
Reference Guided De Novo Genome Assembly of Transformation Pliable Solanum lycopersicum cv. Pusa Ruby

 ,
,
,
,
,
,
Abstract
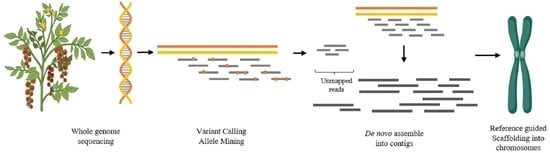
:
1. Introduction
2. Materials and Methods
2.1. Plant Materials and DNA Isolation
2.2. The Whole Genome Paired-End Library Preparation and Sequencing
2.3. Quality Control and Read Processing
2.4. K-mer Analysis and Heterozygosity Estimation
2.5. Reference Guided De Novo Genome Assembly
2.5.1. De Novo Genome Assembly
2.5.2. Filtering Chloroplastic, Mitochondrial and rDNA Genes
2.5.3. Scaffolding
2.6. The Repetitive Elements in the Sequence
2.7. Assessing Assembly Completeness and Quality Control
2.8. Assembly Packaging
2.9. Structural and Functional Annotation of the Pusa Ruby Genome
2.10. Mapping of Illumina PE Short Reads to S. lycopersicum Assembly (Build 4.0)
2.11. Analyses of SNPs, InDels, and Structural Variants
2.12. Whole Genome Comparison of Pusa Ruby against Heinz1706, M82, and Fla
2.13. Identification of Orthologous Genes
2.14. Allele Mining of Putative Genes Involved in Regeneration and Fruit Quality
2.15. Construct Preparation and Plant Transformation
3. Results
3.1. K-mer and Genome Heterozygosity Estimation
3.2. Reference Guided Genome Assembly of S. lycopersicum cv. Pusa Ruby and Quality Assessment
3.3. Genome-Wide Analysis of Variants
3.4. Structural and Functional Annotation of Pusa Ruby Genome
3.5. Whole Genome Comparison of Pusa Ruby with Fla.8924, M82 and Heinz1706
3.6. Comparative Evolutionary Genomics with Other Solanaceae Crops
3.7. Pusa Ruby Has Substantially High Regeneration and Transformation Efficiencies
3.8. Allele Mining of Genes Involved in Regeneration Efficiency
3.9. Characterization of Variants Associated with Fruit Quality-Related and Other Domestication-Related Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tranchida-Lombardo, V.; Aiese Cigliano, R.; Anzar, I.; Landi, S.; Palombieri, S.; Colantuono, C.; Bostan, H.; Termolino, P.; Aversano, R.; Batelli, G. Whole-genome re-sequencing of two Italian tomato landraces reveals sequence variations in genes associated with stress tolerance, fruit quality and long shelf-life traits. DNA Res. 2018, 25, 149–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peralta, I.E.; Knapp, S.; Spooner, D.M. Nomenclature for wild and cultivated tomatoes. Tomato Genet. Coop. Rep. 2006, 56, 6–12. [Google Scholar]
- Giovannoni, J.J. Genetic regulation of fruit development and ripening. Plant Cell 2004, 16, S170–S180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Consortium, T.G.S.; Aflitos, S.; Schijlen, E.; de Jong, H.; de Ridder, D.; Smit, S.; Finkers, R.; Wang, J.; Zhang, G.; Li, N. Exploring genetic variation in the tomato (81 section Lycopersicon) clade by whole-genome sequencing. Plant J. 2014, 80, 136–148. [Google Scholar]
- Jenkins, J.A. The origin of the cultivated tomato. Econ. Bot. 1948, 2, 379–392. [Google Scholar] [CrossRef]
- Lin, T.; Zhu, G.; Zhang, J.; Xu, X.; Yu, Q.; Zheng, Z.; Zhang, Z.; Lun, Y.; Li, S.; Wang, X. Genomic analyses provide insights into the history of tomato breeding. Nat. Genet. 2014, 46, 1220–1226. [Google Scholar] [CrossRef]
- Siva, R.; Bhakta, D. Determination of lycopene and its antioxidant activities in Indian tomatoes. J. Pharm. Res. 2010, 3, 933–937. [Google Scholar]
- Kaur, C.; Walia, S.; Nagal, S.; Walia, S.; Singh, J.; Singh, B.B.; Saha, S.; Singh, B.; Kalia, P.; Jaggi, S. Functional quality and antioxidant composition of selected tomato (Solanum lycopersicon L.) cultivars grown in Northern India. LWT-Food Sci. Technol. 2013, 50, 139–145. [Google Scholar] [CrossRef]
- Chandra, H.M.; Shanmugaraj, B.M.; Srinivasan, B.; Ramalingam, S. Influence of genotypic variations on antioxidant properties in different fractions of tomato. J. Food Sci. 2012, 77, C1174–C1178. [Google Scholar] [CrossRef]
- Naveen, B.; Reddy, K.R.; Saidaiah, P. Genetic divergence for yield and yield attributes in tomato (Solanum lycopersicum). Indian J. Agric. Sci. 2018, 88, 1018–1023. [Google Scholar] [CrossRef]
- Singh, V.; Bhattacherjee, A.; Soni, M.K.; Yadav, A.K.; Dikshit, A. Performance of Tomato (Solanum lycopersicum Mill) Cultivars for Quality Production under Protected Cultivation in Subtropics. Int. J. Curr. Microbiol. App. Sci. 2020, 9, 1127–1135. [Google Scholar] [CrossRef]
- Sharma, M.K.; Solanke, A.U.; Jani, D.; Singh, Y.; Sharma, A.K. A simple and efficient Agrobacterium-mediated procedure for transformation of tomato. J. Biosci. 2009, 34, 423. [Google Scholar] [CrossRef]
- Madhu, S.; Savithramma, D. Standardization of high efficient and rapid regeneration protocol for Agrobacterium mediated transformation of tomato (Solanum lycopersicum L.) cv. Pusa Ruby, Vaibhav and ArkaMeghali. Int. J. Agron. Agric. Res 2014, 5, 161–169. [Google Scholar]
- Kaur, P.; Bansal, K. Efficient production of transgenic tomatoes via Agrobacterium-mediated transformation. Biol. Plant. 2010, 54, 344–348. [Google Scholar] [CrossRef]
- Sarker, R.H.; Islam, K.; Hoque, M. In vitro regeneration and Agrobacterium-mediated genetic transformation of tomato (Lycopersicon esculentum Mill.). Plant Tissue Cult. Biotechnol. 2009, 19, 101–111. [Google Scholar] [CrossRef]
- Patil, R.; Davey, M.; Power, J.; Cocking, E. Effective Protocol for Agrobacterium-mediated Leaf Disc Transformation in Tomato (Lycopersicon esculetum Mill.). Indian J. Biotechnol. 2002, 1, 339–343. [Google Scholar]
- Afroz, A.; Chaudhry, Z.; Rashid, U.; Ali, G.M.; Nazir, F.; Iqbal, J.; Khan, M.R. Enhanced resistance against bacterial wilt in transgenic tomato (Lycopersicon esculentum) lines expressing the Xa21 gene. Plant Cell Tissue Organ Cult. (PCTOC) 2011, 104, 227–237. [Google Scholar] [CrossRef]
- Girhepuje, P.; Shinde, G. Transgenic tomato plants expressing a wheat endochitinase gene demonstrate enhanced resistance to Fusarium oxysporum f. sp. lycopersici. Plant Cell Tissue Organ Cult. (PCTOC) 2011, 105, 243–251. [Google Scholar] [CrossRef]
- Raj, S.; Singh, R.; Pandey, S.; Singh, B. Agrobacterium-mediated tomato transformation and regeneration of transgenic lines expressing Tomato leaf curl virus coat protein gene for resistance against TLCV infection. Curr. Sci. 2005, 88, 1674–1679. [Google Scholar]
- Vidya, C.S.; Manoharan, M.; Kumar, C.R.; Savtthri, H.; Sita, G.L. Agrobacterium-mediated transformation of tomato (Lycopersicon esculentum var. Pusa Ruby) with coat-protein gene of Physalis mottle tymovirus. J. Plant Physiol. 2000, 156, 106–110. [Google Scholar] [CrossRef]
- Vinutha, T.; Vanchinathan, S.; Bansal, N.; Kumar, G.; Permar, V.; Watts, A.; Ramesh, S.; Praveen, S. Tomato auxin biosynthesis/signaling is reprogrammed by the geminivirus to enhance its pathogenicity. Planta 2020, 252, 51. [Google Scholar] [CrossRef] [PubMed]
- Yadav, P.K.; Gupta, N.; Verma, V.; Gupta, A.K. Overexpression of SlHSP90. 2 leads to altered root biomass and architecture in tomato (Solanum lycopersicum). Physiol. Mol. Biol. Plants 2021, 27, 713–725. [Google Scholar] [CrossRef] [PubMed]
- Bhagat, Y.S.; Bhat, R.S.; Kolekar, R.M.; Patil, A.C.; Lingaraju, S.; Patil, R.; Udikeri, S. Remusatia vivipara lectin and Sclerotium rolfsii lectin interfere with the development and gall formation activity of Meloidogyne incognita in transgenic tomato. Transgenic Res. 2019, 28, 299–315. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.; Prasad, M. Silencing AC1 of Tomato leaf curl virus using artificial microRNA confers resistance to leaf curl disease in transgenic tomato. Plant Cell Rep. 2020, 39, 1565–1579. [Google Scholar] [CrossRef]
- Roy, R.; Purty, R.S.; Agrawal, V.; Gupta, S.C. Transformation of tomato cultivar ‘Pusa Ruby’with bspA gene from Populus tremula for drought tolerance. Plant Cell Tissue Organ Cult. 2006, 84, 56–68. [Google Scholar] [CrossRef]
- Pal, H.; Sahu, R.; Sethi, A.; Hazra, P.; Chatterjee, S. Unraveling the metabolic behavior in tomato high pigment mutants (hp-1, hp-2dg, ogc) and non ripening mutant (rin) during fruit ripening. Sci. Hortic. 2019, 246, 652–663. [Google Scholar] [CrossRef]
- Balyan, S.; Rao, S.; Jha, S.; Bansal, C.; Das, J.R.; Mathur, S. Characterization of novel regulators for heat stress tolerance in tomato from Indian sub-continent. Plant Biotechnol. J. 2020, 18, 2118–2132. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Ramanjini Gowda, P.; Saikia, B.; Debbarma, J.; Velmurugan, N.; Chikkaputtaiah, C. Screening of tomato genotypes against bacterial wilt (Ralstonia solanacearum) and validation of resistance linked DNA markers. Australas. Plant Pathol. 2018, 47, 365–374. [Google Scholar] [CrossRef]
- Nanjundappa, A.; Bagyaraj, D.J.; Saxena, A.K.; Kumar, M.; Chakdar, H. Interaction between arbuscular mycorrhizal fungi and Bacillus spp. in soil enhancing growth of crop plants. Fungal Biol. Biotechnol. 2019, 6, 23. [Google Scholar] [CrossRef] [Green Version]
- Khanna, K.; Kohli, S.K.; Ohri, P.; Bhardwaj, R.; Al-Huqail, A.A.; Siddiqui, M.H.; Alosaimi, G.S.; Ahmad, P. Microbial fortification improved photosynthetic efficiency and secondary metabolism in Lycopersicon esculentum plants under Cd stress. Biomolecules 2019, 9, 581. [Google Scholar] [CrossRef] [Green Version]
- Clarke, J.D. Cetyltrimethyl ammonium bromide (CTAB) DNA miniprep for plant DNA isolation. Cold Spring Harb. Protoc. 2009, 2009, pdb–prot5177. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2017. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 25 April 2022).
- Marçais, G.; Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 2011, 27, 764–770. [Google Scholar] [CrossRef] [Green Version]
- Vurture, G.W.; Sedlazeck, F.J.; Nattestad, M.; Underwood, C.J.; Fang, H.; Gurtowski, J.; Schatz, M.C. GenomeScope: Fast reference-free genome profiling from short reads. Bioinformatics 2017, 33, 2202–2204. [Google Scholar] [CrossRef] [Green Version]
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; He, G.; Chen, Y.; Pan, Q.; Liu, Y. SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler. Gigascience 2012, 1, 18. [Google Scholar] [CrossRef]
- Alonge, M.; Lebeigle, L.; Kirsche, M.; Aganezov, S.; Wang, X.; Lippman, Z.B.; Schatz, M.C.; Soyk, S. Automated assembly scaffolding elevates a new tomato system for high-throughput genome editing. bioRxiv 2021. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Alonge, M.; Lebeigle, L.; Kirsche, M.; Jenike, K.; Ou, S.; Aganezov, S.; Wang, X.; Lippman, Z.B.; Schatz, M.C.; Soyk, S. Automated assembly scaffolding using RagTag elevates a new tomato system for high-throughput genome editing. Genome Biol. 2022, 23, 258. [Google Scholar] [CrossRef]
- Chen, N. Using Repeat Masker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinform. 2004, 5, 4.10.11–14.10.14. [Google Scholar] [CrossRef]
- Ou, S.; Su, W.; Liao, Y.; Chougule, K.; Agda, J.R.; Hellinga, A.J.; Lugo, C.S.B.; Elliott, T.A.; Ware, D.; Peterson, T. Benchmarking transposable element annotation methods for creation of a streamlined, comprehensive pipeline. Genome Biol. 2019, 20, 275. [Google Scholar] [CrossRef] [Green Version]
- Seppey, M.; Manni, M.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness. Gene Predict. Methods Protoc. 2019, 1962, 227–245. [Google Scholar]
- Levy Karin, E.; Mirdita, M.; Söding, J. MetaEuk—Sensitive, high-throughput gene discovery, and annotation for large-scale eukaryotic metagenomics. Microbiome 2020, 8, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shumate, A.; Salzberg, S.L. Liftoff: Accurate mapping of gene annotations. Bioinformatics 2021, 37, 1639–1643. [Google Scholar] [CrossRef] [PubMed]
- Stanke, M.; Steinkamp, R.; Waack, S.; Morgenstern, B. AUGUSTUS: A web server for gene finding in eukaryotes. Nucleic Acids Res. 2004, 32, W309–W312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pertea, G.; Pertea, M. GFF Utilities: GffRead and GffCompare [version 1; peer review: 2 approved]. F1000Research 2020, 9, 304. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Pozo, N.; Menda, N.; Edwards, J.D.; Saha, S.; Tecle, I.Y.; Strickler, S.R.; Bombarely, A.; Fisher-York, T.; Pujar, A.; Foerster, H. The Sol Genomics Network (SGN)—From genotype to phenotype to breeding. Nucleic Acids Res. 2015, 43, D1036–D1041. [Google Scholar] [CrossRef]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [Green Version]
- Marçais, G.; Delcher, A.L.; Phillippy, A.M.; Coston, R.; Salzberg, S.L.; Zimin, A. MUMmer4: A fast and versatile genome alignment system. PLoS Comput. Biol. 2018, 14, e1005944. [Google Scholar] [CrossRef] [Green Version]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef] [Green Version]
- Huson, D.H.; Richter, D.C.; Rausch, C.; Dezulian, T.; Franz, M.; Rupp, R. Dendroscope: An interactive viewer for large phylogenetic trees. BMC Bioinform. 2007, 8, 460. [Google Scholar] [CrossRef] [Green Version]
- Čermák, T.; Curtin, S.J.; Gil-Humanes, J.; Čegan, R.; Kono, T.J.Y.; Konečná, E.; Belanto, J.J.; Starker, C.G.; Mathre, J.W.; Greenstein, R.L.; et al. A Multipurpose Toolkit to Enable Advanced Genome Engineering in Plants. Plant Cell 2017, 29, 1196–1217. [Google Scholar] [CrossRef] [Green Version]
- Eck, J.V.; Kirk, D.D.; Walmsley, A.M. Tomato (Lycopersicum esculentum). Agrobacterium Protoc. 2006, 343, 459–474. [Google Scholar]
- Manni, M.; Berkeley, M.R.; Seppey, M.; Simão, F.A.; Zdobnov, E.M. BUSCO update: Novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Mol. Biol. Evol. 2021, 38, 4647–4654. [Google Scholar] [CrossRef]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [Green Version]
- Blanca, J.; Montero-Pau, J.; Sauvage, C.; Bauchet, G.; Illa, E.; Díez, M.J.; Francis, D.; Causse, M.; van Der Knaap, E.; Cañizares, J. Genomic variation in tomato, from wild ancestors to contemporary breeding accessions. BMC Genom. 2015, 16, 257. [Google Scholar] [CrossRef] [Green Version]
- Alonge, M.; Soyk, S.; Ramakrishnan, S.; Wang, X.; Goodwin, S.; Sedlazeck, F.J.; Lippman, Z.B.; Schatz, M.C. RaGOO: Fast and accurate reference-guided scaffolding of draft genomes. Genome Biol. 2019, 20, 224. [Google Scholar] [CrossRef] [Green Version]
- Altpeter, F.; Springer, N.M.; Bartley, L.E.; Blechl, A.E.; Brutnell, T.P.; Citovsky, V.; Conrad, L.J.; Gelvin, S.B.; Jackson, D.P.; Kausch, A.P. Advancing crop transformation in the era of genome editing. Plant Cell 2016, 28, 1510–1520. [Google Scholar] [CrossRef] [Green Version]
- Ikeuchi, M.; Sugimoto, K.; Iwase, A. Plant callus: Mechanisms of induction and repression. Plant Cell 2013, 25, 3159–3173. [Google Scholar] [CrossRef] [Green Version]
- Fehér, A. Somatic embryogenesis—Stress-induced remodeling of plant cell fate. Biochim. Et Biophys. Acta (BBA)-Gene Regul. Mech. 2015, 1849, 385–402. [Google Scholar] [CrossRef]
- Sakamoto, T.; Deguchi, M.; Brustolini, O.J.B.; Santos, A.A.; Silva, F.F.; Fontes, E.P.B. The tomato RLK superfamily: Phylogeny and functional predictions about the role of the LRRII-RLK subfamily in antiviral defense. BMC Plant Biol. 2012, 12, 229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuste-Lisbona, F.J.; Fernández-Lozano, A.; Pineda, B.; Bretones, S.; Ortíz-Atienza, A.; García-Sogo, B.; Müller, N.A.; Angosto, T.; Capel, J.; Moreno, V.; et al. ENO regulates tomato fruit size through the floral meristem development network. Proc. Natl. Acad. Sci. USA 2020, 117, 8187–8195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boutilier, K.; Offringa, R.; Sharma, V.K.; Kieft, H.; Ouellet, T.; Zhang, L.; Hattori, J.; Liu, C.-M.; van Lammeren, A.A.; Miki, B.L. Ectopic expression of BABY BOOM triggers a conversion from vegetative to embryonic growth. Plant Cell 2002, 14, 1737–1749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jha, P.; Kumar, V. BABY BOOM (BBM): A candidate transcription factor gene in plant biotechnology. Biotechnol. Lett. 2018, 40, 1467–1475. [Google Scholar] [CrossRef] [PubMed]
- Hill, K.; Mathews, D.E.; Kim, H.J.; Street, I.H.; Wildes, S.L.; Chiang, Y.-H.; Mason, M.G.; Alonso, J.M.; Ecker, J.R.; Kieber, J.J. Functional characterization of type-B response regulators in the Arabidopsis cytokinin response. Plant Physiol. 2013, 162, 212–224. [Google Scholar] [CrossRef] [Green Version]
- Hill, K.; Schaller, G.E. Enhancing plant regeneration in tissue culture: A molecular approach through manipulation of cytokinin sensitivity. Plant Signal. Behav. 2013, 8, 212–224. [Google Scholar] [CrossRef] [Green Version]
- Huang, M.; Hu, Y.; Liu, X.; Li, Y.; Hou, X. Arabidopsis LEAFY COTYLEDON1 controls cell fate determination during post-embryonic development. Front. Plant Sci. 2015, 6, 955. [Google Scholar] [CrossRef] [Green Version]
- Mu, J.; Tan, H.; Zheng, Q.; Fu, F.; Liang, Y.; Zhang, J.; Yang, X.; Wang, T.; Chong, K.; Wang, X.J.; et al. LEAFY COTYLEDON1 is a key regulator of fatty acid biosynthesis in Arabidopsis. Plant Physiol. 2008, 148, 1042–1054. [Google Scholar] [CrossRef] [Green Version]
- Shohat, H.; Cheriker, H.; Cohen, A.; Weiss, D. ABA-IMPORTING TRANSPORTER 1.1 activity in the endosperm of tomato seeds restrains germination under salinity stress. bioRxiv 2022. [Google Scholar] [CrossRef]
- Huang, F.-C.; Fu, B.-J.; Liu, Y.-T.; Chang, Y.-R.; Chi, S.-F.; Chien, P.-R.; Huang, S.-C.; Hwang, H.-H. Arabidopsis RETICULON-LIKE3 (RTNLB3) and RTNLB8 Participate in Agrobacterium-Mediated Plant Transformation. Int. J. Mol. Sci. 2018, 19, 638. [Google Scholar] [CrossRef] [Green Version]
- Abe-Hara, C.; Yamada, K.; Wada, N.; Ueta, R.; Hashimoto, R.; Osakabe, K.; Osakabe, Y. Effects of the sliaa9 Mutation on Shoot Elongation Growth of Tomato Cultivars. Front. Plant Sci. 2021, 12, 627832. [Google Scholar] [CrossRef]
- Hu, G.; Wang, K.; Huang, B.; Mila, I.; Frasse, P.; Maza, E.; Djari, A.; Hernould, M.; Zouine, M.; Li, Z.; et al. The auxin-responsive transcription factor SlDOF9 regulates inflorescence and flower development in tomato. Nat. Plants 2022, 8, 419–433. [Google Scholar] [CrossRef]
- De Jong, M.; Wolters-Arts, M.; Feron, R.; Mariani, C.; Vriezen, W.H. The Solanum lycopersicum auxin response factor 7 (SlARF7) regulates auxin signaling during tomato fruit set and development. Plant J. 2009, 57, 160–170. [Google Scholar] [CrossRef]
- Willits, M.G.; Kramer, C.M.; Prata, R.T.; De Luca, V.; Potter, B.G.; Steffens, J.C.; Graser, G. Utilization of the genetic resources of wild species to create a nontransgenic high flavonoid tomato. J. Agric. Food Chem. 2005, 53, 1231–1236. [Google Scholar] [CrossRef]
- Lippman, Z.B.; Cohen, O.; Alvarez, J.P.; Abu-Abied, M.; Pekker, I.; Paran, I.; Eshed, Y.; Zamir, D. The making of a compound inflorescence in tomato and related nightshades. PLoS Biol. 2008, 6, e288. [Google Scholar] [CrossRef]
- Xu, C.; Liberatore, K.L.; MacAlister, C.A.; Huang, Z.; Chu, Y.-H.; Jiang, K.; Brooks, C.; Ogawa-Ohnishi, M.; Xiong, G.; Pauly, M. A cascade of arabinosyltransferases controls shoot meristem size in tomato. Nat. Genet. 2015, 47, 784–792. [Google Scholar] [CrossRef]
- Frary, A.; Nesbitt, T.C.; Frary, A.; Grandillo, S.; Knaap, E.v.d.; Cong, B.; Liu, J.; Meller, J.; Elber, R.; Alpert, K.B. fw2. 2: A quantitative trait locus key to the evolution of tomato fruit size. Science 2000, 289, 85–88. [Google Scholar] [CrossRef] [Green Version]
- Powell, A.L.; Nguyen, C.V.; Hill, T.; Cheng, K.L.; Figueroa-Balderas, R.; Aktas, H.; Ashrafi, H.; Pons, C.; Fernández-Muñoz, R.; Vicente, A. Uniform ripening encodes a Golden 2-like transcription factor regulating tomato fruit chloroplast development. Science 2012, 336, 1711–1715. [Google Scholar] [CrossRef] [Green Version]
- Cookson, P.; Kiano, J.; Shipton, C.; Fraser, P.; Romer, S.; Schuch, W.; Bramley, P.; Pyke, K. Increases in cell elongation, plastid compartment size and phytoene synthase activity underlie the phenotype of the high pigment-1 mutant of tomato. Planta 2003, 217, 896–903. [Google Scholar] [CrossRef]
- Mao, L.; Begum, D.; Chuang, H.-W.; Budiman, M.A.; Szymkowiak, E.J.; Irish, E.E.; Wing, R.A. JOINTLESS is a MADS-box gene controlling tomato flower abscissionzone development. Nature 2000, 406, 910–913. [Google Scholar] [CrossRef]
- Fridman, E.; Carrari, F.; Liu, Y.-S.; Fernie, A.R.; Zamir, D. Zooming in on a quantitative trait for tomato yield using interspecific introgressions. Science 2004, 305, 1786–1789. [Google Scholar] [CrossRef] [PubMed]
- Ronen, G.; Carmel-Goren, L.; Zamir, D.; Hirschberg, J. An alternative pathway to β-carotene formation in plant chromoplasts discovered by map-based cloning of Beta and old-gold color mutations in tomato. Proc. Natl. Acad. Sci. USA 2000, 97, 11102–11107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seymour, G.B.; Chapman, N.H.; Chew, B.L.; Rose, J.K. Regulation of ripening and opportunities for control in tomato and other fruits. Plant Biotechnol. J. 2013, 11, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Van Eck, J.; Cong, B.; Tanksley, S.D. A new class of regulatory genes underlying the cause of pear-shaped tomato fruit. Proc. Natl. Acad. Sci. USA 2002, 99, 13302–13306. [Google Scholar] [CrossRef] [Green Version]
- Lifschitz, E.; Eviatar, T.; Rozman, A.; Shalit, A.; Goldshmidt, A.; Amsellem, Z.; Alvarez, J.P.; Eshed, Y. The tomato FT ortholog triggers systemic signals that regulate growth and flowering and substitute for diverse environmental stimuli. Proc. Natl. Acad. Sci. USA 2006, 103, 6398–6403. [Google Scholar] [CrossRef] [Green Version]
- Royo, J.; Kowyama, Y.; Clarke, A.E. Cloning and nucleotide sequence of two S-RNases from Lycopersicon peruvianum (L.) Mill. Plant Physiol. 1994, 105, 751. [Google Scholar] [CrossRef] [Green Version]
- Uluisik, S.; Chapman, N.H.; Smith, R.; Poole, M.; Adams, G.; Gillis, R.B.; Besong, T.M.; Sheldon, J.; Stiegelmeyer, S.; Perez, L. Genetic improvement of tomato by targeted control of fruit softening. Nat. Biotechnol. 2016, 34, 950–952. [Google Scholar] [CrossRef] [Green Version]
- DellaPenna, D.; Alexander, D.C.; Bennett, A.B. Molecular cloning of tomato fruit polygalacturonase: Analysis of polygalacturonase mRNA levels during ripening. Proc. Natl. Acad. Sci. USA 1986, 83, 6420–6424. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Yuan, B.; Leng, P. Cloning of 9-cis-epoxycarotenoid dioxygenase (NCED) gene and the role of ABA on fruit ripening. Plant Signal. Behav. 2009, 4, 460–463. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Zhou, L.; Xu, H.; Wang, L.; Liu, H.; Zhang, C.; Li, Q.; Gu, M.; Wang, C.; Liu, Q.; et al. The qSAC3 locus from indica rice effectively increases amylose content under a variety of conditions. BMC Plant Biol. 2019, 19, 275. [Google Scholar] [CrossRef] [Green Version]
- Project, I.R.G.S. The map-based sequence of the rice genome. Nature 2005, 436, 793–800. [Google Scholar]
- Jiao, Y.; Zhao, H.; Ren, L.; Song, W.; Zeng, B.; Guo, J.; Wang, B.; Liu, Z.; Chen, J.; Li, W. Corrigendum: Genome-wide genetic changes during modern breeding of maize. Nat. Genet. 2014, 46, 1039–1040. [Google Scholar] [CrossRef] [Green Version]
- Ma, Z.; He, S.; Wang, X.; Sun, J.; Zhang, Y.; Zhang, G.; Wu, L.; Li, Z.; Liu, Z.; Sun, G. Resequencing a core collection of upland cotton identifies genomic variation and loci influencing fiber quality and yield. Nat. Genet. 2018, 50, 803–813. [Google Scholar] [CrossRef] [PubMed]
- Mata-Nicolás, E.; Montero-Pau, J.; Gimeno-Paez, E.; Garcia-Carpintero, V.; Ziarsolo, P.; Menda, N.; Mueller, L.A.; Blanca, J.; Cañizares, J.; Van der Knaap, E. Exploiting the diversity of tomato: The development of a phenotypically and genetically detailed germplasm collection. Hortic. Res. 2020, 7, 66. [Google Scholar] [CrossRef]
- Finkers, R.; van Heusden, S. The 150+ tomato genome (re-) sequence project; lessons learned and potential applications. Tomato Breed. Roundtable 2013, 14, 791. [Google Scholar]
- Wu, S.; Zhang, B.; Keyhaninejad, N.; Rodríguez, G.R.; Kim, H.J.; Chakrabarti, M.; Illa-Berenguer, E.; Taitano, N.K.; Gonzalo, M.J.; Díaz, A.; et al. A common genetic mechanism underlies morphological diversity in fruits and other plant organs. Nat. Commun. 2018, 9, 4734. [Google Scholar] [CrossRef] [Green Version]
- Florentin, A.; Damri, M.; Grafi, G. Stress induces plant somatic cells to acquire some features of stem cells accompanied by selective chromatin reorganization. Dev. Dyn. 2013, 242, 1121–1133. [Google Scholar] [CrossRef]
- Grafi, G.; Barak, S. Stress induces cell dedifferentiation in plants. Biochim. Et Biophys. Acta (BBA)-Gene Regul. Mech. 2015, 1849, 378–384. [Google Scholar] [CrossRef]
- Rodríguez, G.R.; Kim, H.J.; van der Knaap, E. Mapping of two suppressors of OVATE (sov) loci in tomato. Heredity 2013, 111, 256–264. [Google Scholar] [CrossRef] [Green Version]
- Zsögön, A.; Čermák, T.; Naves, E.R.; Notini, M.M.; Edel, K.H.; Weinl, S.; Freschi, L.; Voytas, D.F.; Kudla, J.; Peres, L.E.P. De novo domestication of wild tomato using genome editing. Nat. Biotechnol. 2018, 36, 1211–1216. [Google Scholar] [CrossRef] [Green Version]
- Mustilli, A.C.; Fenzi, F.; Ciliento, R.; Alfano, F.; Bowler, C. Phenotype of the tomato high pigment-2 mutant is caused by a mutation in the tomato homolog of DEETIOLATED1. Plant Cell 1999, 11, 145–157. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chromosome | Chromosome Length (bp) Heinz1706 | Chromosome Length (bp) Pusa Ruby | Number of Contigs from the Pusa Ruby De Novo Assembly Ordered and Oriented by RagTag |
|---|---|---|---|
| chr 1 | 90,863,682 | 84,547,471 | 5703 |
| chr 2 | 53,473,368 | 49,336,188 | 33,805 |
| chr 3 | 65,298,490 | 60,101,449 | 44,527 |
| chr 4 | 64,459,972 | 60,396,827 | 41,165 |
| chr 5 | 65,269,487 | 60,896,774 | 46,821 |
| chr 6 | 47,258,699 | 43,868,379 | 30,453 |
| chr 7 | 67,883,646 | 62,983,648 | 47,809 |
| chr 8 | 63,995,357 | 59,251,915 | 45,563 |
| chr 9 | 68,513,564 | 64,410,981 | 48,037 |
| chr 10 | 64,792,705 | 59,814,738 | 46,567 |
| chr 11 | 54,379,777 | 49,956,557 | 39,443 |
| chr 12 | 66,688,036 | 60,856,427 | 47,439 |
| Sum | 772,876,783 | 716,421,354 |
| Statistics without Reference | Heinz1706 Reference | PR |
|---|---|---|
| # contigs or scaffolds | 13 | 96,632 |
| # contigs or scaffolds (≥0 bp) | 13 | 96,632 |
| # contigs or scaffolds (≥1000 bp) | 13 | 4568 |
| # contigs (≥25,000 bp) | 13 | 25 |
| # contigs (≥50,000 bp) | 13 | 15 |
| Largest contig or scaffolds | 90,863,682 | 84,547,471 |
| Total length (≥0 bp) | 782,520,033 | 753,700,435 |
| Total length (≥1000 bp) | 782,520,033 | 729,632,876 |
| Total length (≥25,000 bp) | 782,520,033 | 718,987,089 |
| Total length (≥50,000 bp) | 782,520,033 | 718,782,531 |
| Total length | 782,520,033 | 753,700,435 |
| N50 | 65,269,487 | 60,396,827 |
| N90 | 53,473,368 | 49,336,188 |
| L50 | 6 | 6 |
| L90 | 11 | 11 |
| GC (%) | 34.34 | 33.84 |
| Heinz1706 | PR | Fla.8924 | PR | M82 | PR | |
|---|---|---|---|---|---|---|
| Sequences | ||||||
| Total Seqs | 13 | 13 | 13 | 13 | 13 | 13 |
| Aligned Seqs | 13 (100.00%) | 13 (100.00%) | 13 (100.00%) | 13 (100.00%) | 13 (100.00%) | 13 (100.00%) |
| Unaligned Seqs | 0 (0.00%) | 0 (0.00%) | 0 (0.00%) | 0 (0.00%) | 0 (0.00%) | 0 (0.00%) |
| Bases | ||||||
| Total Bases | 782,520,033 | 718,639,073 | 796,004,315 | 718,639,073 | 792,934,937 | 718,639,073 |
| Aligned Bases | 726,051,384 (92.78%) | 690,758,192 (96.12%) | 715,955,700 (89.9437%) | 679,673,622 (94.58%) | 717,409,934 (90.48%) | 681,068,925 (94.77%) |
| Unaligned Bases | 56,468,649 (7.22%) | 27,880,881 (3.88%) | 80,048,615 (10.0563) | 38,965,451 (5.42%) | 75,525,003 (9.52%) | 37,570,148 (5.23%) |
| Alignments | ||||||
| one-to-one | 232,149 | 216,975 | 224,643 | |||
| Total Length (one-to-one) | 681,763,285 | 681,795,255 | 663,171,553 | 664,463,584 | 665,697,245 | 665,915,093 |
| Avg Length (one-to-one) | 2936.75 | 2936.89 | 3056.725 | 3062.67705 | 2964.6299 | 2902.57 |
| Avg Identity (one-to-one) | 99.84 | 99.5656 | 99.5777 | |||
| M-to-M | 399,293 | 393,605 | 401,181 | |||
| Total Length (M-to-M) | 771,135,016 | 771,128,110 | 781,072,565 | 783,869,191 | 774,217,622 | 774,798,581 |
| Avg Length (M-to-M) | 1931.25 | 1931.23 | 1985.6105 | 1992.7126 | 1930.32895 | 1931.7748 |
| Avg Identity (M-to-M) | 99.15 | 98.5888 | 98.66545 | |||
| Feature Estimates | ||||||
| Breakpoints | 798,577 | 798,553 | 787,208 | 787,189 | 802,352 | 802,339 |
| Relocations | 26,309 | 17,453 | 28,508 | 27,573 | 28,697 | 27,028 |
| Translocations | 7328 | 7221 | 16,357 | 14,667 | 12,041 | 11,130 |
| Inversions | 714 | 718 | 3173 | 2644 | 2422 | 2112 |
| Insertions | 242,155 | 319,388 | 235,641 | 323,561 | 247,574 | 323,754 |
| Insertion Sum | 130,014,304 | 49,843,243 | 162,162,183 | 80,350,739 | 156,691,957 | 70,683,091 |
| Insertion Avg | 536.91 | 156.06 | 688.5165 | 248.3442 | 633.373 | 218.5611 |
| Tandem Ins | 321 | 42 | 374 | 61 | 400 | 110 |
| Sum of Tandem Ins | 92,910 | 2859 | 59,361 | 4491 | 76,900 | 9232 |
| Tandem Ins Avg | 289.44 | 68.07 | 157.92195 | 72.5741 | 192.08725 | 83.38 |
| SNPs | ||||||
| Total SNPs | 335,439 | 786,423 | 1,452,890 | |||
| Total GSNPs | 167,484 | 266,642 | 734,607 | |||
| Total InDels | 356,419 | 1,572,447 | 880,972 | |||
| Total GInDels | 94,663 | 54,287 | 56,923 | |||
| Gene | Arabidopsis Homologue | GENE ID | ITAG 4.0 Positions | Number of Variants in PR | High Impact Variant | Reference |
|---|---|---|---|---|---|---|
| SOMATIC EMBRYOGENESIS RECEPTOR LIKE KINASE | NA | Solyc04g072570.3 | 57484887…57491796 | 13 | N | [59,60,61,62] |
| CLAVATA 1 | NA | Solyc11g071380 | 52945095…52945649 | 3 | N | [59,60,61,62,63] |
| WUSCHEL | NA | Solyc02g083950.3.1 | 45191157…45192582 | 11 | N | |
| SHOOT MERISTEMLESS | NA | Solyc02g081120.4 | 43175431…43179388 | 2 | N | |
| BABY BOOM | NA | Solyc11g008560.2 | 2782134…2787131 | 4 | N | [59,64,65] |
| Cytokinin type-B ARABIDOPSIS RESPONSE REGULATORs | At4g31920 | Solyc12g010330.2 | 3431027…3436636 | 5 | N | [66,67] |
| LEAFY COTYLEDON1 (LEC1) | At1g21970 | Solyc04g015060.3 | 5289310…5292118 | 0 | N | [68,69,70] |
| AGAMOUS-LIKE15 | NA | Solyc01g087990.3 | 75071660…75077872 | 51 | Y; c.562C > T; p.Arg188 *; STOP GAINED | |
| RETICULON-LIKE3 (RTNLB3) | AT1G64090 | Solyc01g095200.3.1; Solyc03g007740.3.1 | 78807148…78810378; 2297766…2300313 | 0; 1 | N | [71] |
| RETICULON-LIKE8 (RTNLB8) | AT3G10260 | Solyc12g006290.2.1 | 809990…814489 | 2 | N | [71] |
| SlIAA9 | NA | Solyc04g076850.3 | 59750087…59755552 | 271 | N | [72] |
| SlDOF9 | NA | Solyc02g090220.3 | 49886648…49887678 | 19 | N | [73] |
| S.No. | Gene | Gene ID | Gene Function | Position in ITAG 4.0 | Number of Variants in PR | High Impact Variant | Reference |
|---|---|---|---|---|---|---|---|
| 1 | AUXIN RESPONSE FACTOR 19 (ARF19) | Solyc07g042260.4 | Negative regulator of fruit set | 55179909…55187268 | 4 | N | [74] |
| 2 | CHALCONE ISOMERASE (CHI) | Solyc06g084260.3 | Contributes in flavonoid biosynthesis | 47032434…47037684 | 1 | N | [75] |
| 3 | COMPOUND INFLORESCENCE (S) | Solyc02g077390.2 | Affects number of flower/fruits per inflorescence | 40324933…40326867 | 22 | N | [76] |
| 4 | FASCIATED (FAS) | Solyc11g071380.1 | Controls number of carpels/locules in the fruit | 52945095…52945649 | 2 | N | [77] |
| 5 | FRUIT WEIGHT 2.2 (FW2.2) | Solyc02g090730.3 | Quantitative variation of fruit size | 50292691…50293481 | 3 | N | [78] |
| 6 | GOLDEN2-LIKE (GLK2) | Solyc10g008160.3 | Controls chloroplast development in fruits | 2169302…2174039 | 0 | N | [79] |
| 7 | HIGH-PIGMENT 1 (HP1) | Solyc02g021650 | Controls anthocyanin accumulation in fruits | 21799951…21820357 | 23 | N | [80] |
| 8 | HIGH-PIGMENT 2 (HP2) | Solyc01g056340 | Controls anthocyanin accumulation in fruits | 46851339…46871997 | 127 | Y; FRAME SHIFT; c.1503dupC; p.His503fs | [80] |
| 9 | JOINTLESS1 (J1) | Solyc11g010570 | Controls abscission zone formation in pedicels | 3671232…3676350 | 148 | N | [81] |
| 10 | LIN5 | Solyc09g010080 | Extracellular invertase expressed in the fruit ovary | 3508156…3512282 | 1 | N | [82] |
| 11 | LYCOPENE β CYCLASE (Cyc-B) | Solyc06g074240 | Conversion of lycopene into B-carotene | 43562526…43564022 | 0 | N | [83] |
| 12 | NON RIPENING (NOR) | Solyc10g006880 | Controls the initiation of the normal fruit ripe program | 1191950…1194846 | 3 | N | [84] |
| 13 | OVATE (O) | Solyc02g085500 | Regulates fruit shape | 46380291…46382023 | 3 | Y; STOP LOST; c.856T > G; p.Ter286Gluext * | [85] |
| 14 | RIPENING INHIBITOR (RIN) | Solyc05g012020 | Controls ripening-related ethylene biosynthesis | 5302348…5308593 | 9 | N | [84] |
| 15 | SINGLE FLOWER TRUSS (SFT) | Solyc03g063100 | Flowering inducer | 29218239…29222055 | 9 | N | [86] |
| 16 | S-RNAse | Solyc01g055200 | Controls Self-incompatibility | 44944259…44945021 | 409 | N | [87] |
| 17 | Pectate Lyase (PL) | Solyc03g111690 | Control pectin degradation in cell wall | 56752862…56755013 | 9 | N | [88] |
| 18 | Plygalacturanase (POLYGAL) | Solyc07g056290 | Cell wall degrading enzyme | 64064076…64067658 | 2 | N | [89] |
| 19 | 9-cis-epoxycarotenoid dioxygenase (NCED) | Solyc07g056570 | Involved in ABA biosysnthesis | 64278530…64280347 | 6 | N | [90] |
| 20 | Β-1-3-galactosyl transferase (GAL1) | Solyc07g052320 | Regulation of fruit galactose levels | 60727212…60731146 | 1 | N |
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.
|
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vats, S.; Kumar, V.; Mandlik, R.; Patil, G.; Sonah, H.; Roy, J.; Sharma, T.R.; Deshmukh, R. Reference Guided De Novo Genome Assembly of Transformation Pliable Solanum lycopersicum cv. Pusa Ruby. Genes 2023, 14, 570. https://doi.org/10.3390/genes14030570
Vats S, Kumar V, Mandlik R, Patil G, Sonah H, Roy J, Sharma TR, Deshmukh R. Reference Guided De Novo Genome Assembly of Transformation Pliable Solanum lycopersicum cv. Pusa Ruby. Genes. 2023; 14(3):570. https://doi.org/10.3390/genes14030570
Chicago/Turabian StyleVats, Sanskriti, Virender Kumar, Rushil Mandlik, Gunvant Patil, Humira Sonah, Joy Roy, Tilak Raj Sharma, and Rupesh Deshmukh. 2023. "Reference Guided De Novo Genome Assembly of Transformation Pliable Solanum lycopersicum cv. Pusa Ruby" Genes 14, no. 3: 570. https://doi.org/10.3390/genes14030570