective intermediates [36].
Furthermore, entropy changes along the reaction pathway need to be taken into consideration. These can stem from vibrational entropy changes of the respective species, as well as from changes in rotational and or/translational entropy of the adsorbates. Similarly, desorption of atomic or molecular species can be associated with a substantial entropy change. Transition state theory provides a way to include the effect of entropy changes on the reaction kinetics [59,60,61,62]. According to the Eyring equation, the rate constant \(k\) can be expressed as [61]:
where \(h\) is Planck’s constant, \(\Delta S^{\ddag }\) is the activation entropy and \(\Delta H^{\ddag }\) is the activation enthalpy for the respective reaction step.
4 Bringing It All Together
The influence of microscopic energy dissipation and entropy changes along the reaction pathway was investigated in detail for the case of the enediyne coupling and cyclization cascade from 1 to 4c [36]. According to the reaction pathway shown in Fig. 5, a system of temperature-dependent kinetic rate equations can be established [see Eqs. (1) and (3)]. The temperature-dependent concentrations of the reactants, intermediates and product species are obtained by numerically solving this system of differential equations (Fig. 6). Different models can be considered:

Adapted by permission from Macmillan Publishers Ltd: Nat. Chem. 8, 678 (2016), copyright 2016
Simulation of reaction kinetics for the reaction pathway from 1 to 4c. a In the adiabatic model only the reactant 1 and the product 4c are stabilized. b Instant thermalization stabilizes intermediates with the highest transformation barriers (2a and 3a). c Taking into account selective dissipation lowers the formation temperature of 4c and leads to the stabilization of intermediate 3b. d A model taking into account selective dissipation and entropy predicts stabilization of the intermediates 2b and 3c in agreement with experimental observations. Solid (dashed) lines represent the relative concentrations of species that are (are not) experimentally observed.
In an adiabatic approximation (Fig. 6a) it is assumed that the chemical energy released in previous reaction steps is not dissipated to the substrate, but instead remains within the molecule. That means that extra energy is available to trigger the subsequent reactions steps. The simulations for this case show that no intermediates are stabilized (contradicting experimental observations). This can directly be rationalized based on the energy landscape shown in Fig. 5: once the first transition state is reached, sufficient energy is available within the molecule to overcome all subsequent transformation barriers. The reaction kinetics are determined by the first reaction barrier.
Instant thermalization (Fig. 6b): This approximation, which is often used in heterogeneous catalysis [63,64,65,66], assumes that the chemical energy released in previous reaction steps is dissipated to the surface before the next reaction step takes place. In this case the intermediates with the highest reaction barriers are stabilized, i.e. 2a and 3a—again contradictory to experimental observations.
Selective dissipation (Fig. 6c): In this approximation the dissipation of the released chemical energy is determined for each reaction step by theoretical simulations. Energy dissipation is strongly dependent on the phonon coupling between vibrational modes of the respective adsorbate and the substrate. Thus the energy dissipation can strongly vary for different intermediates. However, this model predicts stabilization of the intermediate 3b and thus also contradicts the experimental observations.
Selective dissipation and entropy (Fig. 6d): In addition to selective dissipation, entropy changes along the reaction pathway are taken into account [see Eq. (3)]. In this case entropy differences are to a large extent determined by the rototranslational mobility of the adsorbates (vibrational contributions play a smaller role). This model correctly predicts the stabilization of the intermediates 2b and 3c—in agreement with experimental observations (Fig. 4e).
Remarkably, the match between experiment and theory was achieved without the need of any free fitting parameters in the kinetic simulations. The simulations are based only on the theoretical calculations of the energy landscape, energy dissipation, and entropy. Thus, the imaging and identification of the chemical structure of intermediate species together with the agreement between simulated and experimentally observed reactions kinetics provides conclusive evidence for the reaction pathway presented in Fig. 5. Importantly, the reaction kinetics are determined not only by the potential-energy landscape, but energy dissipation to the substrate and entropy changes also need to be taken into account.
5 Outlook
The use of scanning probe microscopy to visualize bond formation and bond rearrangements in individual molecules along a reaction pathway from reactants via intermediates to the reaction products provides a tantalizing opportunity to explore mechanisms and driving forces of complex chemical reactions at the nanoscale.
It is even possible to extract three-dimensional information by atomic force microscopy [67,68,69,70], as well as information about the chemical nature of atoms [71,72,73,74]. Tip-functionalization [18, 52, 75,76,77] with organic molecules can be envisioned, which can serve as sensors that are sensitive to specific functional groups (such as radicals or other reactive centers) thereby allowing to directly assess the local chemical reactivity of organic species or certain surface terminations.
Furthermore, recent studies show how scanning probe setups can be combined with optical pump-probe techniques facilitating time-resolved measurements down to the femtosecond regime [78, 79]. Such new developments bring the realm of real-time observation of molecular movement and chemical transformations at the single-molecule level within reach.
There are exciting times ahead.
References
Wintterlin, J., Volkening, S., Janssens, T.V.W., Zambelli, T., Ertl, G.: Atomic and macroscopic reaction rates of a surface-catalyzed reaction. Science 278, 1931–1934 (1997)
Laidler, K.J.: Chemical kinetics. Harper & Row, New York (1987)
Chamberlain, T.W., Biskupek, J., Skowron, S.T., Markevich, A.V., Kurasch, S., Reimer, O., Walker, K.E., Rance, G.A., Feng, X., Müllen, K., Turchanin, A., Lebedeva, M.A., Majouga, A.G., Nenajdenko, V.G., Kaiser, U., Besley, E., Khlobystov, A.N.: Stop-frame filming and discovery of reactions at the single-molecule level by transmission electron microscopy. ACS Nano 11, 2509–2520 (2017)
Riss, A., Wickenburg, S., Gorman, P., Tan, L.Z., Tsai, H.-Z., de Oteyza, D.G., Chen, Y.-C., Bradley, A.J., Ugeda, M.M., Etkin, G., Louie, S.G., Fischer, F.R., Crommie, M.F.: Local electronic and chemical structure of oligo-acetylene derivatives formed through radical cyclizations at a surface. Nano Lett. 14, 2251–2255 (2014)
Koshino, M., Niimi, Y., Nakamura, E., Kataura, H., Okazaki, T., Suenaga, K., Iijima, S.: Analysis of the reactivity and selectivity of fullerene dimerization reactions at the atomic level. Nat Chem. 2, 117–124 (2010)
Harano, K., Takenaga, S., Okada, S., Niimi, Y., Yoshikai, N., Isobe, H., Suenaga, K., Kataura, H., Koshino, M., Nakamura, E.: Conformational analysis of single perfluoroalkyl chains by single-molecule real-time transmission electron microscopic imaging. J. Am. Chem. Soc. 136, 466–473 (2014)
Khlobystov, A.N., Porfyrakis, K., Kanai, M., Britz, D.A., Ardavan, A., Shinohara, H., Dennis, T.J.S., Briggs, G.A.D.: Molecular motion of endohedral fullerenes in single-walled carbon nanotubes. Angew. Chemie Int. Ed. 43, 1386–1389 (2004)
Ke, X., Bittencourt, C., van Tendeloo, G.: Possibilities and limitations of advanced transmission electron microscopy for carbon-based nanomaterials. Beilstein J. Nanotechnol. 6, 1541–1557 (2015)
Sloan, J., Matthewman, G., Dyer-Smith, C., Sung, A.-Y., Liu, Z., Suenaga, K., Kirkland, A.I., Flahaut, E.: Direct imaging of the structure, relaxation, and sterically constrained motion of encapsulated tungsten polyoxometalate Lindqvist ions within carbon nanotubes. ACS Nano 2, 966–976 (2008)
Grill, L., Dyer, M., Lafferentz, L., Persson, M., Peters, M.V., Hecht, S.: Nano-architectures by covalent assembly of molecular building blocks. Nat Nano. 2, 687–691 (2007)
Hla, S.-W., Bartels, L., Meyer, G., Rieder, K.-H.: Inducing all steps of a chemical reaction with the scanning tunneling microscope tip: towards single molecule engineering. Phys. Rev. Lett. 85, 2777–2780 (2000)
Lindner, R., Kühnle, A.: On-surface reactions. ChemPhysChem 16, 1582–1592 (2015)
Mali, K.S., Pearce, N., De Feyter, S., Champness, N.R.: Frontiers of supramolecular chemistry at solid surfaces. Chem. Soc. Rev. 46, 2520–2542 (2017)
Shen, Q., Gao, H.-Y., Fuchs, H.: Frontiers of on-surface synthesis: from principles to applications. Nano Today. 13, 77–96 (2017)
Held, P.A., Fuchs, H., Studer, A.: Covalent-bond formation via on-surface chemistry. Chem. A Eur. J. 1–20 (2017)
Klappenberger, F., Zhang, Y.Q., Björk, J., Klyatskaya, S., Ruben, M., Barth, J.V.: On-surface synthesis of carbon-based scaffolds and nanomaterials using terminal alkynes. Acc. Chem. Res. 48, 2140–2150 (2015)
Gross, L., Mohn, F., Moll, N., Liljeroth, P., Meyer, G.: The chemical structure of a molecule resolved by atomic force microscopy. Science 325, 1110–1114 (2009)
Jarvis, S.P.: Resolving intra- and inter-molecular structure with non-contact atomic force microscopy. Int. J. Mol. Sci. 16, 19936–19959 (2015)
Iwata, K., Yamazaki, S., Mutombo, P., Hapala, P., Ondrácek, M., Jelínek, P., Sugimoto, Y.: Chemical structure imaging of a single molecule by atomic force microscopy at room temperature. Nat. Commun. 6, 7766 (2015)
Huber, F., Matencio, S., Weymouth, A.J., Ocal, C., Barrena, E., Giessibl, F.J.: Intramolecular force contrast and dynamic current-distance measurements at room temperature. Phys. Rev. Lett. 115, 66101 (2015)
Palma, C.A., Diller, K., Berger, R., Welle, A., Björk, J., Cabellos, J.L., Mowbray, D.J., Papageorgiou, A.C., Ivleva, N.P., Matich, S., Margapoti, E., Niessner, R., Menges, B., Reichert, J., Feng, X., Räder, H.J., Klappenberger, F., Rubio, A., Müllen, K., Barth, J.V.: Photoinduced C-C reactions on insulators toward photolithography of graphene nanoarchitectures. J. Am. Chem. Soc. 136, 4651–4658 (2014)
Colazzo, L., Sedona, F., Moretto, A., Casarin, M., Sambi, M.: Metal-free on-surface photochemical homocoupling of terminal alkynes. J. Am. Chem. Soc. 138, 10151–10156 (2016)
Basagni, A., Colazzo, L., Sedona, F., Marino, M.Di, Carofiglio, T., Lubian, E., Forrer, D., Vittadini, A., Casarin, M., Verdini, A., Cossaro, A., Floreano, L., Sambi, M.: Stereoselective photopolymerization of tetraphenylporphyrin derivatives on Ag(110) at the sub-monolayer level. Chem. A Eur. J. 20, 14296–14304 (2014)
Basagni, A., Ferrighi, L., Cattelan, M., Nicolas, L., Handrup, K., Vaghi, L., Papagni, A., Sedona, F., Valentin, C.Di, Agnoli, S., Sambi, M.: On-surface photo-dissociation of C-Br bonds: towards room temperature Ullmann coupling. Chem. Commun. 51, 12593–12596 (2015)
Gao, H.-Y., Zhong, D., Mönig, H., Wagner, H., Held, P.-A., Timmer, A., Studer, A., Fuchs, H.: Photochemical Glaser coupling at metal surfaces. J. Phys. Chem. C 118, 6272–6277 (2014)
Riss, A., Wickenburg, S., Tan, L.Z., Tsai, H.-Z., Kim, Y., Lu, J., Bradley, A.J., Ugeda, M.M., Meaker, K.L., Watanabe, K., Taniguchi, T., Zettl, A., Fischer, F.R., Louie, S.G., Crommie, M.F.: Imaging and tuning molecular levels at the surface of a gated graphene device. ACS Nano 8, 5395–5401 (2014)
Wickenburg, S., Lu, J., Lischner, J., Tsai, H.-Z., Omrani, A.A., Riss, A., Karrasch, C., Bradley, A., Jung, H.S., Khajeh, R., Wong, D., Watanabe, K., Taniguchi, T., Zettl, A., Neto, A.H.C., Louie, S.G., Crommie, M.F.: Tuning charge and correlation effects for a single molecule on a graphene device. Nat. Commun. 7, 13553 (2016)
Morgenstern, K.: Switching individual molecules by light and electrons: From isomerisation to chirality flip. Prog. Surf. Sci. 86, 115–161 (2011)
Schuler, B., Fatayer, S., Mohn, F., Moll, N., Pavliček, N., Meyer, G., Peña, D., Gross, L.: Reversible Bergman cyclization by atomic manipulation. Nat. Chem. 8, 220–224 (2016)
Pavlicek, N., Schuler, B., Collazos, S., Moll, N., Pérez, D., Guitián, E., Meyer, G., Peña, D., Gross, L.: On-surface generation and imaging of arynes by atomic force microscopy. Nat. Chem. 7, 623–628 (2015)
Mohn, F., Repp, J., Gross, L., Meyer, G., Dyer, M.S., Persson, M.: Reversible bond formation in a gold-atom-organic-molecule complex as a molecular switch. Phys. Rev. Lett. 105, 266102 (2010)
Albrecht, F., Neu, M., Quest, C., Swart, I., Repp, J.: Formation and characterization of a molecule-metal-molecule bridge in real space. J. Am. Chem. Soc. 135, 9200–9203 (2013)
Ladenthin, J.N., Frederiksen, T., Persson, M., Sharp, J.C., Gawinkowski, S., Waluk, J., Kumagai, T.: Force-induced tautomerization in a single molecule. Nat. Chem. 8, 1–6 (2016)
Lafferentz, L., Eberhardt, V., Dri, C., Africh, C., Comelli, G., Esch, F., Hecht, S., Grill, L.: Controlling on-surface polymerization by hierarchical and substrate-directed growth. Nat. Chem. 4, 215–220 (2012)
Kawai, S., Haapasilta, V., Lindner, B.D., Tahara, K., Spijker, P., Buitendijk, J.A., Pawlak, R., Meier, T., Tobe, Y., Foster, A.S., Meyer, E.: Thermal control of sequential on-surface transformation of a hydrocarbon molecule on a copper surface. Nat. Commun. 7, 12711 (2016)
Riss, A., Paz, A.P., Wickenburg, S., Tsai, H.-Z., De Oteyza, D.G., Bradley, A.J., Ugeda, M.M., Gorman, P., Jung, H.S., Crommie, M.F., Rubio, A., Fischer, F.R.: Imaging single-molecule reaction intermediates stabilized by surface dissipation and entropy. Nat. Chem. 8, 678–683 (2016)
Zint, S., Ebeling, D., Schlöder, T., Ahles, S., Mollenhauer, D., Wegner, H.A., Schirmeisen, A.: Imaging successive intermediate states of the on-surface Ullmann reaction on Cu(111): role of the metal coordination. ACS Nano. 11, 4183–4190 (2017)
Papageorgiou, A.C., Fischer, S., Oh, S.C., Sağlam, Ö., Reichert, J., Wiengarten, A., Seufert, K., Vijayaraghavan, S., Écija, D., Auwärter, W., Allegretti, F., Acres, R.G., Prince, K.C., Diller, K., Klappenberger, F., Barth, J.V.: Self-terminating protocol for an interfacial complexation reaction in vacuo by metal-organic chemical vapor deposition. ACS Nano 7, 4520–4526 (2013)
Wiengarten, A., Lloyd, J.A., Seufert, K., Reichert, J., Auwärter, W., Han, R., Duncan, D.A., Allegretti, F., Fischer, S., Oh, S.C., Saglam, Ö., Jiang, L., Vijayaraghavan, S., Écija, D., Papageorgiou, A.C., Barth, J.V.: Surface-assisted cyclodehydrogenation; break the symmetry, enhance the selectivity. Chem. A Eur. J. 21, 12285–12290 (2015)
van Vörden, D., Wortmann, B., Schmidt, N., Lange, M., Robles, R., Brendel, L., Bobisch, C.A., Möller, R.: Following the steps of a reaction by direct imaging of many individual molecules. Chem. Commun. 52, 1–4 (2016)
Blake, M.M., Nanayakkara, S.U., Claridge, S.A., Fernández-Torres, L.C., Sykes, E.C.H., Weiss, P.S.: Identifying reactive intermediates in the Ullmann coupling reaction by scanning tunneling microscopy and spectroscopy. J. Phys. Chem. A. 113, 13167–13172 (2009)
Wang, W., Shi, X., Wang, S., Van Hove, M.A., Lin, N.: Single-molecule resolution of an organometallic intermediate in a surface-supported ullmann coupling reaction. J. Am. Chem. Soc. 133, 13264–13267 (2011)
Treier, M., Pignedoli, C.A., Laino, T., Rieger, R., Müllen, K., Passerone, D., Fasel, R.: Surface-assisted cyclodehydrogenation provides a synthetic route towards easily processable and chemically tailored nanographenes. Nat Chem. 3, 61–67 (2011)
Onishi, H.: Reaction intermediates on TiO2(110) identified by time-lapse scanning tunneling microscopy. Catal. Surv. Jpn 6, 1–8 (2002)
Matthiesen, J., Wendt, S., Hansen, J., Madsen, G., Lira, E., Galliker, P., Vestergaard, E., Schaub, R., Lægsgaard, E., Hammer, B., Besenbacher, F.: Observation of all the intermediate steps of a chemical reaction on an oxide surface by scanning tunneling microscopy. ACS Nano 3, 517–526 (2009)
Björk, J.: Reaction mechanisms for on-surface synthesis of covalent nanostructures. J. Phys.: Condens. Matter 28, 83002 (2016)
Xi, M., Bent, B.E.: Iodobenzene on Cu(111): formation and coupling of adsorbed phenyl groups. Surf. Sci. 278, 19–32 (1992)
Xi, M., Bent, B.E.: Mechanisms of the Ullmann coupling reaction in adsorbed monolayers. J. Am. Chem. Soc. 115, 7426–7433 (1993)
Weiss, P.S., Kamna, M.M., Graham, T.M., Stranick, S.J.: Imaging benzene molecules and phenyl radicals on Cu(111). Langmuir 14, 1284–1289 (1998)
Dong, L., Liu, P.N., Lin, N.: Surface-activated coupling reactions confined on a surface. Acc. Chem. Res. 48, 2765–2774 (2015)
Fan, Q., Gottfried, J.M., Zhu, J.: Surface-catalyzed C-C covalent coupling strategies toward the synthesis of low-dimensional carbon-based nanostructures. Acc. Chem. Res. 48, 2484–2494 (2015)
Bartels, L., Meyer, G., Rieder, K.-H.: Controlled vertical manipulation of single CO molecules with the scanning tunneling microscope: a route to chemical contrast. Appl. Phys. Lett. 71, 213 (1997)
Heinrich, B.W., Ahmadi, G., Müller, V.L., Braun, L., Pascual, J.I., Franke, K.J.: Change of the magnetic coupling of a metal-organic complex with the substrate by a stepwise ligand reaction. Nano Lett. 13, 4840–4843 (2013)
de Oteyza, D.G., Gorman, P., Chen, Y.-C., Wickenburg, S., Riss, A., Mowbray, D.J., Etkin, G., Pedramrazi, Z., Tsai, H.-Z., Rubio, A., Crommie, M.F., Fischer, F.R.: Direct imaging of covalent bond structure in single-molecule chemical reactions. Science 340, 1434–1437 (2013)
de Oteyza, D.G., Pérez Paz, A., Chen, Y.-C., Pedramrazi, Z., Riss, A., Wickenburg, S., Tsai, H.-Z., Fischer, F.R., Crommie, M.F., Rubio, A.: Noncovalent dimerization after enediyne cyclization on Au(111). J. Am. Chem. Soc. 138, 10963–10967 (2016)
Arrhenius, S.: Über die Reaktionsgeschwindigkeit bei der Inversion von Rohrzucker durch Säuren. Z. Phys. Chem. 4, 226–248 (1889)
Winzor, D., Jackson, C.: Interpretation of the temperature dependence of equilibrium and rate constants. J. Mol. Recognit. 19, 389–407 (2006)
Pollak, E., Talkner, P.: Reaction rate theory: what it was, where is it today, and where is it going? Chaos. 15, 26116 (2005)
Marbach, H., Steinrück, H.-P.: Studying the dynamic behaviour of porphyrins as prototype functional molecules by scanning tunnelling microscopy close to room temperature. Chem. Commun. 50, 9034 (2014)
Ditze, S., Stark, M., Buchner, F., Aichert, A., Jux, N., Luckas, N., Görling, A., Hieringer, W., Hornegger, J., Steinrück, H.P., Marbach, H.: On the energetics of conformational switching of molecules at and close to room temperature. J. Am. Chem. Soc. 136, 1609–1616 (2014)
Eyring, H.: The activated complex in chemical reactions. J. Chem. Phys. 3, 107–115 (1935)
Laidler, K.J., King, M.C.: Development of transition-state theory. J. Phys. Chem. 87, 2657–2664 (1983)
Reuter, K., Scheffler, M.: First-principles kinetic Monte Carlo simulations for heterogeneous catalysis: application to the CO oxidation at RuO2(110). Phys. Rev. B. 73, 45433 (2006)
Stoltze, P., Nørskov, J.K.: Theoretical modelling of catalytic reactions. In: Handbook of Heterogeneous Catalysis. Wiley-VCH Verlag GmbH & Co. KGaA (2008)
Meyer, J., Reuter, K.: Modeling heat dissipation at the nanoscale: an embedding approach for chemical reaction dynamics on metal surfaces. Angew. Chemie Int. Ed. 53, 4721–4724 (2014)
Neurock, M.: The microkinetics of heterogeneous catalysis. By J. A. Dumesic, D. F. Rudd, L. M. Aparicio, J. E. Rekoske, and A. A. Treviño, ACS Professional Reference Book, American Chemical Society, Washington, DC, 1993, 315p (1994)
Schuler, B., Liu, W., Tkatchenko, A., Moll, N., Meyer, G., Mistry, A., Fox, D., Gross, L.: Adsorption geometry determination of single molecules by atomic force microscopy. Phys. Rev. Lett. 111, 106103 (2013)
Moreno, C., Stetsovych, O., Shimizu, T.K., Custance, O.: Imaging three-dimensional surface objects with submolecular resolution by atomic force microscopy. Nano Lett. 15, 2257–2262 (2015)
Albrecht, F., Pavliček, N., Herranz-Lancho, C., Ruben, M., Repp, J.: Characterization of a surface reaction by means of atomic force microscopy. J. Am. Chem. Soc. 137, 7424–7428 (2015)
Albrecht, F., Bischoff, F., Auwärter, W., Barth, J.V., Repp, J.: Direct identification and determination of conformational response in adsorbed individual nonplanar molecular species using noncontact atomic force microscopy. Nano Lett. 16, 7703–7709 (2016)
Schmid, M., Stadler, H., Varga, P.: Direct observation of surface chemical order by scanning tunneling microscopy. Phys. Rev. Lett. 70, 1441–1444 (1993)
Sugimoto, Y., Pou, P., Abe, M., Jelinek, P., Pérez, R., Morita, S., Custance, O.: Chemical identification of individual surface atoms by atomic force microscopy. Nature 446, 64 (2007)
van der Heijden, N.J., Hapala, P., Rombouts, J.A., van der Lit, J., Smith, D., Mutombo, P., Švec, M., Jelinek, P., Swart, I.: Characteristic contrast in Δfmin maps of organic molecules using atomic force microscopy. ACS Nano 10, 8517–8525 (2016)
Onoda, J., Ondráček, M., Jelínek, P., Sugimoto, Y.: Electronegativity determination of individual surface atoms by atomic force microscopy. Nat. Commun. 8, 15155 (2017)
Mohn, F., Schuler, B., Gross, L., Meyer, G.: Different tips for high-resolution atomic force microscopy and scanning tunneling microscopy of single molecules. Appl. Phys. Lett. 102, 73109 (2013)
Okabayashi, N., Gustafsson, A., Peronio, A., Paulsson, M., Arai, T., Giessibl, F.J.: Influence of atomic tip structure on the intensity of inelastic tunneling spectroscopy data analyzed by combined scanning tunneling spectroscopy, force microscopy, and density functional theory. Phys. Rev. B Condens. Matter Mater. Phys. 93, 1–6 (2016)
Xin, X., Gan, L.-Y., Van Hove, M.A., Ren, X., Wang, H., Guo, C.-S., Zhao, Y.: Exploring molecules beyond CO as tip functionalizations in high-resolution noncontact atomic force microscopy: a first principles approach. ACS Omega. 1, 1004–1009 (2016)
Cocker, T.L., Peller, D., Yu, P., Repp, J., Huber, R.: Tracking the ultrafast motion of a single molecule by femtosecond orbital imaging. Nature 539, 263–267 (2016)
Peplow, M.: The next big hit in molecule Hollywood. Nature 544, 408–410 (2017)
Acknowledgements
I want to thank everyone. Support from the European Research Council Consolidator Grant NanoSurfs (No. 615233) is acknowledged. A.R. acknowledges fellowship support by the Austrian Science Fund (FWF): J3026-N16.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made. The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2018 The Author(s)
About this paper

Cite this paper
Riss, A. (2018). Mechanistic Insights into Surface-Supported Chemical Reactions. In: de Oteyza, D., Rogero, C. (eds) On-Surface Synthesis II. Advances in Atom and Single Molecule Machines. Springer, Cham. https://doi.org/10.1007/978-3-319-75810-7_1
Download citation
DOI: https://doi.org/10.1007/978-3-319-75810-7_1
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-75809-1
Online ISBN: 978-3-319-75810-7
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)