
Abstract
Bats play an essential role in maintaining ecosystems. Their unique characteristics increase the likelihood of interactions with various species, making them a potential source for the emergence and spread of infectious diseases. Hantaviruses are continuously expanding their range of hosts. This study presents the identification of a partial genome associated with Hantavirus in samples collected from neotropical bats. We conducted a metagenomic study using samples from Carollia perspicillata in Maranhão, Brazil. Tissue fragments were used for RNA extraction and subsequent sequencing. The resulting data was subjected to bioinformatic analysis. A sequence showing an identity of 72.86% with the L gene in the reference genome was obtained. The phylogenetic analysis revealed the study sequence, denoted as Buritiense, clustering within the Mobatvirus clade. The intragroup analysis showed a broader dispersion and were markedly asymmetric. This observation suggests the possibility that Buritiense could potentially represent a new species within the bat-borne hantaviruses, but further analyses are needed to provide additional insights if bats plays a role as reservoirs and the potential for transmission to human populations.
Similar content being viewed by others
Introduction
Bats play an essential role within ecosystems, such as insect population control and plant pollination. The adaptation of bats to a variety of environments and their flying capabilities afford them the opportunity to come into contact with various species, including rodents, birds, and other mammals, rendering them a potential source for the emergence and dissemination of infectious diseases. Their immunological and physiological characteristics of bats may enable them to harbor viruses in a manner that does not induce illnesses within themselves, but these viruses could potentially be transmitted1,2.
Hantaviruses are one of the various types of emerging viruses that have presented serious public health issues in the last decades. In humans, Hantavirus (HV) infections can lead to two significant clinical syndromes, Hemorrhagic Fever with Renal Syndrome (HFRS) and Hantavirus Cardiopulmonary Syndrome (HCPS). HFRS, characterized by fever, hemorrhage, and renal complications, is primarily found in Asian countries. On the other hand, HCPS is more prevalent in the Americas, manifests with severe respiratory and cardiovascular symptoms, often leading to a critical condition3. Recent cases of hantaviruses have been confirmed in South America, in countries such as Argentina, Panama, and Brazil4.
The first description of HV emerged from studies on the origin of hemorrhagic fever in over 3000 soldiers during the Korean conflict (1950–1953). However, the causative agent of the disease remained unknown until the early 1980s when the Hantaan virus was isolated, found in the lungs of the striped field mouse (Apodemus agrarius), its natural reservoir3,5.
As the costs of Next-Generation Sequencing (NGS) have declined, obtaining complete viral sequences or draft genomes has become more attainable without the need for cell culture. The characterization of the virome in various host species now serves as a valuable tool for zoonotic surveillance. The metagenomic approach not only facilitates the detection of well-known viruses of significance, but also opens the door to the identification of novel viral species6. HV are primarily transmitted by rodents, but bats in Brazil have also been identified as hosts for different HV species7,8,9.
Members of the family Hantaviridae are enveloped, spherical with a diameter from 80 to 120 nm and have segmented negative-stranded RNA linear genome. The genome encodes 3 segments L, M and S. The segment L encodes RNA-dependent RNA polymerase (RdRp), the M segment encodes the glycoprotein precursor (GPC) which is later cleaved into G1 and G2 and the S segment encodes the nucleoprotein (NP). These proteins are essential in the virus life cycle, replication, and interactions with host cells. Among the four subfamilies, Mammahantavirinae have a particular significance, as it includes important genera, such as Orthohantavirus and Mobatvirus, which are known to be hosted by rodents and bats respectively3,10.
The increasing number of highly impact viral diseases in public health is directly linked to environmental and climatic changes. These impacts contribute to the spread of zoonotic diseases, altering the endemic habitats of wildlife, leading to changes in species distribution, as well as a reduction in the barrier between wildlife and the human population. This, in turn, disrupts the ecosystem and exposes humans to emerging pathogens. In the Brazilian forest, this scenario is particularly concerning, given the incidences of deforestation and disturbance of the natural fauna11,12.
HV is among the viruses extending its connections to new hosts, having also been identified in domestic animals. Environmental and economic factors are intensifying human interaction with the virus hosts, giving rise to scenarios where new strains with potentially heightened infectivity and pathogenicity can emerge12,13. For this reason, we may be facing new challenges in disease prevention and control, as we understand that the HV type is intimately linked to host selection and regional adaptation, continually evolving through genetic recombination13,14. This study reports the discovery of a partial genome related to HV in samples obtained from neotropical bats captured in the Brazilian biome.
Results
After bioinformatic analysis, a contig of 6291 nucleotides (nt) has been recovered, which encodes a protein comprising 2096 amino acids (aa). The 5’ and 3’ ends are missing in this genome. The BlastX results showed 99% query coverage and 73.21% identity with Dakrong virus (GenBankID: MG663536), corresponding to segment L, which encodes RNA polymerase. This novel sequence has been named Buritiense virus.
The sequences were translated and aligned to facilitate phylogenetic inference analysis based on the partial L segment of HV, encompassing positions between sites 137–6398, related to the RdRp domain Fig. 1.

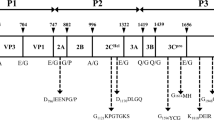
Conserved domains of Mobatvirus. Demonstration of the different functional domains of proteins from various viruses belonging to the genus Mobatvirus. The yellow arrow corresponds to the coding region of the proteins. The different domains are shown, such as RNA endonuclease, cap-snatching (TIGR04202) in red, RNA-dependent RNA polymerase (PF12426) in green, Bunyavirus RNA-dependent RNA polymerase (PF04196) in purple, and RdRp of negative ssRNA viruses with segmented genomes catalytic domain profile (PS50525) in blue.
The nt and aa identity matrices were consolidated in Table S1. At Mobatvirus genus, the aa matrix showed a reduced genetic distance between the recently sequenced Buritiense compared to the Dakrong and Robinaense HV species, with values of 0.2672 and 0.2676, respectively. When assessed at the nt level, the distances were 0.4214 and 0.4369 in comparison to Dakrong and Robinaense, respectively.
On the other hand, at Orthohantavirus genus, the species Boweense and Jejuense exhibited higher genetic distances in relation to Buritiense, with values of 0.3651 and 0.3642 for the aa matrix and 0.5443 and 0.5290 for the nt matrix. The aa tree was used to represent the phylogeny.
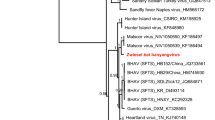
Assessment of the phylogenetic signal in the sequence set used to reconstruct the Hantaviridae phylogeny revealed that 98.7% of quartets were resolved (see Supplementary Fig. S1). In the phylogenetic inference, three HV genera are observed, with the Buritiense strain grouping within the Mobatvirus clade, which in turn exhibited two distinct subclades: one for bat hosts and another for rodents with a support of 85%. The Buritiense strain formed an isolated branch of clade within the bat subclade supported at 99%, closely related to the Quenzonense and Robinaense species as illustrated in Fig. 2.

Phylogenetic tree. Reconstruction of the phylogeny of Hantaviridae family representative viruses was performed using the Maximum Likelihood method. The bootstraping values defined for a run of 1.000 repetitions are displayed on each node. Colored circles indicate the clades of the main taxonomic groups, with the newly sequenced taxon highlighted in red. The study sequence, Buritiense, forms a distinct branch within bats clade, near to Quenzonense and Robinaense.
Two boxplot graphs were generated to illustrate the intergroup and intragroup genetic divergence for the genera Orthohantavirus, Mobatvirus, and Thottimvirus. On the graph A, the bat subclade Mobatvirus I intergroup demonstrates a narrow interquartile range, with most of the data clustered around the median, that indicates minimal data dispersion within the box plot. This observation persists despite the presence of an outlier. The data exhibit a similar distance range to that of the rodent subclade Mobatvirus II intergroup (see Supplementary Fig. S2).
When comparing each formed clade in relation to the others, Mobatvirus demonstrates low intergroup dispersion, indicating a small distance. In intragroup analysis, the values had a larger dispersion and were entirely asymmetric. These data imply that Buritiense could potentially belong to a new species within bat-borne HVs.
Discussion
A significant objective of infectious disease research is to understand and characterize the unidentified pathogens present in reservoirs before they manifest in humans. Bats are involved in the transmission cycle of various emerging and re-emerging diseases6. Brazil has become a recurrent location for research due to the vast variety of bat species across its different biomes15. All known human pathogenic HV are rodent-borne and belong to the Orthohantavirus. The HCPS presents a significant public health concern in Brazil, given the numerous reported cases. Infections are often linked to precarious agricultural conditions that foster rodent infestations, as well as inadequate housing in rural areas, rendering individuals susceptible to rodent excretions16. Furthermore, ecosystem alterations, such as deforestation, contribute to an imbalance in the proximity dynamics with humans. In urban settings, the virus can also proliferate, as reservoirs thrive in informal urban settlements, easily drawn by food sources and inadequate hygiene practices17,18.
Bats also begin to have a different interaction with humans, potentially allowing the virus to encounter permissive cells for replication and the ability to disseminate it.
A study conducted with Phyllostomidae, Molossidae and Vespertilionidae bats captured in the southeastern region of Brazil, between 2012 and 2014, detected IgG antibodies for Araraquara hantavirus (ARAQV) using an indirect enzyme-linked immunosorbent assay (ELISA). ARAQV is a pathogenic HV genotype, inserted within the Andes orthohantavirus genus, which causes diseases in humans19. This supports the idea that these bats become infected with the HV at some point in their lifetime. In another report, HV was detected in Carollia perspicillata and Desmodus rotundus, both from the Phyllostomidae family, by indirect ELISA using a recombinant N protein of ARAQV as antigen. Furthermore, blood samples and tissue from the kidneys, liver, heart, spleen, and lungs were subjected to RT-PCR, confirming the presence of HV in D. rotundus. Immunohistochemical analysis revealed positive brown immune-staining for the HV nucleocapsid in hepatocyte cells and striated cardiac muscle cells7.
Most HV cases occur in the southern and southeastern regions of Brazil17, and due to their higher vulnerability, virus monitoring studies are more frequent in these areas7,8,9,19. However, all reports of HV in bats in Brazil are related to Orthohantavirus, here we report a sequence belonging to the Mobatvirus genus.
There are studies that discuss the possibility of other mammals could be the source of infection, given the close phylogenetic relationships among certain HV taxa across large geographic areas, supporting the occurrence of long-term virus-host co-divergence among the mammalian orders Rodentia, Chiroptera, and Soricomorpha20. Sequences related to Mobatvirus genus, including Xuan son virus, Laibin virus, Drakong virus and Quenzon virus, that have complete genomes, are described in frugivorous and insectivororus bats from Asia21, and some in bats from Africa22,23. The limited reports in South America highlight the need for further in-depth studies on the virome of Chiroptera fauna in this region.
At Maranhão, only human serologic evidence has indicated the circulation of HV in six municipalities. Notably, no cases of HCPS were officially declared in any of these areas. These findings imply that either mild or asymptomatic cases were taking place, but were not recognized as HCPS, or that cases of HV infection were not being reported. In the state of Ceará, also situated in the Northeastern of Brazil, blood samples were collected from patients suspected of Dengue virus infection and subjected to ELISA, using the recombinant N protein of ARAQV as antigen, three tested positive. One presented IgM antibodies HV, while the other two exhibited IgG antibodies to HV24. This emphasizes the importance of virological surveillance in other regions, where HV may also circulate in bats, suggesting that moderate or asymptomatic cases may be occurring in these locations.
In the North and Northeastern region of Brazil, there are many rural areas characterized by abundant native plantations used for extractive activities and the local population frequents these areas to derive their livelihood from nature25,26. This interaction places them in direct proximity to bats, potentially facilitating viral transmission if these animals are carriers of pathogenic HV.
It is known that frugivorous bats are responsible for the transmission of many RNA viruses2,6. In Brazil, a variety of bat species primarily act as reservoirs for the Rabies virus. Reports of HV in bats are recent, and it is unknown whether the animals develop the disease or not27. Therefore, the feeding dynamics may be related to the viral source, frugivorous bats, such as Carollia perspicillata, may share the same food sources with other wildlife, enabling viral transmission between them.
Molecular biology techniques have led to the identification of numerous bat-borne viruses across the globe. Nevertheless, NGS methods have significantly expedited the discovery of novel bat viruses2. In the analysis of bat tissue from East Africa, a novel virus named Kiwira virus was identified within the genus Mobatvirus. This discovery revealed a closer phylogenetic proximity to the Quezon virus and Robina virus23. Just as in our analyses, it’s also observed the formation of a branch close to Robinaense and Quezonense viruses with Buritiense in the phylogenetic tree.
The recent discovery was Brno virus in the European bats collected in the Czech Republic during 2008–2013, sequence comparison revealed that this virus showed 54.7–78.3% nt and 44.5–81.7% aa sequence identity with other bat-borne HV. Interestingly, it clustered more closely with the Longquan virus, found in Chinese bats20,28. In our findings, the obtained sequences showed a closer affinity with HVs from Southeast Asian bats.
Nevertheless, the genomes of bat-borne hantaviruses are generally described as incomplete. The limited detection may be attributed to the high divergence of their genomes, as well as the localized nature of infection, small sample sizes, primer incompatibilities, suboptimal PCR cycling conditions, and variable tissue preservation with degraded RNA21,29. The study sequence is part of a viral metagenomic analysis for surveillance purposes, making it crucial to investigate whether there are sequences related to viral families that cause diseases in humans. Therefore, even though we have not been able to obtain the complete sequence of this HV, it is a significant discovery indicating the presence of Mobatvirus circulating in Brazilian bats.
Methods
Site and sample collection
A metagenomic study was conducted for the virological surveillance of RNA hosted in bats inhabiting specific areas in the state of Maranhão, located in Northeastern Brazil. The capture was authorized by the Ethics Committee on Animal Experimentation of State University of Maranhão, number 025/2019 and adhered to the prescribed research protocols in accordance with the ARRIVE guidelines30. All methods were conducted in accordance with the guidelines and regulations outlined in the American Veterinary Medical Association (AVMA) Guidelines for the Euthanasia of Animals (2020).
Three frugivorous bats, Carollia perspicillata, were captured using mist nets in a district of Timon City, Maranhão, named Buriti Cortado (5\(^\circ \) 09’ 21.5” S 43\(^\circ \) 11’ 33.4” W) in October 2021 Fig. 3. They were anesthetized with ketamine/xylazine and euthanized by exsanguination. The tissues (liver, lung, heart, and kidney) were collected and stored in liquid nitrogen until transportation to the Institute, where they were stored at \(-80^\circ \hbox {C}\). The samples were grouped in three tubes for analysis.

Map of the collection site in Timon, Maranhão The region is situated in the Northeastern of Brazil. The figure was generated using QGIS v.3.10 software (available at https://qgis.org/en/site/), which utilized satellite approximations from the Google Maps application in conjunction with the IBGE 2022 cartographic database (available at https://www.ibge.gov.br/).
RNA extraction, cDNA synthesis and sequencing
In this study, a pool of heart, lung, and kidney was used in a unique tube for the isolation of total RNA. The process was initiated using the TRIzolTM Plus RNA Purification Kit and a 5 mm tungsten bead using the TissueLyser II system (Qiagen, Hilden, Germany) for 2 min at 25 Hz, adhering to the prescribed protocols provided by the manufacturer. Following this step, the concentration of RNA was quantified employing the Qubit® RNA HS Assay Kit in conjunction with the Qubit® 2.0 Fluorometer (Invitrogen), while RNA integrity was assessed through the application of the Agilent RNA 6000 Pico Kit on the Bioanalyzer 2100 system (Agilent Technologies).
The synthesis of complementary DNA (cDNA) was carried out, utilizing the SuperScriptTM IV VILOTM Master Mix (Invitrogen) for the initial strand synthesis, and the Second Strand cDNA Synthesis Kit (Invitrogen) for the synthesis of the second strand. The resultant cDNA was subsequently subjected to a purification process using the PureLinkTM PCR Purification Kit (Invitrogen).
The generation of sequencing libraries was facilitated by the Nextera XT DNA Library Preparation Kit designed for Illumina® platforms, meticulously following the manufacturer’s recommended procedures. Accurate quantification of the library was performed employing the Qubit® dsDNA HS Assay kit, while fragment sizes were precisely evaluated with the High Sensitivity DNA Analysis Kit on the Bioanalyzer 2100 (Agilent Technologies).
High-throughput sequencing was conducted on an Illumina® NextSeq 500 platform, employing the NextSeq 500/550 High Output v2.5 (300 cycles) kit, thus generating paired-end sequencing reads for comprehensive data analysis.
Bioinformatic analysis
Fastp was employed to assess the quality of raw reads and eliminate short reads, those with low quality, and undetermined bases31. The generated results were assembled using the De Novo method in the MEGAHIT v1.2.9 assembler32 and aligned against non-redundant protein and nucleotide databases using DIAMOND33 and KRAKEN234 respectively, with a significance threshold set at \(10^{-5}\).
Contigs were further examined using MEGAN6, KronaTools v2.8.1, and the Pavian v1.0 software, which provided taxonomic classifications corresponding to the viral contigs35,36,37. Subsequently, the contig was mapped against the raw data and aligned to a reference sequence using Geneious v.9.1.838.
Identity/divergence analysis
The partial genome obtained was compared with 37 RefSeq genomes from the subfamily Mammahantavirinae and an outgroup from the subfamily Repantavirinae. All sequences are available in Table S2. The multiple alignment of the dataset and the sequences of this study were performed using Clustal W for nucleotide and amino acid analysis39. The partial genome sequence of HV was deposited in GenBank under the accession number OR684449.
The identity and divergence matrices were generated using Geneious v.9.1.8 and MEGA X40. The amino acid identity matrix, generated by MEGA X using the amino acid substitution p-distances, was converted, using the R programming language, into a boxplot graphs illustrating intragroup and intergroup distances/divergence between members of the same genus when compared to each other and in relation to new sequence of HV.
Phylogenetic analysis
Phylogenetic inference was based on the amino acid sequences of various virus strains belonging to the Hantaviridae family, available in the National Center for Biotechnology Information database (http://www.ncbi.nlm.nih.gov). The coding regions of the L segment were utilized for analysis.The set of sequences, including the sample from the present study, underwent Multiple Sequence Alignment (MSA), followed by manual inspection using Geneious v.9.1.8 software (available at https://www.geneious.com/).
Phylogenetic inferences were conducted using the Maximum Likelihood (ML) method, implemented in the IQ-TREE v.241, employing the LG+F+R5 substitution model and employing 1000 bootstrap replicates for statistical support42. The visualization and editing of the resulting trees were conducted using FigTree (available at http://tree.bio.ed.ac.uk/software/figtree/) and Inkscape (https://inkscape.org/), respectively.
Ethical approval and informed consent
The project was authorized by the Ethics Committee on Animal Experimentation of State University of Maranhão, number 025/2019 and adhered to the prescribed research protocols in accordance with the ARRIVE guidelines.
Conclusion
The report of HV in bats from Northeastern Brazil is intriguing. Further analysis regarding the viral load in different tissues and viral isolation, followed by infection experiments, are necessary to provide additional insights and confirm whether bats indeed act as reservoirs. This study raises concerns about its potential transmission to the human population. Understanding the viral diversity carried by these species can aid in staying alert about potential spillover occurrences.
Data availability
The consensus sequence of this research have been submitted to the National Center for Biotechnology Information under accession OR684449 and it’s available at December 13/2023.
References
O’Shea, T. J. et al. Bat flight and zoonotic viruses. Emerg. Infect. Dis. 20, 741–745. https://doi.org/10.3201/eid2005.130539 (2014).
Letko, M., Seifert, S. N., Olival, K. J., Plowright, R. K. & Munster, V. J. Bat-borne virus diversity, spillover and emergence. Nat. Rev. Microbiol. 18, 461–471. https://doi.org/10.1038/s41579-020-0394-z (2020).
Mir, M. A. Hantaviruses. Clin. Lab. Med. 30, 67–91. https://doi.org/10.1016/j.cll.2010.01.004 (2010).
Sah, R. et al. Recent outbreaks of hantavirus-a very lethal and zoonotic virus: An update and counteracting strategies. Int. J. Surg. Open 49, 100582. https://doi.org/10.1016/j.ijso.2022.100582 (2022).
Kuhn, J. H. & Schmaljohn, C. S. A brief history of bunyaviral family hantaviridae. Diseases 11, 38. https://doi.org/10.3390/diseases11010038 (2023).
Brussel, K. V. & Holmes, E. C. Zoonotic disease and virome diversity in bats. Curr. Opin. Virol. 52, 192–202. https://doi.org/10.1016/j.coviro.2021.12.008 (2022).
Sabino-Santos, G. Jr. et al. Natural infection of neotropical bats with hantavirus in Brazil. Sci. Rep. 8, 9018. https://doi.org/10.1038/s41598-018-27442-w (2018).
Bueno, L. M., Melo, D. M., Azevedo, R. D., de Souza, W. M. & Figueiredo, L. T. M. Serological evidence of hantavirus infection in neotropical bats in an urban area of São Paulo state, Brazil. Trans. R. Soc. Trop. Med. Hyg. 117, 297–300. https://doi.org/10.1093/trstmh/trac111 (2022).
de Araujo, J. et al. Detection of hantavirus in bats from remaining rain forest in São Paulo, Brazil. BMC Res. Notes 5, 1–5. https://doi.org/10.1186/1756-0500-5-690 (2012).
Lefkowitz, E. J. et al. Virus taxonomy: the database of the International Committee on Taxonomy of Viruses (ICTV). Nucleic Acids Res. 46, D708–D717. https://doi.org/10.1093/nar/gkx932 (2017).
Smith, I. & Wang, L.-F. Bats and their virome: An important source of emerging viruses capable of infecting humans. Curr. Opin. Virol. 3, 84–91. https://doi.org/10.1016/j.coviro.2012.11.006 (2013).
Ellwanger, J. H. et al. Beyond diversity loss and climate change: Impacts of amazon deforestation on infectious diseases and public health. An. Acad. Bras. Ciên.https://doi.org/10.1590/0001-3765202020191375 (2020).
Williams, E. P. et al. Common themes in zoonotic spillover and disease emergence: Lessons learned from bat- and rodent-borne RNA viruses. Viruses 13, 1509. https://doi.org/10.3390/v13081509 (2021).
Wang, N. et al. Genetic evolution analysis and host characteristics of hantavirus in Yunnan province, China. Int. J. Environ. Res. Public Health 19, 13433. https://doi.org/10.3390/ijerph192013433 (2022).
Delgado-Jaramillo, M., Aguiar, L. M. S., Machado, R. B. & Bernard, E. Assessing the distribution of a species-rich group in a continental-sized megadiverse country: Bats in Brazil. Divers. Distrib. 26, 632–643. https://doi.org/10.1111/ddi.13043 (2020).
da Saúde. Secretaria de Vigilância em Saúde. Departamento de Vigilância Epidemiológica, B. M. Manual de vigilância, prevenção e controle das hantaviroses (Ministério da Saúde, Brasília, 2013).
de Oliveira, S. V. et al. Vulnerability of Brazilian municipalities to hantavirus infections based on multi-criteria decision analysis. Emerg. Themes Epidemiol. 12, 1–8. https://doi.org/10.1186/s12982-015-0036-5 (2015).
Willemann, M. C. A. & de Oliveira, S. V. Risk factors associated with hantavirosis fatality: A regional analysis from a case-control study in Brazil. Rev. Soc. Bras. Med. Trop. 47, 47–51. https://doi.org/10.1590/0037-8682-0243-2013 (2014).
Sabino-Santos, G. et al. Evidence of hantavirus infection among bats in Brazil. Am. J. Trop. Med. Hyg. 93, 404–406. https://doi.org/10.4269/ajtmh.15-0032 (2015).
Guo, W.-P. et al. Phylogeny and origins of hantaviruses harbored by bats, insectivores, and rodents. PLoS Pathog. 9, e1003159. https://doi.org/10.1371/journal.ppat.1003159 (2013).
Arai, S. & Yanagihara, R. Genetic diversity and geographic distribution of bat-borne hantaviruses. Curr. Issues Mol. Biol. 39, 1–28. https://doi.org/10.21775/cimb.039.001 (2020).
Weiss, S. et al. Hantavirus in bat, Sierra Leone. Emerg. Infect. Dis. 18, 159–161. https://doi.org/10.3201/eid1801.111026 (2012).
Weiss, S. et al. Kiwira virus, a newfound hantavirus discovered in free-tailed bats (Molossidae) in East and Central Africa. Viruses 14, 2368. https://doi.org/10.3390/v14112368 (2022).
Lima, D. M. et al. Hantavirus infection in suspected dengue cases from state of Ceará, Brazil. Rev. Soc. Bras. Med. Trop. 44, 795–796. https://doi.org/10.1590/s0037-86822011000600031 (2011).
Camilotti, V. L., Pinho, P., Brondízio, E. S. & Escada, M. I. S. The importance of forest extractive resources for income generation and subsistence among caboclos and colonists in the Brazilian amazon. Hum. Ecol. 48, 17–31. https://doi.org/10.1007/s10745-020-00127-7 (2020).
Brandão, D. O., Barata, L. E. S., Nobre, I. & Nobre, C. A. The effects of amazon deforestation on non-timber forest products. Reg. Environ. Change 21, 1–13. https://doi.org/10.1007/s10113-021-01836-5 (2021).
Castelo-Branco, D. et al. Role of Brazilian bats in the epidemiological cycle of potentially zoonotic pathogens. Microb. Pathog. 177, 106032. https://doi.org/10.1016/j.micpath.2023.106032 (2023).
Straková, P. et al. Novel hantavirus identified in European bat species Nyctalus noctula. Infect. Genet. Evol. 48, 127–130. https://doi.org/10.1016/j.meegid.2016.12.025 (2017).
Gu, S. et al. Molecular phylogeny of hantaviruses harbored by insectivorous bats in Côte d’ivoire and Vietnam. Viruses 6, 1897–1910. https://doi.org/10.3390/v6051897 (2014).
du Sert, N. P. et al. The ARRIVE guidelines 2.0: Updated guidelines for reporting animal research. PLOS Biol. 18, e3000410. https://doi.org/10.1371/journal.pbio.3000410 (2020).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890. https://doi.org/10.1093/bioinformatics/bty560 (2018).
Li, D., Liu, C.-M., Luo, R., Sadakane, K. & Lam, T.-W. Megahit: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31, 1674–1676. https://doi.org/10.1093/bioinformatics/btv033 (2015).
Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60. https://doi.org/10.1038/nmeth.3176 (2014).
Wood, D. E., Lu, J. & Langmead, B. Improved metagenomic analysis with kraken 2. Genome Biol. 20, 1–13. https://doi.org/10.1186/s13059-019-1891-0 (2019).
Huson, D. H., Auch, A. F., Qi, J. & Schuster, S. C. MEGAN analysis of metagenomic data. Genome Res. 17, 377–386. https://doi.org/10.1101/gr.5969107 (2007).
Ondov, B. D., Bergman, N. H. & Phillippy, A. M. Interactive metagenomic visualization in a web browser. BMC Bioinform. 12, 1–10. https://doi.org/10.1186/1471-2105-12-385 (2011).
Breitwieser, F. P. & Salzberg, S. L. Pavian: Interactive analysis of metagenomics data for microbiome studies and pathogen identification. Bioinformatics 36, 1303–1304. https://doi.org/10.1093/bioinformatics/btz715 (2019).
Kearse, M. et al. Geneious basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649. https://doi.org/10.1093/bioinformatics/bts199 (2012).
Thompson, J. D., Higgins, D. G. & Gibson, T. J. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22, 4673–4680. https://doi.org/10.1093/nar/22.22.4673 (1994).
Kumar, S., Stecher, G., Li, M., Knyaz, C. & Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549. https://doi.org/10.1093/molbev/msy096 (2018).
Nguyen, L.-T., Schmidt, H. A., von Haeseler, A. & Minh, B. Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. https://doi.org/10.1093/molbev/msu300 (2014).
Felsenstein, J. Confidence limits on phylogenies: An approach using the bootstrap. Evolution 39, 783–791. https://doi.org/10.1111/j.1558-5646.1985.tb00420.x (1985).
Acknowledgements
This study was supported by the workers and students of the Evandro Chagas Institute and State University of Maranhão who work in the fields of virology, molecular biology, and bioinformatics. We appreciate the dedication of each individual in contributing to the development of these results.
Funding
This research was funded by the Coordination for Improvement of Higher Education Personnel Foundation (CAPES) Grant Nos. 88887.510220/2020-00 and 1703/2018. This study was supported by Brazilian Council for Scientific and Technologic Development Agency (CNPq) for financial support to ACRC Grant No. 314522/2021-2) and Evandro Chagas Institute, Ministry of Health, Brazil.
Author information
Authors and Affiliations
Contributions
The project was conceived by A.C.R.C. The capture of bats was carried out by T.F.S.B.C. The samples were collected by M.C.B and S.B.M. Molecular biology aspects were carried out by M.B.S. and S.P.S. Bioinformatics aspects were conducted by F.S.S., S.P.S., and D.D.D. The manuscript was prepared by N.K.A., S.S.P and M.B.S with figures created by S.P.S., F.S.S., N.K.A, and D.D.D. under the supervision of A.C.R.C.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information


Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Barbosa dos Santos, M., Koide Albuquerque, N., Patroca da Silva, S. et al. A novel hantavirus identified in bats (Carollia perspicillata) in Brazil. Sci Rep 14, 6346 (2024). https://doi.org/10.1038/s41598-024-56808-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-56808-6
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.

{kind=link}
{kind=link}