
Abstract
Finding the active sites of catalysts and photo-catalysts is crucial for an improved fundamental understanding and the development of efficient catalytic systems. Here we have studied the photo-activated dehydrogenation of ethanol on reduced and oxidized rutile TiO2(110) in ultrahigh vacuum conditions. Utilizing scanning tunnelling microscopy, various spectroscopic techniques and theoretical calculations we found that the photo-reaction proceeds most efficiently when the reactants are adsorbed on regular Ti surface sites, whereas species that are strongly adsorbed at surface defects such as O vacancies and step edges show little reaction under reducing conditions. We propose that regular Ti surface sites are the most active sites in photo-reactions on TiO2.
Similar content being viewed by others


Introduction
Catalysis and photo-catalysis are fields of paramount importance both with a view on the demands on the chemical industry and the challenges in future renewable energy generation as well as to sustain our environment. To fulfil all these demands it is crucial to enhance the efficiency of designated chemical processes. One of the most promising materials in photo-catalysis is Titania (TiO2)1,2,3,4,5,6. Besides photo-catalysis, TiO2 is used in thermal heterogeneous catalysis, solar cells, gas sensors and biocompatible materials.
An increasingly important field is the photo-catalytic and renewable generation of fuels and particularly ethanol (CH3CH2OH or EtOH) holds great promise in this regard5,6. In addition, EtOH is an essential solvent and it could be used as feedstock in a possible green chemistry in the future7,8,9. These expectations and the fact that alcohols serve as model molecules of catalytic and photo-catalytic processes4,10,11,12, have stimulated considerable research efforts towards the thermal and photo-catalytic oxidation of EtOH on TiO2. Numerous studies have been conducted using TiO2 powders, where TiO2 nanoparticles (often a mix of the two most common polymorphs, anatase and rutile) were used to oxidize EtOH condensed as liquid or in the gas phase13,14,15,16,17,18,19,20,21. While other work has addressed photo-catalytic reactions of methanol (MeOH)4,6,11,22,23,24,25,26 it is worth indicating two fundamental differences between MeOH and EtOH. First, because of the carbon-carbon bond the chemistry of EtOH is akin to higher hydrocarbons and different from that of C1 hydrocarbons. Second, MeOH is made from syngas and therefore not a renewable feedstock.
In surface science, where single crystalline surfaces can be studied at ultrahigh vacuum (UHV) conditions, particular focus has been on the interaction between EtOH and the rutile TiO2(110)–(1 × 1) surface27,28,29,30,31,32,33,34,35,36,37,38,39, because this surface is the most stable one of rutile and often serves as a model for transition metal surfaces1,2,3,4,10,11,12. The TiO2(110)–(1 × 1) surface consists of alternating rows of fivefold-coordinated titanium (5f-Ti) atoms (the Ti troughs) and protruding, twofold coordinated bridge-bonded oxygen (Obr) atoms. Following cycles of Ar+ sputtering and vacuum-annealing, the TiO2(110) crystals are reduced, leading to the creation of bulk defects and Obr vacancies on the surface1,10,12,40,41. This leads to changes in electronic properties, where the empty Ti3d orbitals become populated, leading to a state within the ~3.1 eV wide band gap ~0.85 eV below the Fermi level (EF)1,40,41,42. This Ti3d defect state can be removed upon oxygen adsorption1,10,40,41,42,43.
The adsorption of EtOH and the thermally activated chemistry of EtOH on rutile TiO2(110)–(1 × 1) have been studied previously by means of photoelectron spectroscopy (PES)28,30,44, temperature-programmed desorption (TPD)27,28,29,32,33,36,37,38,39, femtosecond two-photon photoemission spectroscopy (2PPE)36 and scanning tunnelling microscopy (STM)32,33,35. In addition, the EtOH–TiO2(110) interaction has been studied using density functional theory (DFT) calculations31,32,33,34,35,36. The adsorption of an EtOH molecule occurs in a manner similar to that of acid–base reaction whereby the oxygen of EtOH is adsorbed on a 5f-Ti site. Ambiguity still exists to which extents EtOH adsorbs molecularly and dissociatively32,36.
Some studies also addressed the photo-chemistry of EtOH on rutile TiO2(110)30,31,36,37,38,39. PES and TPD studies suggest that EtOH converts to acetaldehyde (CH3CHO) and, finally, acetate (CH3COO−) under UV light illumination in an oxygen background30,31. A 2PPE study also revealed the formation of CH3CHO and reports on a photo-induced excited state ~2.4 eV above EF for EtOH/TiO2(110) that was associated with the dissociation of EtOH on 5f-Ti sites36. Finally, applying pump–probe laser ionization techniques, it has been suggested that, in the presence of oxygen, methyl radicals are produced during the photo-catalytic oxidation of EtOH37.
Here we studied the photo-activated dehydrogenation of EtOH on TiO2(110) in different oxidation states by combining several surface science techniques and DFT calculations. The active sites on the terraces and at step edges are identified by high-resolution STM, whereas the chemical identity of the products is unravelled by TPD and isothermal mass spectrometry (ISOMS) measurements. Utilizing synchrotron-radiation PES, we compare the rates of EtOH dehydrogenation at 290 K for TiO2(110) surfaces in different oxidation states. A reaction mechanism is tested with the help of DFT calculations that explains the experimental observations.
Results
Photo-Reactions of EtOH on Rutile TiO2(110) studied by STM
To test the influence of the oxidation state and the surface structure on the photo-reactivity of EtOH we prepared TiO2(110) samples with different characteristic point defects on the surface. Among the reduced TiO2(110) surfaces we distinguish between clean, reduced TiO2(110) surfaces [r-TiO2] characterized by the presence of Obr vacancies and hydrogenated TiO2(110) surfaces [h-TiO2] characterized by H adatoms (Had), capping some of the Obr atoms. Starting with a clean r-TiO2(110) surface, an h-TiO2(110) surface can be easily produced by letting water dissociate at the Obr vacancies43,45,46,47,48. Additionally, we prepared oxidized TiO2(110) surfaces [o-TiO2] by exposing h-TiO2(110) surfaces to 200 L O2 [1 L (Langmuir) = 1.33 × 10−6 mbar · s] at 300 K10,41. Such o-TiO2(110) surfaces are characterized by a diminished Ti3d gap state, by perfect Obr rows (no Obr vacancies) and by a number of O adatoms (Oot) located on-top of 5f-Ti sites in the Ti troughs (Fig. 1c, inset). The Oot adatoms show up in the STM images as protrusions distributed along the Ti troughs10,41,46,49,50. In empty-state STM images of TiO2(110), the Ti troughs appear bright, whereas geometrically protruding Obr atoms appear dark1,10,46. Accordingly, the Obr vacancies on r-TiO2(110) appear as protrusions along the dark rows, “connecting” bright Ti troughs (Fig. 1b, inset).

STM study of EtOH photo-reactions on r- and o-TiO2(110) surfaces.
(a) STM image (420 Å × 420 Å) obtained after exposing an r-TiO2(110) surface to EtOH at 300 K. (b) Zoom-in STM image (130 Å × 130 Å) corresponding to the data in (a) that allows to distinguish between EtOHTi/EtOTi and EtObr species. (c) Zoom-in STM image (130 Å × 130 Å) obtained after exposing an o-TiO2(110) surface to EtOH at 300 K. Insets in (b,c) display (40 Å × 40 Å) areas on the pristine r- and o-TiO2(110) surfaces. (d–f) Corresponding STM images obtained after illumination of the EtOH-covered surfaces with UV-light for 11 min at 290 K in UHV. Insets in (a,d) show the indicated areas (40 Å × 40 Å) enlarged. (g–i) Corresponding STM images obtained after subsequent annealing of the TiO2(110) crystals for 2 min at ~550 K (g,h) and ~630 K (i), respectively. Symbols indicate Obr vacancies (squares), an Oot adatom (circle), EtObr ethoxides (dotted circles), EtOS ethoxides (arrows), isolated Had species (hexagons) and rows of Had species (rectangles), respectively. The Ti troughs are indicated in (b,c) and in the insets of (a,d) by thin white lines. STM images were collected with a tunnelling current ≤0.1 nA and a tunnelling voltage of ~1.2 V. The STM images are shown enlarged in the Supplementary Information (Figs S1–S9).
Following the saturation of an r-TiO2(110) surface with EtOH at 300 K [Fig. 1a,b: EtOH/r-TiO2(110)] we found bright protrusions on (34 ± 2)% of the 5f-Ti sites that arise both from EtOHTi molecules and EtOTi ethoxides (EtOHTi/EtOTi)32. The EtOHTi molecules and EtOTi ethoxides appear in the STM images with very similar contrast32, allowing their distinction solely at low coverage. In addition, adsorbates occurred in the Obr rows (marked by dotted circles in Fig. 1b) that are ascribed to EtObr ethoxides, resulting from the dissociation of EtOHTi molecules at Obr vacancies32. New adsorbates also occurred along the step edges running parallel to  and
and  directions (Fig. 1a, inset), which are ascribed as EtOS ethoxides (“S” stands for step)33. The EtOS ethoxides are adsorbed at the upper terraces and located at the end of the Obr rows33. Both ethoxide species (EtObr and EtOS) are strongly bound at O vacancies32,33.
directions (Fig. 1a, inset), which are ascribed as EtOS ethoxides (“S” stands for step)33. The EtOS ethoxides are adsorbed at the upper terraces and located at the end of the Obr rows33. Both ethoxide species (EtObr and EtOS) are strongly bound at O vacancies32,33.
When starting instead with an o-TiO2(110) surface, we again obtained EtOHTi molecules and EtOTi ethoxides in the Ti troughs [Fig. 1c: EtOH/o-TiO2(110)]. However, this time no EtObr ethoxides were formed, because no Obr vacancies existed at which the EtOH molecules could dissociate. In addition, on EtOH/o-TiO2(110), the density of EtOS ethoxides was considerably lower than on EtOH/r-TiO2(110). On EtOH/o-TiO2(110), the density of Oot adatoms was (10.1 ± 0.5)%ML [1 ML (monolayer) is the density of the (1 × 1) unit cells, 5.2 × 1014/cm2] and the coverage of EtOHTi/EtOTi species was (24.1 ± 0.8)% ML. Because the EtOHTi and EtOTi species protrude much more from the surface than the Oot adatoms32,50 the Oot adatoms cannot be seen in the STM images. However, before the sample was exposed to EtOH, the Oot adatoms were well-resolved in the STM images (Fig. 1c, inset). Thus, we analysed the images acquired on o-TiO2(110) before the EtOH exposure to estimate the Oot adatom density.
Following UV-light illumination for 11 min at 290 K [365 nm; ~2 × 1016 photons/(s · cm2)] we obtained clear changes on both EtOH-covered TiO2(110) surfaces, as illustrated by the STM images shown in Fig. 1d–f. Importantly, all the adsorbates in the Ti troughs, EtOHTi and EtOTi species, disappeared on EtOH/r-TiO2 (Fig. 1d,e) and EtOH/o-TiO2 (Fig. 1f). Solely the EtObr and EtOS ethoxides on r-TiO2(110) did not (or rarely) photo-react (in Fig. 1b,e all the EtObr species are marked by white dotted circles). In addition, Had species were evident on both surfaces in very high densities [r-TiO2: (27 ± 1)%ML; o-TiO2: (35 ± 1)%ML]. Single Had species appear brighter than Obr vacancies10,12,46,47 (some are marked in Fig. 1e,f,i by white hexagons). Note that Had species adsorbed closely to each other within the Obr rows appear as extra-bright, elongated protrusions (see the examples indicated by the rectangles in Fig. 1e,f). Regarding the EtOH/o-TiO2(110) surface, notice that no Oot adatoms remained on the surface after illumination with UV light (Fig. 1f). This can be explained through the recombination of Had species (created in the course of the photo-reaction) with Oot adatoms10,43,51. Thus, water molecules are formed in the photo-reaction. These water molecules greatly facilitate the diffusion of Had species47,48, which explains why all the Oot adatoms were reacted off. Because we observed only few water monomers52 in the STM images in Fig. 1d–f, many of them must have desorbed from the surface: 2Had+Oot → H2O ↑.
Subsequent annealing of the two TiO2(110) samples at 550–630 K for 2 min led to r-TiO2(110) surfaces without any Had species, regardless whether we started the experiment with r- or o-TiO2(110), see Fig. 1g–i. Interestingly, the r-TiO2(110) surfaces obtained after brief annealing at this high temperature were characterized by Obr vacancy densities that were about twice as high as those found on the clean r-TiO2(110) surfaces at the very beginning of the experiments. These enhanced Obr vacancy densities originate from the recombinative desorption of Had species – a reaction that includes the formation of water molecules and the consumption of Obr atoms: 2Had+Obr → H2O ↑43,45. The formation of Had species within the photo-reaction of EtOHTi and EtOTi species is confirmed by these annealing-experiments. Additional STM studies (Fig. S10 in the Supplementary Information) and water-TPD measurements (Fig. S11) further corroborate the formation of Had species in the photo-reaction on EtOH/r-TiO2(110).
Evidence for acetaldehyde formation by mass spectroscopy
When illuminating EtOH/r-TiO2(110) and EtOH/o-TiO2(110) surfaces in UHV, the Had species are the sole detectable reaction product on the surfaces (Fig. 1a–f). This finding implies that all other products are ejected into the vacuum immediately after their formation. To identify these products we conducted time-resolved ISOMS experiments (Fig. 2). In such experiments, the reaction products are detected with a mass spectrometer in-situ as a function of time and at a given sample temperature. The experimental setup is sketched in the inset of Fig. 2. During the ISOMS experiments the sample was held at 290 K (as in the STM experiments) and masses m/z = 18; 26; 29; 31; 43; 44 were recorded. Regardless whether we started with EtOH-saturated r-, h- or o-TiO2(110) surfaces, no m/z = 31 peak was detected, but we always detected a peak for m/z = 29 when the light was switched on. Because the base peak of EtOH is m/z = 31, simple photo-desorption of the EtOH species in the Ti troughs can be ruled out. Instead, on the basis of the known cracking pattern of acetaldehyde53, we conclude that the m/z = 29 signals originate from acetaldehyde (CH3CHO). The m/z = 29 signal obtained for EtOH/o-TiO2(110) was more intense than that obtained for EtOH-covered h- and r-TiO2(110) surfaces [Fig. 2, data for EtOH/h-TiO2(110) (not shown) are very similar to those found for EtOH/r-TiO2(110)]. In each case the m/z = 29 signals dropped down to the base line in less than 1 min illumination time, indicating that the EtOH photo-reactions have completed.

ISOMS data acquired during EtOH photo-reaction at 290 K.
Black curve: r-TiO2(110); red curve: o-TiO2(110). Prior to the experiments both TiO2(110) surfaces were saturated with EtOH at 300 K. The inset displays the experimental setup. The quadrupole mass spectrometer (QMS), the TiO2(110) crystal (light brown block) and the direction of the incident light are indicated.
EtOH photo-reaction on r-TiO2(110) in the presence of oxygen
In contrast to EtOHTi and EtOTi species, the EtObr and EtOS ethoxides strongly bound to O vacancies on EtOH/r-TiO2(110) did barely photo-react in UHV (Fig. 1b,e). An analysis of STM images acquired before and after UV-light illumination for 11 min revealed no clear changes in the densities of EtObr and EtOS ethoxides. Nevertheless, a 30 min illumination with UV light decreased the density of EtObr ethoxides by ~34%. To photo-react more of the EtObr and EtOS ethoxide species we conducted another STM experiment wherein the photo-reaction was carried out in an O2 background. This experiment was started with a TiO2(110) surface with EtObr/EtOS ethoxides and Had species prepared by illuminating an EtOH/r-TiO2(110) surface with UV-light for 7 min (Fig. 3a,b). The STM images in Fig. 3a,b are alike those presented in Fig. 1d,e because the corresponding sample preparations were almost identical. Subsequently, the sample was illuminated in ~5 × 10−8 mbar O2 for 25 min at 290 K (Fig. 3c,d) and another 20 min at the same O2 pressure and temperature (Fig. 3e,f). In O2, the EtObr ethoxide species photo-reacted indeed more efficiently, as was evident from there diminished occurrence. For example, within 25 min the density of EtObr ethoxides decreased from (6.1 ± 0.2) to (2.2 ± 0.1)%ML. At the same time, we found that two new types of adsorbates (denoted “A” and “B”) had appeared in the Ti troughs. In Fig. 3d,f, we indicate some of the newly formed adsorbates in the Ti troughs. Along the [001] direction, these reaction products are centred in between two 5f-Ti sites. After a total illumination time of 45 min only (0.5 ± 0.1)%ML EtObr ethoxides remained. The densities of the new reaction products in the Ti troughs (sum of both types) were (4.0 ± 0.3)%ML and (6.3 ± 0.4)%ML for 25 and 45 min total illumination time, respectively. Clearly, the photo-reactivity of the EtObr species is considerably higher in an O2 background than in UHV and new products are formed in an O2 background that are stable on the TiO2(110) surface at 290 K.

STM study of the photo-reaction on EtOH/r-TiO2(110) in oxygen (fate of EtObr ethoxides).
(a,b) STM images (240 Å × 240 Å and 68 Å × 68 Å, respectively) of an illuminated EtOH/r-TiO2(110) surface. The UV-light illumination was accomplished at 290 K in UHV. (c,d) STM images of the same sample after illumination in 5 × 10−8 mbar O2 at 290 K for 25 min. (e,f) STM images of the same sample after additional illumination in 5 × 10−8 mbar O2 at 290 K for 20 min. Symbols indicate Had species (hexagons), EtOS ethoxides (white arrows) and EtObr ethoxides (black crosses). Occupied adsorption sites of unidentified products “A” (dotted black circle) and “B” (black circle) are indicated by green and pink dots, respectively. Lattice grids in (b,d,f) are centred on-top of 5f-Ti sites.
The EtOS ethoxides, however, are even more difficult to photo-react than the EtObr ethoxides (Fig. 4). In the experiment corresponding to Fig. 4a,b, we illuminated an EtOH/r-TiO2(110) surface with UV light in ~5 × 10−8 mbar O2 for 45 min. It can be seen that the species adsorbed along the step edges running in  and
and  directions (some are marked by white arrows) appear in between the Ti troughs, i.e. at the end of the Obr rows on the upper terraces. This observation confirms the assignment of these protrusions to EtOS ethoxides33. Notably, the occurrence of the EtOS ethoxides did not decline, in spite of the long illumination time in the presence of oxygen.
directions (some are marked by white arrows) appear in between the Ti troughs, i.e. at the end of the Obr rows on the upper terraces. This observation confirms the assignment of these protrusions to EtOS ethoxides33. Notably, the occurrence of the EtOS ethoxides did not decline, in spite of the long illumination time in the presence of oxygen.

STM study of the photo-reaction on EtOH/r-TiO2(110) in oxygen (fate of EtOS ethoxides).
(a) STM image (220 Å × 220 Å) of an illuminated EtOH/r-TiO2(110) surface. The UV-light illumination was accomplished at 290 K in UHV. Few EtOS ethoxides are indicated by white arrows. (b) STM image of the same sample after illumination in 5 × 10−8 mbar O2 at 290 K for 45 min. White lines are superimposed on the Ti troughs. These STM images are shown with higher contrast than the images in the other figures.
Another aspect regarding the EtOH photo-reaction experiments in O2 worth noting is the lowered coverage of Had species. The STM images depicted in Fig. 3 and 4 show this quite clearly. Apparently, oxygen reacts the Had species off, forming water. Not all of the Had species reacted at the chosen conditions (see Fig. 3f and 4b).
EtOH photo-reactions on o- and h-TiO2(110) surfaces studied by PES
To compare the photo-assisted dehydrogenation of EtOHTi and EtOTi species on the terraces of reduced and oxidized TiO2(110) surfaces in more depth, we collected PE C1s spectra at the ASTRID storage ring in Aarhus54. In the first series of PES experiments we started with an o-TiO2(110) surface and in the second series with an h-TiO2(110) surface (Fig. 5). Starting with h-TiO2(110) was preferred over r-TiO2(110), because the acquisition of the PES data took long time and thus an r-TiO2(110) surface would gradually be changed into h-TiO2(110)40. Each series of experiments was conducted in UHV and repeated two times. The sample temperature was throughout kept at 290 K. The two C1s signals in the PE spectra arise from the carbon atoms in the –CH2O– and –CH3 groups28,30,44. As shown in Fig. 5a for o-TiO2(110), the C1s signals attenuated upon UV-light illumination. However, the C1s signal did not reach zero, even after UV-light illumination for 11 min. Because we know from our STM studies that the EtOS ethoxides do not photo-react and no EtObr species exist on EtOH/o-TiO2(110), we consider the C1s signal detected after 11 min illumination time to be exclusively due to the EtOS ethoxide species. Before the illumination of the EtOH/o-TiO2(110) surface, the C1s signal originates from EtOHTi and EtOTi species on the terraces (ITi), as well as EtOS species bound along the step edges (IS).

Photo-reactions on EtOH/o-TiO2(110) and EtOH/h-TiO2(110) surfaces studied by PES.
(a) C1s PES data (dots) collected with photon energy (hν) of 350 eV on EtOH/o-TiO2(110) following UV light illumination at 290 K for 0 and 11 min, respectively. Spectra are offset for clarity. Gaussian fits to the data are shown as red curves. (b) Logarithmic plot of the normalized integrated areas of the C1s spectra as function of illumination time (filled green and red dots) and X-rays (open green and red dots), respectively. Starting points in these experiments were h- (green) and o-TiO2(110) surfaces (red) that were saturated with EtOH at 290 K. Full lines are exponential fits to the data points and the shaded areas display the standard deviations of these fits. Contributions of the non-reactive EtOS ethoxides (IS) have been subtracted from the integrated intensity Im of the measured C1s spectra. Accordingly, the plot shows the contributions to the C1s signals arising exclusively from the EtOHTi and EtOTi species, ITi (C1s).
Figure 5b shows the attenuation of the C1s signals for EtOH/o- and EtOH/h-TiO2(110) surfaces as function of the illumination time (filled red and green dots). To extract the contributions to the C1s signals arising solely from the EtOHTi and EtOTi species, ITi (C1s), we subtracted the signal arising from the EtOS ethoxides, IS (C1s) from the measured signal, Im(C1s). It can be seen that ITi(C1s) attenuates exponentially on both TiO2(110) surfaces, with the attenuation on o-TiO2 being more than two times faster than on h-TiO2 (see the solid lines in Fig. 5b). Thus, also the PES data show that o-TiO2(110) surfaces are advantageous for the photo-assisted dehydrogenation of EtOH. Figure 5b further shows that the influence of the X-rays on the C1s signals (open dots) is measurable but negligibly small with the used low-intensity X-ray beam.
DFT modelling of the EtOH photo-reaction on TiO2(110)
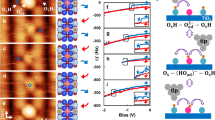
To further explore the photo-oxidation of EtOH on TiO2(110) in different oxidation states we performed first-principles DFT calculations (Fig. 6). In our theoretical analysis we assume that the photo-reaction of EtOH on TiO2 proceeds via oxidation by photo-generated holes and that the photo-electrons are trapped at the surface2,3,4. As possible trapping sites for electrons and holes we considered the splitting of a neutral water molecule in OH and H fragments that adsorb on 5f-Ti sites and Obr atoms, respectively. The two fragments act as trapping sites for electrons (OHTi)55,56 and holes (Had)55, respectively.

DFT modelling of the acetaldehyde formation on TiO2(110).
Energy profiles for EtOHTi adsorbed on  (a) and [TiO2(110)]0 (d) supercells. The formation of EtOTi ethoxides is considered in (b,e) and the formation of acetaldehyde species (CH3CHOTi) is addressed in (c,f). All adsorption energies are given in eV. Small red-brown balls represent 5f-Ti atoms, large dark-grey balls Obr atoms and large light-grey balls in-plane O atoms. Atoms of the adsorbates are displayed as follows: C atoms: black balls; Had species: small yellow balls; O atoms: large pink balls. Corresponding band schemes, each consisting of the conduction (CB) and valence band (VB), are shown in the insets of (b,c,e,f). Filling of the bands is indicated by dark yellow colour and positions of the Fermi level (EF) are indicated by dashed lines.
(a) and [TiO2(110)]0 (d) supercells. The formation of EtOTi ethoxides is considered in (b,e) and the formation of acetaldehyde species (CH3CHOTi) is addressed in (c,f). All adsorption energies are given in eV. Small red-brown balls represent 5f-Ti atoms, large dark-grey balls Obr atoms and large light-grey balls in-plane O atoms. Atoms of the adsorbates are displayed as follows: C atoms: black balls; Had species: small yellow balls; O atoms: large pink balls. Corresponding band schemes, each consisting of the conduction (CB) and valence band (VB), are shown in the insets of (b,c,e,f). Filling of the bands is indicated by dark yellow colour and positions of the Fermi level (EF) are indicated by dashed lines.
First, we studied the effect of the hole on the photo-reaction of EtOH (Fig. 6a–c). To this end, we used a stoichiometric TiO2(110) supercell with an OHTi group adsorbed on one side, denoted  . In order to maintain the hole created by the addition of the OHTi group, it is essential to use a stoichiometric rather than a reduced supercell57. In agreement with previous computational results31,32,34,36 and TPD data27,28,29,39 we found that the adsorption strength of molecularly (−0.79 eV) and dissociatively (−0.71 eV) adsorbed ethanol is strongest on-top on 5f-Ti sites and almost degenerated. Because the barrier for EtOHTi dissociation via O–H bond scission is only ~0.28 eV32 we consider the EtOTi ethoxide (Fig. 6b) as the given intermediate for acetaldehyde (CH3CHOTi) formation (Fig. 6c). The reaction of the EtOTi ethoxide to CH3CHOTi is initiated by breaking of the C1–H bond, followed by H transfer to the Obr row and completed upon formation of the C1 = O double bond. On the
. In order to maintain the hole created by the addition of the OHTi group, it is essential to use a stoichiometric rather than a reduced supercell57. In agreement with previous computational results31,32,34,36 and TPD data27,28,29,39 we found that the adsorption strength of molecularly (−0.79 eV) and dissociatively (−0.71 eV) adsorbed ethanol is strongest on-top on 5f-Ti sites and almost degenerated. Because the barrier for EtOHTi dissociation via O–H bond scission is only ~0.28 eV32 we consider the EtOTi ethoxide (Fig. 6b) as the given intermediate for acetaldehyde (CH3CHOTi) formation (Fig. 6c). The reaction of the EtOTi ethoxide to CH3CHOTi is initiated by breaking of the C1–H bond, followed by H transfer to the Obr row and completed upon formation of the C1 = O double bond. On the  supercell (Fig. 6a), this reaction is barrier-less (i.e., it occurs spontaneously) and extremely favourable (−1.50 eV). The alternative pathway, the desorption of EtOHTi into the gas phase, is hindered by a barrier of ~0.79 eV [cf.
supercell (Fig. 6a), this reaction is barrier-less (i.e., it occurs spontaneously) and extremely favourable (−1.50 eV). The alternative pathway, the desorption of EtOHTi into the gas phase, is hindered by a barrier of ~0.79 eV [cf.  (EtOH) in Fig. 6a]. Thus, the photo-induced dehydrogenation of EtOTi ethoxide to CH3CHOTi is the preferred reaction on
(EtOH) in Fig. 6a]. Thus, the photo-induced dehydrogenation of EtOTi ethoxide to CH3CHOTi is the preferred reaction on  . Once it has formed, a CH3CHOTi molecule cannot recombine again to EtOTi ethoxide, because this process is hindered by a ~1 eV high barrier. Apparently, this computational approach leads to results that are in good agreement with the experimental results on the EtOH photo-reaction.
. Once it has formed, a CH3CHOTi molecule cannot recombine again to EtOTi ethoxide, because this process is hindered by a ~1 eV high barrier. Apparently, this computational approach leads to results that are in good agreement with the experimental results on the EtOH photo-reaction.
For comparison, we also studied the interaction of EtOHTi and EtOTi species with a stoichiometric supercell without an OHTi group, which is denoted [TiO2(110)]0 (Fig. 6d–f). This was done to simulate the thermally activated dehydrogenation reaction as opposed to the photo-catalytic reaction. For this supercell we found that the reaction of EtOTi to CH3CHOTi is hindered by a high barrier, ~1.42 eV [cf.  (EtO-ald.) in Fig. 6d], which is in good general agreement with experimental results for dehydrogenation reactions on metal oxides7. Accordingly, on [TiO2(110)]0, desorption of EtOHTi is more favourable than the formation of CH3CHOTi [cf.
(EtO-ald.) in Fig. 6d], which is in good general agreement with experimental results for dehydrogenation reactions on metal oxides7. Accordingly, on [TiO2(110)]0, desorption of EtOHTi is more favourable than the formation of CH3CHOTi [cf.  (EtOH)]. On both supercells considered, [TiO2(110)]0 and
(EtOH)]. On both supercells considered, [TiO2(110)]0 and  , the CH3CHOTi molecules are rather weakly bound to 5f-Ti sites via the oxygen lone pairs (Fig. 6c,f). With both supercells we computed adsorption energies of −0.64 eV [cf.
, the CH3CHOTi molecules are rather weakly bound to 5f-Ti sites via the oxygen lone pairs (Fig. 6c,f). With both supercells we computed adsorption energies of −0.64 eV [cf.  (ald.) and
(ald.) and  (ald.), respectively] and the ejection of CH3CHOTi into the gas phase is more favourable than the reconversion to the initial EtOTi state. Accordingly, acetaldehyde is expected to desorb at 290 K, the temperature at which we studied the photo-reaction of EtOH. Thus, these DFT calculations are in good agreement with the experimental observations.
(ald.), respectively] and the ejection of CH3CHOTi into the gas phase is more favourable than the reconversion to the initial EtOTi state. Accordingly, acetaldehyde is expected to desorb at 290 K, the temperature at which we studied the photo-reaction of EtOH. Thus, these DFT calculations are in good agreement with the experimental observations.
To elucidate the reason why an EtOTi species on a  supercell reacts favourably to CH3CHOTi but not on [TiO2(110)]0 we studied the electronic structure of the considered EtOTi and acetaldehyde species, the results of which are shown in the insets of Fig. 6b,c,e,f. When an EtOTi reacts to CH3CHOTi, the overall electronic effect is the transfer of two electrons from the EtOTi ethoxide to TiO2 states. Thus, for each photon two electrons are created – an effect that is well documented in the literature and often referred to as the “current doubling effect”58,59,60. On the
supercell reacts favourably to CH3CHOTi but not on [TiO2(110)]0 we studied the electronic structure of the considered EtOTi and acetaldehyde species, the results of which are shown in the insets of Fig. 6b,c,e,f. When an EtOTi reacts to CH3CHOTi, the overall electronic effect is the transfer of two electrons from the EtOTi ethoxide to TiO2 states. Thus, for each photon two electrons are created – an effect that is well documented in the literature and often referred to as the “current doubling effect”58,59,60. On the  supercell (insets in Fig. 6b,c), one of the two electron is captured by the (photo-generated) hole, leading to electron transfer to the top of the valence band (VB) and the other is transferred to the TiO2 conduction band (CB). In contrast, on [TiO2(110)]0, both electrons are transferred to the TiO2-CB (insets in Fig. 6e,f), which costs more energy. That is, the reaction is favoured on
supercell (insets in Fig. 6b,c), one of the two electron is captured by the (photo-generated) hole, leading to electron transfer to the top of the valence band (VB) and the other is transferred to the TiO2 conduction band (CB). In contrast, on [TiO2(110)]0, both electrons are transferred to the TiO2-CB (insets in Fig. 6e,f), which costs more energy. That is, the reaction is favoured on  because energy is gained when one of the electrons is transferred to the top of the TiO2-VB rather than to the much higher level at the TiO2-CB.
because energy is gained when one of the electrons is transferred to the top of the TiO2-VB rather than to the much higher level at the TiO2-CB.
Discussion
The experimental results addressing the photo-reaction of EtOH on rutile TiO2(110) presented above are consistent with the following scheme: First, EtOTi species  react with holes (h+), leading to the formation of ethoxide radicals:
react with holes (h+), leading to the formation of ethoxide radicals:

The ethoxide radicals react further via proton and electron transfer, leading to the formation of acetaldehyde and Had species:

Such a direct oxidation of EtOH on TiO2(110) by the holes is fully supported by our DFT modelling. Further, this scheme is in line with previous spectroscopic studies, wherein EtOH and MeOH are reported to act as effective hole traps in photo-catalysed reactions58,59,60,61,62. Our results obtained on model surfaces in UHV are thus in remarkable agreement with TiO2 powder studies where TiO2 nanoparticles (anatase and rutile) were used to oxidize EtOH condensed as liquid or in the gas phase13,14,15,16,17,18,19,20,21. Most interestingly, two active sites have been proposed in the EtOH photo-reaction on the TiO2 particles, leading to the formation of acetaldehyde and formate/acetate, respectively13,14,15,16,17,18,19,20,21. These two active sites are distinct because of large differences in the observed reaction rates. This comparison suggests that the rutile TiO2(110) surface may serve as a good model for TiO2 nanoparticles that are used in many applications.
Another important result of our study is that the photo-reaction proceeds faster when the EtOH/EtO species are bound to regular surface Ti sites – the 5f-Ti sites. In fact, our STM studies uncover large differences in photo-reactivity between the various EtOH-related species on the TiO2(110) surface (Figs 1, 3 and 4). Whereas the EtOHTi/EtOTi species were so highly reactive that even the low-intensity X-rays led to the removal of some adsorbates (Fig. 5b), the EtObr species photo-reacted only in oxygen (Fig. 3) and the EtOS species did not react at all at the tested conditions (Fig. 4). We note that such differences, although very clearly seen by STM, are very difficult to recognize in spectroscopic studies.
The observed low photo-reactivity of the ethoxides strongly bound at Obr vacancies and step edges may suggest that these adsorption sites constitute only a minor channel within the overall photo-reaction of EtOH on TiO2(110). Previous work63 on the photo-oxidation of trimethyl acetate [TMA or (CH3)3CCOO] on r-TiO2(110) showed an inhibition of hole-mediated photo-chemistry when the molecules are bound to Obr vacancies. These two examples [the TMA/r-TiO2(110)63 and EtOH/r-TiO2(110) – this work] indicate that the site requirement is independent of the specific organic molecule. We thus propose that regular surface Ti sites are mostly the active sites for hole-mediated photo-reactions on TiO2. Moreover, the step sites may contribute in the photo-reactivity in cases where the adsorption of the molecules is the limiting factor.
Among the studied TiO2(110) surfaces the EtOHTi and EtOTi species photo-reacted best on o-TiO2(110), in agreement with previous reports on other photo-catalysed reactions on rutile TiO2(110)4,11,64. From PES studies it is well-established that the adsorption of oxygen on reduced TiO2 surfaces leads to the depopulation of the Ti3d defect state in the band gap and an upward bending of the VB and CB’s1,41,65. This upward band bending near the TiO2(110) surface facilitates the charge carrier separation and drives the photo-generated holes towards the surface, thereby increasing the photo-reaction rate of hole-mediated reactions at the surface65. Furthermore, the depopulation of the defect state decreases the number of electrons in the near surface region – thus the rate for the recombination of holes and electrons is decreased. In addition, we note that the presence of Oot adatoms on the TiO2(110) surface probably shifts the mix of EtOHTi/EtOTi species more towards the EtOTi side, which should also facilitate the formation of acetaldehyde17.
Comparing the presented data addressing EtOH/TiO2(110) to those reported recently for MeOH/TiO2(110)22,23,24,25,26 we find both similarities and differences. Common in these studies and in our work is that the 5f-Ti sites are considered as important sites for photo-catalytic reactions on TiO2(110). In addition, methoxy groups are reported to be active species in photochemical hole scavenging reactions of MeOH on TiO2(110)22, similarly as we find that ethoxides in the Ti troughs are photo-active in case of EtOH/TiO2(110). However, Zhou et al. reported time-dependent 2PPE measurements, indicating that defects have a strong positive effect on the photocatalytic kinetic process25. Specifically, it was suggested that “surface defects and/or subsurface defects on the TiO2(110) surface enhance the photo-catalyzed MeOH dissociation rate by significantly lowering the photoreaction barrier”25. This conclusion appears to be opposite to what we found for EtOH/TiO2(110). Further studies are needed to explore the similarities and differences between these two systems.
Conclusions
We have studied site-specific photo-reactions of EtOH species on TiO2(110) in different oxidation states and showed that the photo-reaction of EtOH on TiO2(110) proceeds most efficiently when the reactants are adsorbed on regular surface Ti sites on TiO2(110) surfaces with Oot adatoms. On the contrary, strongly bound ethoxides, either adsorbed in Obr vacancies or at defects sites associated with the step edges, photo-react very slowly. When the photo-reaction was carried out in UHV at 290 K, acetaldehyde desorbed into the gas phase, whereas the second product, Had species, stayed on the surface. On the basis of the presented atomistic insights and literature data we propose that regular surface Ti sites are the most active sites in photo-reactions on TiO2 surfaces. In contrast, low-coordinated defects sites on the terraces and at the step edges are least active and the presence of oxygen is required to induce photo-reactivity of molecules at these sites. The rutile TiO2(110) surface, providing both regular Ti sites and defect sites, may serve as a good model for studying relevant processes on TiO2 nanoparticles, since loosely and strongly bound adsorbates can be studied simultaneously.
Methods
Experimental details
The STM and TPD experiments were carried out in an UHV chamber with a base pressure in the low 10−11 Torr range equipped with a homebuilt, variable-temperature Aarhus STM, a QMS and standard facilities for sample preparation and characterization49. The Aarhus STM used in this study is capable of fast scanning at high resolution in a temperature range between 100 and 400 K66. Electrochemically etched tungsten tips were used in all the STM experiments. The differentially pumped QMS (Balzers) was connected to the main chamber via a closed cone with a small aperture (d ~ 3 mm) facing the sample at a distance of ~1 mm. This design ensures that only molecules released from the sample can reach the QMS. The temperature of the sample could be varied from 100 K using liquid nitrogen to 1200 K by heating the back side of the sample with a filament and electron bombardment. The sample temperature (measured using a K-type thermocouple) was controlled and recorded with a Eurotherm temperature controller that contains an automatic compensation of ambient temperature changes.
To obtain clean TiO2(110) surfaces, the crystals were Ar+ sputtered at 300 K and vacuum annealed at 880–950 K several times. A short flash to ~600 K was applied when the sample reached 300 K after vacuum annealing in order to free the sample from water and Had species that result from water adsorption during the cooling of the sample after the 20 min anneal46. The used TiO2(110) samples were characterized by Obr vacancy densities between 6.1 and 10.8% ML. All the STM images presented in this study were acquired in the constant current mode with a tunnelling voltage of ~+1.25 V and a tunnelling current of ~0.1 nA. Throughout, the sample was kept between 110 and 135 K during scanning.
The analysis of the STM data corresponding to Figs 1 and 3 rely on scanned areas of at least ~3000 nm2 for each given density and the given error bars represent the standard error  , with
, with  being the standard deviation of the counted adsorbates, or Obr vacancies and n being the number of the analysed STM images of the same size.
being the standard deviation of the counted adsorbates, or Obr vacancies and n being the number of the analysed STM images of the same size.
The PES C1s measurements were conducted at the ASTRID synchrotron radiation facility in Aarhus54 at the SGM1/Scienta beamline, using a low-intense synchrotron beam and a Scienta 200 analyser. The photon energy, hν, was 350 eV and the pass energy, Epass, was set to 75 eV. The PE C1s spectra were recorded at normal emission with the sample at 290 K. Great care was taken to avoid beam damage, which we identified to be a serious problem for this particular system and the chosen sample temperature. For each surface, h- and o-TiO2(110), respectively, three series of UV light experiments were carried out. Additionally, we conducted for each surface two series of control experiments to check for beam damage. The positions of the C1s peaks were calibrated using a gold reference. The C1s spectra were normalized to the photon flux and a linear background was subtracted. Subsequently, the spectra were fit with two Gaussian peaks to extract the integrated intensity Im of the measured C1s spectra (the area of the Gaussian fits). For each surface, the three data series were fit with a global exponential function I(t) = It(t)+Is, where It(t) = It,0 exp(−t/τ) and It,0 and Is were allowed to vary for each series, whereas the lifetime τ was constrained to be the same. The fits were weighted with the standard deviations of the C1s peak area.
For the EtOH exposures we used directional dosers, containing a 10 μm glass capillary array disk of ~8 mm diameter. For the exposures, the samples were placed in front of the doser at a distance of ~1 mm. When using such a microcapillary array doser, the local EtOH pressure at the TiO2(110) surface is unknown, but unwanted exchange reactions at the chamber walls are kept at a minimum.
The UV-light illumination was accomplished in all the experiments with an UV-A LED source (Optimax 365) with a peak emission at 365 nm (~3.4 eV) and a full-width-at-half-maximum (FWHM) of 10 nm. The LED source was mounted outside of the UHV chambers at a CF35 viewport and the light intensity in front of the sample was estimated to be ~2 × 1016 photons/(s · cm2), which corresponds to ~20 mW/cm2.
Computational details
The DFT calculations were performed using the GPAW program67,68, where the electrons are described using the projector augmented wave (PAW) method in the frozen core approximation69. The generalized gradient approximation (GGA) with the Perdew–Burke–Ernzerhof (PBE) functional70 was used to describe the exchange-correlation effects. The TiO2(110) surface was modelled using periodic slabs of four TiO2 tri-layers with a c(4 × 2) surface unit cell. All four tri-layers and the adsorbates were fully relaxed. The climbing nudged elastic band (NEB) method71 was used to calculate diffusion and dissociation barriers.
The presence of a photo-induced hole at the surface is modelled by introducing a co-adsorbed OH on one side of the slab. The OH is a spectator molecule and does not break or form any bonds during the reaction of EtOH. However, because of its strong electronegativity, the OH attracts an electron from the top of the VB on the stoichiometric slab, thereby producing a slab that is akin to that in the presence of a photo-induced hole55. This method has been used successfully in describing the photo-oxidation of methyl chloride over TiO2(110) surfaces57.
Additional Information
How to cite this article: Hansen, J. Ø. et al. Unravelling Site-Specific Photo-Reactions of Ethanol on Rutile TiO2(110). Sci. Rep. 6, 21990; doi: 10.1038/srep21990 (2016).
References
Diebold, U. The surface science of titanium dioxide. Surf. Sci. Rep. 48, 53–229 (2003).
Thompson, T. L. & Yates, Jr., J. T. Surface science studies of the photoactivation of TiO2-new photochemical processes. Chem. Rev. 106, 4428–4453 (2006).
Fujishima, A., Zhang, X. & Tryk, D. A. TiO2 photocatalysis and related surface phenomena. Surf. Sci. Rep. 63, 515–582 (2008).
Henderson, M. A. A surface science perspective on TiO2 photocatalysis. Surf. Sci. Rep. 66, 185–297 (2011).
Murdoch, M. et al. The effect of gold loading and particle size on photocatalytic hydrogen production from ethanol over Au/TiO2 nanoparticles. Nat. Chem. 3, 489–492 (2011).
Ma, Y. et al. Titanium dioxide-based nanomaterials for photocatalytic fuel generations. Chem. Rev. 114, 9987–10043 (2014).
Idriss, H. & Seebauer, E. G. Reactions of ethanol over metal oxides. J. Mol. Catal. A 152, 201–212 (2000).
Rass-Hansen, J., Falsig, H., Jorgensen, B. & Christensen, C. H. Bioethanol: Fuel or feedstock? J. Chem. Technol. Biotechnol. 82, 329–333 (2007).
Surisetty, V. R., Dalai, A. K. & Kozinski, J. Alcohols as alternative fuels: An overview. Appl. Catal. A Gen. 404, 1–11 (2011).
Dohnálek, Z., Lyubinetsky, I. & Rousseau, R. Thermally-driven processes on rutile TiO2(110) – (1 × 1): A direct view at the atomic scale. Prog. Surf. Sci. 85, 161–205 (2010).
Henderson, M. A. & Lyubinetsky, I. Molecular-level insights into photocatalysis from scanning probe microscopy studies on TiO2(110). Chem. Rev. 113, 4428–4455 (2013).
Pang, C. L., Lindsay, R. & Thornton, G. Chemical reactions on rutile TiO2(110). Chem. Soc. Rev. 37, 2328–2353 (2008).
Sauer, M. L. & Ollis, D. F. Photocatalyzed oxidation of ethanol and acetaldehyde in humidified air. J. Catal. 158, 570–582 (1996).
Nimlos, M. R., Wolfrum, E. J., Brewer, M. L., Fennell, J. A. & Bintner, G. Gas-phase heterogeneous photocatalytic oxidation of ethanol: Pathways and kinetic modeling. Environ. Sci. Technol. 30, 3102–3110 (1996).
Muggli, D. S., McCue, J. T. & Falconer, J. L. Mechanism of the photocatalytic oxidation of ethanol on TiO2 . J. Catal. 173, 470–483 (1998).
Muggli, D. S., Lowery, K. H. & Falconer, J. L. Identification of adsorbed species during steady-state photocatalytic oxidation of ethanol on TiO2 . J. Catal. 180, 111–122 (1998).
Hwang, S. J. & Raftery, D. In situ solid-state NMR studies of ethanol photocatalysis: Characterization of surface sites and their reactivities. Catal. Today 49, 353–361 (1999).
Wu, W. C., Chuang, C. C. & Lin, J. L. Bonding geometry and reactivity of methoxy and ethoxy groups adsorbed on powdered TiO2 . J. Phys. Chem. B 104, 8719–8724 (2000).
Liao, L. F., Wu, W. C., Chen, C. Y. & Lin, J. L. Photooxidation of formic acid vs formate and ethanol vs ethoxy on TiO2 and effect of adsorbed water on the rates of formate and formic acid photooxidation. J. Phys. Chem. B 105, 7678–7685 (2001).
Coronado, J. M., Kataoka, S., Tejedor-Tejedor, I. & Anderson, M. A. Dynamic phenomena during the photocatalytic oxidation of ethanol and acetone over nanocrystalline TiO2: simultaneous FTIR analysis of gas and surface species. J. Catal. 219, 219–230 (2003).
Yu, Z. Q. & Chuang, S. S. C. In situ IR study of adsorbed species and photogenerated electrons during photocatalytic oxidation of ethanol on TiO2 . J. Catal. 246, 118–126 (2007).
Shen, M. M. & Henderson, M. A. Identification of the active species in photochemical hole scavenging reactions of methanol on TiO2 . J. Phys. Chem. Lett. 2, 2707–2710 (2011).
Shen, M. M. & Henderson, M. A. Role of water in methanol photochemistry on rutile TiO2(110). J. Phys. Chem. C 116, 18788–18795 (2012).
Zhou, C. Y. et al. Site-specific photocatalytic splitting of methanol on TiO2(110). Chem. Sci. 1, 575–580 (2010).
Zhou, C. Y. et al. Effect of defects on photocatalytic dissociation of methanol on TiO2(110). Chem. Sci. 2, 1980–1983 (2011).
Guo, Q. et al. Stepwise photocatalytic dissociation of methanol and water on TiO2(110). J. Am. Chem. Soc. 134, 13366–13373 (2012).
Gamble, L., Jung, L. S. & Campbell, C. T. Decomposition and protonation of surface ethoxys on TiO2(110). Surf. Sci. 348, 1–16 (1996).
Farfan-Arribas, E. & Madix, R. J. Role of defects in the adsorption of aliphatic alcohols on the TiO2(110) surface. J. Phys. Chem. B 106, 10680–10692 (2002).
Kim, Y. K., Kay, B. D., White, J. M. & Dohnálek, Z. Alcohol chemistry on rutile TiO2(110): the influence of alkyl substituents on reactivity and selectivity. J. Phys. Chem. C 111, 18236–18242 (2007).
Jayaweera, P. M., Quah, E. L. & Idriss, H. Photoreaction of ethanol on TiO2(110) single-crystal surface. J. Phys. Chem. C 111, 1764–1769 (2007).
Nadeem, A. M. et al. Ethanol photo-oxidation on a rutile TiO2(110) single crystal surface. Phys. Chem. Chem. Phys. 13, 7637–7643 (2011).
Hansen, J. Ø. et al. Direct evidence for ethanol dissociation on rutile TiO2(110). Phys. Rev. Lett. 107, 136102 (2011).
Martinez, U. et al. Reduced step edges on rutile TiO2(110) as competing defects to oxygen vacancies on the terraces and reactive sites for ethanol dissociation. Phys. Rev. Lett. 109, 155501 (2012).
Muir, J. N., Choi, Y. & Idriss, H. Computational study of ethanol adsorption and reaction over rutile TiO2(110) surfaces. Phys. Chem. Chem. Phys. 14, 11910–11919 (2012).
Huo, P. et al. Ethanol diffusion on rutile TiO2(110) mediated by H adatoms. J. Phys. Chem. Lett. 3, 283–288 (2012).
Ma, Z. B. et al. Photocatalytic dissociation of ethanol on TiO2(110) by near-band-gap excitation. J. Phys. Chem. C 117, 10336–10344 (2013).
Kershis, M. D. & White, M. G. Photooxidation of ethanol and 2-propanol on TiO2(110): evidence for methyl radical ejection. Phys. Chem. Chem. Phys. 15, 17976–17982 (2013).
Kundu, S. et al. Ethanol photoreaction on RuOx/Ru-modified TiO2(110). J. Phys. Chem. C 117, 11149–11158 (2013).
Walenta, C. A. et al. Ethanol photocatalysis on rutile TiO2(110): the role of defects and water. Phys. Chem. Chem. Phys. 17, 22809–22814 (2015).
Kurtz, R. L., Stockbauer, R., Madey, T. E. & Román, E. & de Segovia J. L. Synchrotron radiation studies of H2O adsorption on TiO2(110). Surf. Sci. 218, 178–200 (1989).
Wendt, S. et al. The role of interstitial sites in the Ti3d defect state in the band gap of titania. Science 320, 1755–1759 (2008).
Thomas, A. G. et al. Comparison of the electronic structure of anatase and rutile TiO2 single-crystal surfaces using resonant photoemission and x-ray absorption spectroscopy. Phys. Rev. B 75, 035105 (2007).
Henderson, M. A., Epling, W. S, Peden, C. H. F. & Perkins, C. L. Insights into photoexcited electron scavenging processes on TiO2 obtained from studies of the reaction of O2 with OH groups adsorbed at electronic defects on TiO2(110). J. Phys. Chem. B 107, 534–545 (2003).
Kim, Y. K. & Hwang, C. C. Photoemission study on the adsorption of ethanol on clean and oxidized rutile TiO2(110)–(1 × 1) surfaces. Surf. Sci. 605, 2082–2086 (2011).
Hugenschmidt, M. B., Gamble, L. & Campbell, C. T. The Interaction of H2O with a TiO2(110) Surface. Surf. Sci. 302, 329–340 (1994).
Wendt, S. et al. Oxygen vacancies on TiO2(110) and their interaction with H2O and O2: A combined high-resolution STM and DFT study. Surf. Sci. 598, 226–245 (2005).
Wendt, S. et al. Formation and splitting of paired hydroxyl groups on reduced TiO2(110). Phys. Rev. Lett. 96, 066107 (2006).
Kristoffersen, H. H. et al. Role of steps in the dissociative adsorption of water on rutile TiO2(110). Phys. Rev. Lett. 110, 146101 (2013).
Lira, E. et al. Dissociative and molecular oxygen chemisorption channels on reduced rutile TiO2(110): An STM and TPD study. Surf. Sci. 604, 1945–1960 (2010).
Lira, E. et al. Effects of the crystal reduction state on the interaction of oxygen with rutile TiO2(110). Catal. Today 182, 25–38 (2012).
Matthiesen, J. et al. Observation of all the intermediate steps of a chemical reaction on an oxide surface by scanning tunneling microscopy. ACS Nano 3, 517–526 (2009).
Matthiesen, J. et al. Formation and diffusion of water dimers on rutile TiO2(110). Phys. Rev. Lett. 102, 226101 (2009).
ISA, Centre for Storage Ring Facilities, Aarhus University, Aarhus: http://www.isa.au.dk.
Martinez, U. & Hammer, B. Adsorption properties versus oxidation states of rutile TiO2(110). J. Chem. Phys. 134, 194703 (2011).
Deskins, N. A., Rousseau, R. & Dupuis, M. Defining the role of excess electrons in the surface chemistry of TiO2 . J. Phys. Chem. C 114, 5891–5897 (2010).
Kristoffersen, H. H., Martinez, U. & Hammer, B. Modeling methyl chloride photo oxidation by oxygen species on TiO2(110). Top. Catal. 57, 171–176 (2014).
Gomes, W. P., Freund, T. & Morrison, S. R. Chemical reactions involving holes at zinc oxide single crystal anode. J. Electrochem. Soc. 115, 818–823 (1968).
Micic, O. I., Zhang, Y. N., Cromack, K. R., Trifunac, A. D. & Thurnauer, M. C. Photoinduced hole transfer from TiO2 to methanol molecules in aqueous-solution studied by electron-paramagnetic-resonance. J. Phys. Chem. 97, 13284–13288 (1993).
Yamakata, A., Ishibashi, T. & Onishi, H. Electron- and hole-capture reactions on Pt/TiO2 photocatalyst exposed to methanol vapor studied with time-resolved infrared absorption spectroscopy. J. Phys. Chem. B 106, 9122–9125 (2002).
Ohno, T., Izumi, S., Fujihara, K., Masaki, Y. & Matsumura, M. Vanishing of current-doubling effect in photooxidation of 2-propanol on TiO2 in solutions containing Fe(III) ions. J. Phys. Chem. B 104, 6801–6803 (2000).
Tamaki, Y. et al. Direct observation of reactive trapped holes in TiO2 undergoing photocatalytic oxidation of adsorbed alcohols: Evaluation of the reaction rates and yields. J. Am. Chem. Soc. 128, 416–417 (2006).
Wang, Z. T., Deskins, N. A., Henderson, M. A. & Lyubinetsky, I. Inhibitive influence of oxygen vacancies for photoactivity on TiO2(110). Phys. Rev. Lett. 109, 266103 (2012).
Cremer, T., Jensen, S. C. & Friend, C. M. Enhanced photo-oxidation of formaldehyde on highly reduced o-TiO2(110). J. Phys. Chem. C 118, 29242–29251 (2014).
Zhang, Z. & Yates Jr., J. T. Band bending in semiconductors: chemical and physical consequences at surfaces and interfaces. Chem. Rev. 112, 5520–5551 (2012).
Lægsgaard, E., Besenbacher, F., Mortensen, K. & Stensgaard, I. A fully automated, thimble-size scanning tunnelling microscope. J. Microsc. 152, 663–669 (1988).
Mortensen, J. J., Hansen, L. B. & Jacobsen, K. W. Real-space grid implementation of the projector augmented wave method. Phys. Rev. B 71, 035109 (2005).
Enkovaara, J. et al. Electronic structure calculations with GPAW: a real-space implementation of the projector augmented-wave method. J. Phys.-Condes. Matter. 22, 253202 (2010).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Henkelman, G., Uberuaga, B. P. & Jónsson, H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 113, 9901–9904 (2000).
Acknowledgements
We acknowledge the financial support by the Danish Research Agency, the Strategic Research Council, the Villum Kahn Rasmussen Foundation, the Carlsberg Foundation, the Danish Center for Scientific Computing and the European Research Council through an Advanced ERC grant (F.B.).
Author information
Authors and Affiliations
Contributions
S.W., J.Ø.H., H.I., B.H. and F.B. designed the research. J.Ø.H., E.L., Y.Y.W., S.W. and L.L. conducted the STM experiments. R.B., J.Ø.H., S.P., S.W. and Z.L. conducted the PES experiments. J.Ø.H. analysed the STM data and R.B., J.Ø.H. and S.P. the PES data. U.M. and B.H. performed the theoretical calculations. S.W. and J.Ø.H. wrote the manuscript. All authors contributed to writing and revising the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Hansen, J., Bebensee, R., Martinez, U. et al. Unravelling Site-Specific Photo-Reactions of Ethanol on Rutile TiO2(110). Sci Rep 6, 21990 (2016). https://doi.org/10.1038/srep21990
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep21990
This article is cited by
Enhanced Photocatalytic Fuel Denitrification over TiO2/α-Fe2O3 Nanocomposites under Visible Light Irradiation
Scientific Reports (2017)
Synthesis and photo-catalytic property of TiO2 nanotube arrays/ZnS
Applied Physics A (2017)
Efficient Synthesis of Ethanol from CH4 and Syngas on a Cu-Co/TiO2 Catalyst Using a Stepwise Reactor
Scientific Reports (2016)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
