
Abstract
The cardiovascular system is frequently affected by coronavirus disease-19 (COVID-19), particularly in hospitalized cases, and these manifestations are associated with a worse prognosis. Most commonly, heart involvement is represented by myocarditis, myocardial infarction, and pulmonary embolism, while arrhythmias, heart valve damage, and pericarditis are less frequent. While the clinical suspicion is necessary for a prompt disease recognition, imaging allows the early detection of cardiovascular complications in patients with COVID-19. The combination of cardiothoracic approaches has been proposed for advanced imaging techniques, i.e., CT scan and MRI, for a simultaneous evaluation of cardiovascular structures, pulmonary arteries, and lung parenchyma. Several mechanisms have been proposed to explain the cardiovascular injury, and among these, it is established that the host immune system is responsible for the aberrant response characterizing severe COVID-19 and inducing organ-specific injury. We illustrate novel evidence to support the hypothesis that molecular mimicry may be the immunological mechanism for myocarditis in COVID-19. The present article provides a comprehensive review of the available evidence of the immune mechanisms of the COVID-19 cardiovascular injury and the imaging tools to be used in the diagnostic workup. As some of these techniques cannot be implemented for general screening of all cases, we critically discuss the need to maximize the sustainability and the specificity of the proposed tests while illustrating the findings of some paradigmatic cases.
Similar content being viewed by others
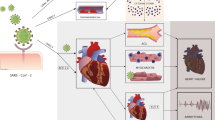


Introduction
The disease associated with an acute infection by SARS-CoV-2 was coined COVID-19 and became a pandemic by March 2020, causing over 5.5 million deaths worldwide as of December 2021 [1, 2]. The clinical manifestations of COVID-19 typically include respiratory involvement with cough and dyspnea, as well as systemic signs and symptoms such as fever, fatigue, myalgia, and arthralgia, among others [1]. Some degree of cardiovascular involvement is observed in 20–30% of hospitalized cases, raising to 55% in the presence of a pre-existing cardiovascular disease [3, 4]. Acute heart injury in COVID-19 may include myocarditis, myocardial infarction, pulmonary embolism, and less frequently arrhythmias, valvular damage, and pericarditis [5, 6]. Data from hospitalized COVID-19 cases in our highly impacted area demonstrated that myocardial injury (defined by elevated troponin and/or elevated B natriuretic peptide — BNP) is associated with a risk of death comparable to the coexistence of renal failure and inferior only to being older than 70 [7].
Prompted by this observation and moving from our clinical experience, we evaluate the literature regarding the heart involvement in COVID-19, to discuss the pathogenic role of the immune system and the use of imaging to better define the myocardial damage for the management and risk stratification of patients with COVID-19.
Immunopathogenetic Mechanisms of COVID-19 Myocardial Injury
Several mechanisms have been proposed to explain the COVID-19-related myocardial injury [8,9,10,11], and among these, an important role is played by the host immune system [12], which is responsible for the aberrant response characterizing severe infections and causing organ damage [13]. Figure 1 illustrates the proposed mechanisms of COVID-19-related injury mediated by the immune system, which are detailed below.

Immunopathogenetic mechanisms of COVID-19 myocardial injury. Mechanisms illustrated are cytokine release syndrome; cardiomyocyte injury mediated by cytotoxic T cells primed with viral antigens; pro-thrombotic status due to cytokine release, up-regulation of adhesion molecules, platelets activation, and antiphospholipid antibody formation; netosis; local endotheliitis of artery vessels leading to plaque rupture; anti-heart antibodies produced by molecular mimicry between viral antigens and human heart epitopes. See text for further details. Legend. SARS-CoV-2, severe acute respiratory syndrome-coronavirus 2; MIS-C, multisystem inflammatory syndrome in children; ACE2, angiotensin-converting enzyme 2; HLA, human leukocyte antigen; NETs, neutrophil extracellular traps
Cytokine Release Syndrome
In a subset of patients with COVID-19, the host immune system is aberrantly activated, resulting in acute systemic inflammation leading to the injury of multiple organs, including the heart [14]. Released cytokines have profound cardiovascular consequences and may cause the onset of tachycardia, hypotension, and left ventricular dysfunction, by the activation of different signalling pathways. In fact, tumor necrosis factor alpha (TNF-α) induces negative inotropic effects in vivo and in vitro, with mechanisms mediated by the sphingomyelinase pathway and by nitric oxide (NO), which reduce β-adrenergic signaling. On the other hand, two major cytokines involved in the cytokine release syndrome, namely interleukin (IL)-1 and IL-6, may also mediate ventricular dysfunction through the production of NO [15] and, indirectly, through the activation and release of IL-18 [16].
Elevated proinflammatory cytokines are involved in the development of cardiac disease, and overexpressed TNF-α, IL-1β, IL-6, and matrix metalloproteinases (MMPs) in the heart can contribute to the onset of myocardial infarction. Furthermore, these changes have been associated with the impaired intracellular calcium and potassium current regulation and altered expression of proteins in cardiomyocytes lining the infarct border zone, promoting electrical disturbances. The cytokine release syndrome has been correlated with the development of atrial fibrillation and more generally the increased risk of cardiac arrhythmias [17, 18]. Further associations have been proposed with the development of sustained cardiac injury, characterized by elevated plasmatic levels of BNP and other myocardial injury markers [17]. We note that there is significant evidence suggesting that chronic inflammation is a pathogenic driver of heart failure, leading to an abnormal thickening and fibrosis of cardiac tissue, and ultimately to cardiac dysfunction [19], as well represented by the cardiovascular risk associated with rheumatoid and psoriatic arthritis [20]. Proinflammatory cytokines induce myocyte hypertrophy, alteration in gene expression, and activation of MMP, resulting in myocardial apoptosis and development of fibrosis [16].
In the pediatric population, COVID-19 generally induces a less severe disease, with lower mortality rates, but in a subgroup of children, fever and signs of systemic inflammation occur, mimicking the feature of Kawasaki disease, a medium-vessel vasculitis associated with infections. Kawasaki disease is an acute and usually self-limiting disease, occasionally complicated by hemodynamic instability, a condition known as Kawasaki disease shock syndrome or by a macrophage activation syndrome (MAS) [21]. The manifestations occurring in some pediatric cases of COVID-19 differ from classical Kawasaki disease, as patients are usually older, with more pronounced signs of cardiac involvement, shock, and MAS and more frequently require adjunctive glucocorticoid treatment [22]. Based on the clinical differences, the syndrome has been coined “pediatric inflammatory multisystem syndrome temporally associated with SARS-CoV-2 infection” (PIMS-TS) in Europe and “multisystem inflammatory syndrome in children” (MIS-C) in the USA. Although it has been demonstrated that hyperinflammation in MIS-C differs from the COVID-19-related cytokine storm [23], children with this syndrome present high levels of IL-1, IL-6, and IL-18, which can be responsible for shock and negative cardiac outcomes [24, 25]. Relevant to the present review article, a similar syndrome has been proposed for adults developing delayed-onset myocarditis after SARS-CoV-2 infection. In a small series, patients with a recent recovery from COVID-19 presented with cardiogenic shock, significant increase of inflammatory markers, fever, pulmonary infiltrates, and mucocutaneous and gastrointestinal involvement, similarly to what occurs in MIS-C [26].
T Cell–Mediated Myocardial Injury
The entry of SARS-CoV-2 into cells is mediated by the binding of the viral spike protein to the membrane protein angiotensin-converting enzyme 2 (ACE2) [27] which is found also on cardiomyocytes [28], beside the epithelial cells of the respiratory tract, type II pneumocytes. ACE2 is upregulated [29] in patients with dilated or ischemic cardiomyopathy independent of COVID-19, and this observation may explain why patients with a preexisting heart disease are at higher risk of developing COVID-19-related complications. Furthermore, the virus directly damages cardiomyocytes, but a T cell–mediated cytotoxicity may also occur as viral antigens expressed by infected cardiomyocytes prime naïve T lymphocytes via antigen-presenting cells, and primed CD8 + T lymphocytes, chemoattracted by the hepatocyte growth factor (HGF) produced by damaged myocardiocytes, may in turn be cytotoxic to the cardiovascular tissues. This process is amplified during the COVID-19 cytokine release syndrome, when proinflammatory cytokines disseminate and T cells become hyperactivated, in a positive feedback loop of immune activation and myocardial damage [28]. T lymphocytes have been found in the damaged myocardium independent of the presence of cardiomyocytes viral infection. [30] In fact, the SARS-CoV-2 genome has been found only in some cases of endomyocardial biopsy or at autopsy [31,32,33], suggesting a potential perpetuation of the damage after the viral clearance, or that T lymphocytes are primed in other sites and subsequently cross-react with myocardial antigens.
Moreover, the presence of myocarditis has been questioned altogether by some authors, as the presence of the virus was not associated with an increased infiltration of mononuclear cells in the myocardium and histology was not fully consistent with the criteria of myocarditis.
On the other hand, the reduced lymphocytic infiltrate found in the hearts of SARS-CoV-2-infected individuals compared to controls may be attributed to the systemic lymphopenia associated with the viral infection. It has been shown that a proportion of patients recovered from COVID-19 had alterations consistent with myocarditis at cardiac magnetic resonance (CMR), including myocardial edema, fibrosis, and impaired ventricular function, suggesting the possibility that cardiac inflammation may persist months after the resolution of the acute infection, with resulting ventricular dysfunction [34,35,36].
Pro-thrombotic Status
The altered cytokine and chemokine levels occurring in COVID-19 may disrupt the endothelial function and integrity and lead to the upregulation of cell adhesion molecules (ICAM-1, integrins, P- and E-selectin), the release of Von Willebrand factor (VWF), inducing endothelial production of cytokines and chemokines, with recruitment of platelets and leukocytes [17].
In this scenario, the activated platelets can release the chemokines stored in their granules, such as PF-4 (platelet factor-4), CXCL1 (chemokine (C-X-C motif) ligand), CXCL5, CXCL7, CCL3 (chemokine (C–C motif) ligand), CCL5, and CCL7. This leads to further leukocyte recruitment, vascular injury, and increased proinflammatory and procoagulant factors [37]. Neutrophil activation can also cause thrombosis, and in the serum of patients with COVID-19, high level of neutrophil extracellular traps (NETs) has been reported. NETs are extracellular networks of nucleic acid wrapped with histones, extruded by neutrophils with a controlled process that allows the mixing of chromatin with granular proteins. The web-like structure formed is involved in virus entrapment and physical containment of the infection. On the other hand, NET release triggers an intense inflammation, with complement activation, formation of complexes of NETs, platelets, and thrombin [38].
COVID-19 is also associated with higher levels of complement, tissue factor–enriched NETs and tissue factor activity. Compared to mild COVID-19, neutrophils from patients requiring ventilatory support are characterized by enhanced NETosis, suggesting that levels of NETs correlate with disease severity [39], as well represented by the highest levels of NETs in patients needing invasive ventilation and correlating with mortality. In a small case series of COVID-19 with myocardial infarction, NETs were detected at high density in the coronary aspirates and thrombi were composed of fibrin and polymorphonuclear cells, without showing fragments of atherosclerotic plaques [40]. Post mortem, microthrombi containing NETs, neutrophils, and platelets were reported in the lung tissue of COVID-19.
COVID-19-related thrombosis can be also linked to the presence of serum antiphospholipid antibodies (aPLs), including anti-cardiolipin (aCL) IgG and IgM, anti-beta2-glycoprotein I (β2-GPI) IgG and IgM, and lupus anticoagulant (LAC), which induce both arterial and venous thrombosis. It has been well established that infections trigger aPLs, either transiently or chronically with or without the development of thrombosis [41], and several cases of aPLs-associated thrombosis occurring in COVID-19 have been reported, occurring both in severe and mild disease. Among these, a few had myocardial infarction, while the majority presented deep vein thrombosis or pulmonary embolism [21, 42, 43].
Local prothrombotic factors have also been advocated in COVID-19 manifestations, and IL-1, IL-6, and other pro-inflammatory cytokines produced by activated leukocytes are known to induce endothelial activation, adhesion molecules, further inflammatory cell infiltration, and vascular inflammation. These processes may be locally enhanced during a viral infection in the adjacent vascular smooth muscle cells, and local macrophages release plasminogen activators as pro-coagulant factors. The altered endothelium becomes activated and pro-coagulant, predisposing to microthrombosis leading to the COVID-19-induced endotheliitis, occurring in several organs as a result of the host inflammatory response to the presence of the virus [17]. This microembolism triggered by the damaged endothelium or hypercoagulation state further leads to the destabilization of the existing coronary artery plaques, thus precipitating a myocardial ischemia [44, 45]. Patients with COVID-19 may also manifest higher complement pathway activation. In a few cases, deposits of C5b-9 and C4d were found in the lung microvasculature [46], indicating the activation of the alternative and lectin-based complement system.
Anti-heart Autoantibody
One report illustrates the correlation between serum anti-heart autoantibodies and COVID-19-related cardiovascular disease. Anti-endothelial cell, anti-cardiomyocytes, anti-smooth muscle, and anti-cardiac conductive tissue antibodies have been measured in a small cohort, and their levels correlated with cardiovascular manifestations and with the presence of systemic inflammation. In particular, anti-cardiomyocyte antibodies correlate with low QRS complex voltage, anti-smooth muscle antibody levels with atrial fibrillation, and antinuclear and anti-cardiomyocyte antibodies with pericardial effusion. Anti-smooth muscle and anti-cardiomyocyte antibodies were associated with poor prognosis [47]. These data suggest a possible involvement of these antibodies in the development of inflammatory cardiac injury, other than being an epiphenomenon of systemic inflammation.
Another hypothesis is that a molecular mimicry mechanism may trigger the adaptive immune response, leading to the development of specific, potentially pathogenic autoantibodies, as discussed in previous studies for other diseases [12, 48, 49]. In an earlier study, it was demonstrated that idiopathic inflammatory cardiomyopathy is caused by the activation of MYH6-specific CD4 + T cells and B cells by a mimic peptide of a commensal gut bacteria, Bacteroides theta, in genetically susceptible individuals [50]. To support this hypothesis, we searched for homologies between the reported immune-dominant epitopes of MYH6 [50, 51], troponin [52], and SARS-CoV-2 proteins (Table 1) and identified relevant alignments for amino acids 6291–6297 of SARS-CoV-2, with homology with MYH6-aa614-629. This sequence also corresponds to a fragment of the ORF1ab protein, an exonuclease which provides error-correcting capability to the RNA synthesis complex of SARS-CoV-2 while other homologies were identified with two sequences of the viral replicase polyprotein 1a and 1ab, encoded by ORF1ab. Furthermore, the predicted binding of SARS-CoV-2 peptides homologous to MYH6 epitopes was relevant for class II HLA, especially for HLA-DRB1*04:05, HLA-DRB3*02:02, and HLA-DQA1*03:01/DQB1*03:02. We believe that our analysis supports the hypothesis that molecular mimicry between SARS-CoV-2 peptides and immune-dominant epitopes of human MYH6 is involved in the pathogenesis of inflammatory cardiomyopathy, which can be presented by HLA-DQ and -DR4 alleles associated with myocarditis [51]. To demonstrate that molecular mimicry is effectively involved in SARS-CoV-2-mediated cardiac damage would require the identification of autoreactive T cells and/or autoantibodies reacting against the specific epitopes of MYH6 and SARS-CoV-2.
Imaging for the Detection of COVID-19 Cardiovascular Injury
Myocardial injury is a frequently observed complication in hospitalized COVID-19 patients, but it has been poorly studied during the first pandemic wave. Advanced cardiac imaging (cardiac MR and cardiac CT) plays a major role both in the early detection of the COVID-19 cardiovascular involvement in a setting of pre-existing cardiovascular conditions (Fig. 2) and in the definition of cardiovascular complications, but its widespread use was challenging during the first months of the pandemic due to often unmanageable organizational issues.

Chest CT scan of a patient before and after COVID-19 infection. The CT scan on panel A was performed during an ER evaluation due to suspected nephrolithiasis; the imaging in panel B was performed on the same patient 18 days later demonstrating bilateral consolidations and ground-glass opacities, septal thickening, mainly located within the upper lobes
Table 2 illustrates the radiological indications and the main imaging findings in COVID-19-related cardiovascular complications. We believe that CMR represents the imaging modality of choice to assess myocardial injury while signs of cardiac failure can be described at chest X-ray, chest computed tomography (CT), and echocardiography [53].
Imaging of Myocardial Injury
The prevalence of acute damage in hospitalized COVID-19 patients is estimated in the range of 12–36%, especially in the presence of prior cardiovascular conditions or severe COVID-19 manifestations [54]. The myocardial injury is defined as an increase in cardiac troponin (cTn) values exceeding the 99th percentile and encompasses both ischemic and non-ischemic causes. Evidence of cardiac involvement in hospitalized COVID-19 patients is significant because this finding is associated with higher mortality. According to cTn variations, the damage is defined as either acute or chronic (the latter defined as a stable elevation with a variation ≤ 20%) [55]. The diagnostic approach includes a careful medical history and ECG, followed by imaging techniques for the evaluation of myocardial morphology and function. Transthoracic echocardiography (TTE) is commonly the first-line imaging test while advanced imaging techniques, i.e., cardiac CT and MR, are less used in acute settings for practical organizational/management issues, despite their usefulness in the differential diagnosis of myocardial conditions for therapeutic decision-making [56].
Specifically, CMR is the gold standard for structural and functional non-invasive evaluation of the myocardium and its high accuracy in the detection of tissue edema allows to discriminate among ischemic and non-ischemic causes of acute myocardial damage according to the distribution pattern [57]. Galea et al. proposed a combined cardiothoracic MRI (CaTh-MRI; overall length of the standard protocol 45–60 min) protocol in COVID-19 patients hospitalized with myocardial damage which allowed a simultaneous evaluation of cardiovascular structures, pulmonary arteries, and lung parenchyma [58]. In the authors’ experience, abnormal T2 mapping was the most commonly observed abnormality and significantly correlated with hs-cTnT values, reflecting the predominant inflammatory nature of myocardial damage observed during the active phase of the disease [59]. The acute or hyperacute inflammatory myocardial damage observed in COVID-19, associated to an elevated myocardial T2 signal at CMR, has been directly related to the presence of virus-mediated cardiomyocyte injury characterized by the physical presence of viral particles in the myocardium [60]. The main limitation of CMR in acute COVID-19 is the scan duration that might not be tolerated in case of hemodynamic and/or respiratory instability. Shorter protocols lasting 10–15 min overall are recommended in patients with active infection and poor functional status for a focused evaluation of cardiac function and structure [61]. Similar to CaTh-MRI, cardiothoracic CT in COVID-19 has been reported in the literature, considering that chest CT has a central role in the diagnosis of COVID-19 pneumonia and is already routinely performed [62]. Pontone et al. introduced the idea of a quadruple rule-out with CT for the simultaneous evaluation of signs of interstitial pneumonia, pulmonary embolism, and ischemic or non-ischemic cardiac disease [63]. Moreover, CT may provide cardiovascular parameters with clinical and prognostic relevance also if acquired without ECG gating, as usually performed in the acute phase. An increased diameter of the pulmonary artery — indirect expression of pulmonary hypertension — and elevated coronary artery calcium (CAC) score — expression of subclinical atherosclerosis and coronary artery disease — can be in fact predictors of myocardial injury and mortality in COVID-19 patients [64, 65]. The obvious advantage of CT to evaluate both the lung parenchyma and the cardiovascular features should not be underestimated particularly in conditions of high clinical burden.
Long COVID
Approximately one in four COVID-19 cases have lingering symptoms, even after recovering from the virus; long-term cardiovascular sequelae of COVID-19, coined “long COVID,” are not completely understood, but, as already reported with SARS-CoV-1 [66], chronic manifestations may occur after recovery. Symptoms are generally aspecific and include chest pain, syncope, palpitation, persistent dyspnea, possibly underlying arrhythmias, ventricular dysfunction, and new-onset heart failure [67]. These conditions have been identified as complications of COVID-19-related immune activation [28] in which chronic forms may manifest with persistent cardiac failure, which may progress to dilated cardiomyopathy [68]. In a recent paper [69], long COVID has been reported as the clinical expression of the presence of “anti-idiotype antibodies” of SARS-CoV-2 infection and/or vaccination. Briefly, the protective antibodies created against the virus can subsequently trigger a new antibody response toward themselves, leading to their disappearance over time. These secondary antibodies, called anti-idiotype antibodies, can bind to — and deplete — the initial protective antibody responses. These have the potential to mirror or act like the original antigen itself, and anti-idiotype responses against the enterovirus coxsackievirus B3 can cause autoimmune myocarditis in mice [70]. Ultimately, this mechanism may be involved in the persistent myocardial inflammation described in earlier reports of long COVID-19 [71] and the implications of cardiac injury in the subacute and chronic COVID-19 forms have been evaluated by CMR which represents the standard reference of cardiac involvement assessment [72].
Myocardial Infarction
Myocardial infarction is a subtype of myocardial injury characterized by clinical evidence of ischemia. Type 1 myocardial infarction is due to coronary athero-thrombosis after plaque disruption, and type 2 myocardial infarction is related to a mismatch between oxygen supply and demand [55].
The relationship between acute infections and myocardial infarction has been already established [73], and the potential mechanisms are similar in COVID-19 patients. The instability of atherosclerotic plaques increases for the higher intraplaque inflammatory activity, and a systemic prothrombotic and procoagulant state raises the risk of coronary thrombosis after plaque rupture. According to the literature, type I myocardial infarction in COVID-19 is less common [54] while type II myocardial infarction may be precipitated by multiple factors related to both systemic inflammation and severe hypoxia (the most common clinical manifestation of COVID-19) [54]. In COVID patients, systemic inflammation, as represented by elevated CRP, has been associated to coagulopathies such as venous thrombo-embolism (VTE) and disease severity such as acute kidney injury (AKI), critical illness, and in-hospital mortality [74]. Consequences include coronary thrombosis without evident atherosclerotic plaque, decreased coronary perfusion because of the combination of tachycardia (increased metabolic needs of peripheral organs), and pneumonia-related hypoxemia.
The definition of a myocardial infarction diagnostic algorithm in COVID-19 is challenging because invasive unnecessary examinations should be avoided to minimize the risk of dissemination [57]. The choice of the appropriate imaging tests is key for the accurate diagnosis and management; in a patient presenting with chest pain, the initial diagnostic workup should include a careful medical history and physical examination, ECG, chest X–ray, and biomarkers to confirm the suspicion of acute coronary syndrome. Apart from patients with a high probability of acute coronary syndrome (STEMI or high-risk non-STEMI) that are immediately evaluated with invasive coronary angiography (ICA), echocardiography should be performed to confirm a regional wall motion abnormality (RWMA), typical of myocardial infarction. Subsequently, advanced imaging techniques can be used to confirm the suspicion or for a differential diagnosis [67]. Coronary CT angiography (CTCA) is associated with a high negative predictive value to rule out clinically significant coronary artery disease and may avoid unnecessary invasive procedures, especially in patients without indications for revascularization [75]. In fact, 40% of ICA performed in patients with COVID-19 and STEMI were not able to identify a culprit lesion, because type 2 myocardial infarction is common in this group of patients [76]. The proposed cardiothoracic CT protocol in COVID-19 with low or intermediate probability of acute coronary syndrome includes a non-enhanced chest CT focused on involvement of lung parenchyma and calculation of CAC score and a CTCA (ECG-gated CT angiography) for the simultaneous evaluation of the patency of pulmonary and coronary arteries. On one hand, the detection of a severe stenosis in coronary arteries may be an indication for ICA while, on the other hand, a delayed CT acquisition after 7–10 min from the contrast injection can be performed to identify other causes of non-obstructive coronary artery disease or myocardial injury [77]. Contrast-enhanced CT scan can demonstrate the presence of intracardiac thrombi that may cause coronary or systemic embolisms and type 2 myocardial infarction [78] and infarcted areas appear initially as focal decreased enhancement and demonstrate myocardial late iodine enhancement [55]. Multiphase CCTA, i.e., acquisition during an entire cardiac cycle, can be useful to evaluate also left ventricular ejection fraction and RWMA [79]. However, elevated heart rate and difficulty to reduce it adequately may be a major limiting factor for CCTA, reducing the global diagnostic accuracy and the potential advantages of CCTA in many hypoxemic patients. CMR should be limited to cases of diagnostic uncertainty after assessment by echocardiography and, if applicable, by cardiac CT. In acute myocardial infarction, CMR can assess myocardial viability and function, microvascular obstruction, and intramyocardial hemorrhage. A typical protocol would include performing (a) short-axis cine imaging to determine cardiac size and function, (b) T2-weighted imaging, (c) delayed post-contrast (after the administration of contrast material) imaging, and (d) T1 and T2 mapping, if available. In T2 sequences, subendocardial or transmural edema is a common finding of myocardial infarction and it has a typical distribution in myocardial segments according to coronary distribution [57]. Cine-imaging with CMR is the gold standard for the measurement of ventricular volumes, mass, and function, and late gadolinium enhancement determines the extent of irreversibly injured myocardium [80]. Moreover, pharmacological stress MRI might be performed in symptomatic patients with known coronary artery disease and abnormal ECG, avoiding exercise tests to minimize the risk of COVID-19 spread (Fig. 3) [67]. Gadolinium-based contrast agents have a late washout from fibrotic areas, and this mechanism enhances myocardial fibrosis [55]. The estimate of transmural extent of an infarcted area is a predictor of the functional recovery, acting as an independent prognostic marker.

Myocardial infarction. Cardiac MRI (panel A: SSFP image, panel B: late gadolinium enhancement image, panel C: native T1 map) illustrating an extensive anterior myocardial infarction (white arrows in panels B and C) involving the anterior and anteroseptal segments, the apex, and the apical inferior segment of left ventricle. Infarcted area is characterized by a low-signal intensity component consistent with the presence of severe microvascular obstruction (black arrows in panel B). There is a concomitant apical thrombus (white star in panel A), appearing as a low-signal intensity non-enhancing mass on post-contrast images (white star in panel B)
Myocarditis
Myocarditis is a non-ischemic cardiac cause of acute myocardial injury [55], already described in previous outbreaks of Coronaviridae [81,82,83], being in some cases the main clinical manifestation of COVID-19 [84]. The clinical presentation is variable, and acute forms can mimic myocardial infarction since patients report chest pain and dyspnea, in some cases associated with troponin elevation. Fulminant forms may also occur and manifest with acute-onset heart failure [68]. ECG is usually abnormal with aspecific changes, such as sinus tachycardia, PR depression, or ST/T-wave anomalies; serum troponin and inflammatory markers (IL-6, CRP, procalcitonin) are usually elevated. Echocardiography is usually the first-line imaging, but findings are generally non-specific; echocardiography allows an exclusion diagnosis with other causes of acute chest pain; usual findings are diffuse wall motion abnormalities, reduced ejection fraction with left ventricular hypokinesis, and pericardial effusion [85]. Endomyocardial biopsy is the diagnostic gold standard but, especially during the early phases of the pandemic [68], was rarely employed for its invasive nature.
CMR is the non-invasive gold standard test for the study of the myocardium [57] and can be performed in the acute forms for immediate management [86] (Fig. 4). The diagnosis of acute myocarditis using CMR should fulfil the Lake Louise Criteria, which allows a 91% specificity and a 67% sensitivity using conventional sequences [87]. More recently, new diagnostic criteria were released based on the introduction of novel acquisition techniques with T1 and T2 mapping, measuring specific relaxometry-based changes characterizing active inflammation [88]. The T1-based criterion refers to hyperemia and capillary leakage or to myocardial necrosis and fibrosis with the former appearing as regional or global enhancement before and shortly after contrast injection (early gadolinium enhancement — EGE) and the latter resulting in late gadolinium enhancement (LGE) of the necrotic/fibrotic area in a non-ischemic distribution pattern after 10 min from contrast injection. The T2-based criterion refers to myocardial edema that appears in a regional or global enhancement, generally subepicardial or middle wall. Pericardial effusion or systolic LV wall motion abnormalities in cine CMR images are included in the supportive criteria (Fig. 4) [88].

Myocarditis. Cardiac MRI of 2 COVID-19 patients with chest pain and troponin elevation during hospitalization. Panel A/B: late gadolinium enhancement (LGE) images of a patient with ECG changes. Subepicardial areas of LGE (white arrows) are visible in the inferior and infero-lateral wall of the left ventricle. In the right ventricle inflow, further areas of LGE indicate bi-ventricular myocardial damage. Panel C/D: late gadolinium enhancement (LGE) images of a patient without ECG changes. White arrows show spotty subepicardial enhancement in the mid-lateral wall; there is a mild pericardial effusion (asterisk): black in PSIR images and white in IR images (panel C/D)
This redefined diagnostic approach proved to be particularly useful in atypical forms of myocarditis, including those with cardiomyopathic or arrhythmic disease onset that have been observed in COVID-19 patients, where subtle tissue changes may be underestimated using a conventional, qualitative-based imaging approach [89]. However, CMR cannot discriminate myocarditis due to direct viral damage and an immune-mediated myocardial damage. ECG-gated cardiac CT may be an alternative to CMR when the latter is not available because iodine- and gadolinium-based contrast media have similar properties. In clinical practice, images acquired 6–8 min after contrast injection can assess the increase in myocardial extracellular volume (ECV) fraction and identify LIE in areas of focal fibrosis. As previously discussed, CT may have advantages in COVID-19 for the simultaneous cardiothoracic evaluation and differential diagnosis of acute myocardial injury — coined as “quadruple-rule out” [77].
In patients with COVID-19-related myocardial injury, the prevalence of myocardial structural abnormalities at CMR is around 56–78%, mainly as non-ischemic myocardial damage. This represents a tenfold increase if compared to age-matched control population [90]. This initial finding deserves more studies, since it is well known that myocardial scars do have prognostic implications over time.
Even if in lower percentage, asymptomatic patients could have encountered abnormal CMR findings. Simultaneous active pericarditis can be detected in areas with peri-epicardial LGE in combination with pericardial effusion [35, 36, 91, 92]. However, in subacute and chronic myocarditis, CMR has a lower sensitivity than in acute forms because edema is less evident. LGE incidence is similar in acute and chronic stages, and only T2 sequences allow the diagnosis of acute myocardial inflammation [68]: Increased native T1 and T2 indicate an active inflammation, while increased native T1 and normal native T2 point to a residual or resolving inflammation [35]. T1-mapping has a high overall negative predictive value in ruling out acute or chronic myocarditis due to a high sensitivity in the detection of increased myocardial free water content [86].
Other authors detected an impaired RV function in cine CMR (right ventricular strain); it can be easily affected by increased pulmonary vascular resistance due to typical lung involvement of COVID-19 [36, 93, 94].
Pulmonary Thromboembolism
Pulmonary thromboembolism is included among the most frequent vascular complications of COVID-19, but data about its incidence are conflicting [95]. According to a recent meta-analysis, pulmonary thromboembolism occurs in 16.5% of COVID-19 cases and exceeds 20% in patients admitted to ICU [96]. Pulmonary thromboembolism is usually a consequence of deep vein thrombosis, but in situ microvascular thrombosis may also coexist, defined as pulmonary artery thrombosis (PAT) [97,98,99]. The pathogenesis of PAT is not completely understood and evidence supports a hypercoagulability state [100], as demonstrated also in other viral respiratory infections [101].
Ciceri et al. encourage the use of the term MicroCLOTS (microvascular COVID-19 lung vessel obstructive thromboinflammatory syndrome) to define a progressive endothelial thromboinflammatory syndrome secondary to alveolar viral damage [102]. This hypothesis is supported by a peculiar arterial localization that has been evaluated on CT pulmonary angiography (CTPA), the current standard of care to show arterial filling defects and to provide a rapid diagnosis of pulmonary thromboembolism (Fig. 5) [103]. A high rate of distal pulmonary thromboembolism in segmental or subsegmental arteries is suggestive of immunothrombosis, especially in the absence of reported DVT [97, 104], and is frequently localized in lung segments with COVID-related parenchymal abnormalities [105].

Pulmonary thromboembolism. CT pulmonary angiography showing pulmonary thromboembolism (white arrows) in a patient with COVID-19 with elevated D-dimer. There is disease mainly involving the inferior right and the upper left superior lobar pulmonary arteries
CT may also detect pulmonary thromboembolism complications, as signs of right-heart failure leading to increased central venous pressure, hepatic congestion, or lung infarction. The typical radiological appearance of an infarcted area is a wedge-shaped parenchymal consolidation, eventually associated with evident filling defects in obstructed vessels, ground-glass opacities in areas of pulmonary hemorrhage and less commonly to a reverse halo sign or cavitation (sign of septic pulmonary embolism). It is important to differentiate ground-glass opacities and consolidations as complications of pulmonary thromboembolism or as primary parenchymal manifestations of COVID-19 [106]. Moreover, CT scan allows the estimation of RV/LV diameter ratio measured on transverse images: Its increase, sign of right ventricular dysfunction, is associated with a fivefold higher risk of pulmonary thromboembolism-related mortality [107]. An additional advantage of CTPA is the possibility to evaluate the volume of perfused and ventilated lungs without further imaging examinations. In fact, a specific analytical software allows to reconstruct parenchymal maps to identify low-perfusion areas, i.e., with pulmonary thromboembolism, resulting in ventilation-perfusion (V/Q) mismatch [108].
There is currently no consensus on the appropriate use of CTPA in COVID-19 and on the D-dimer threshold to perform diagnostic imaging. In fact, D-dimer values are routinely elevated in COVID-19 as an acute phase reactant [109] and may also have a prognostic role in the prediction of in-hospital mortality [110]. Elevated D-dimer values do not justify testing for pulmonary thromboembolism in absence of suspicious clinical symptoms/signs — cough, dyspnea, hemoptysis, tachycardia, and chest pain — or additional supportive information — history data, sonographic evidence, or previous deep vein thrombosis [95]. These parameters are included in the Wells score to determine the pretest probability of PE presence: A total score ≤ 4 indicates that PE is unlikely and warrants D-dimer testing; a total score > 4 indicates that PE is likely and needs rapid imaging evaluation with CTPA as first-line, especially in patients with a high bleeding risk that cannot start a full therapeutic anticoagulation therapy [111]. The accuracy of Well score in COVID-19 patients seems lower due to the possibility of immunologically driven thrombosis and the absence of the classical risk factors for DVT [112]. For this reason, different studies have focused on the identification of an adjusted D-dimer threshold for pulmonary thromboembolism imaging in COVID-19 to account for the inflammatory confounders. According to Suh et al., the conventional cutoff values of 500 if age-adjusted or 1000 μg/L can be used also in COVID-19 with high sensitivity, 96% and 91%, respectively, taking into consideration also the pretest clinical probability [96]. Higher threshold values (> 1000 μg/L) reduce the sensitivity of D-dimer, decreasing its usefulness [113] while obviously increasing its specificity. A peak D-dimer value > 6000 μg/L during hospitalization may represent an independent risk factor for pulmonary thromboembolism, especially if correlated to high values of CRP for a systemic inflammatory state [114]. Daily monitoring of D-dimers levels may identify patients at risk of pulmonary thromboembolism [115], and significant increases during hospitalization (> 2000–4000 μg/L) should be an indication for CTPA, especially in the presence of worsening clinical conditions (dyspnea, refractory hypoxemia, or unexplained new-onset tachycardia or hypotension) [116]. According to the European Society of Radiology and the European Society of Thoracic imaging, CTPA may rule out pulmonary thromboembolism in COVID-19 in need for supplementary oxygen and with limited parenchymal disease extension seen in a previous non-enhanced CT scan [117].
It is uncertain if pulmonary thromboembolism develops in the earliest phases of COVID-19 as Whyte et al. asserted that almost half of positive CTPA have been performed in the first 72 h of admission, indirectly suggesting that pulmonary thromboembolism may predate hospitalization [118]. Other authors described a higher pulmonary thromboembolism risk in the second or third week of symptomatic disease (11–20 days from onset) [119, 120]. In fact, Birk et al. demonstrated that PTD detection rate is low if CTPA is performed in all patients admitted to ER undergoing non-enhanced chest CT for COVID-related parenchymal evaluation; this finding may reflect a lower pulmonary thromboembolism prevalence in the early stages of the disease [121].
Pulmonary scintigraphy is the traditional nuclear scan test for the study of lung V/Q, but it cannot be performed in COVID-19 patients for the risk of airborne aerosol contamination and increased infectious risk for healthcare workers Nuclear medicine evaluation of PE is still important in case of contraindication or non-diagnostic CTPA: Perfusion SPECT/CT has been proposed as an alternative imaging test to rule out pulmonary thromboembolism without increased risk of contagious droplets [122, 123].
Conclusions
The predominant clinical involvement of acute SARS-CoV-2 infection manifests as acute lung injury of varying severity, but the cardiovascular system can also be significant involved. The aberrant immune response to the viral infection accounts for many pathogenic pathways leading to heart damage, as in the case of myocarditis or pulmonary thromboembolism. Cardiovascular involvement can be underrecognized, and heart and lung multimodality imaging plays a central role in different clinical settings and is essential for diagnosis, risk stratification, and management of patients with COVID-19.
References
Ghosh S, Deshwal H, Saeedan MB et al (2020) Imaging algorithm for COVID-19: a practical approach. Clin Imaging 72:22–30. https://doi.org/10.1016/j.clinimag.2020.11.022
WHO Coronavirus (COVID-19) Dashboard | WHO coronavirus (COVID-19) dashboard with vaccination data. https://covid19.who.int/. Accessed 10 May 2021
Shi S, Qin M, Shen B et al (2020) Association of cardiac injury with mortality in hospitalized patients with COVID-19 in Wuhan, China. JAMA Cardiol 5:802–810. https://doi.org/10.1001/jamacardio.2020.0950
Guo T, Fan Y, Chen M et al (2020) Cardiovascular implications of fatal outcomes of patients with coronavirus disease 2019 (COVID-19). JAMA Cardiol 5:811–818. https://doi.org/10.1001/jamacardio.2020.1017
Nishiga M, Wang DW, Han Y et al (2020) COVID-19 and cardiovascular disease: from basic mechanisms to clinical perspectives. Nat Rev Cardiol 17:543–558. https://doi.org/10.1038/s41569-020-0413-9
Long B, Brady WJ, Koyfman A, Gottlieb M (2020) Cardiovascular complications in COVID-19. Am J Emerg Med 38:1504–1507. https://doi.org/10.1016/j.ajem.2020.04.048
Stefanini GG, Chiarito M, Ferrante G et al (2020) Early detection of elevated cardiac biomarkers to optimise risk stratification in patients with COVID-19. Heart 106:1512–1518. https://doi.org/10.1136/heartjnl-2020-317322
Xu Z, Shi L, Wang Y et al (2020) Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir Med 8:420–422. https://doi.org/10.1016/S2213-2600(20)30076-X
Zheng YY, Ma YT, Zhang JY, Xie X (2020) COVID-19 and the cardiovascular system. Nat Rev Cardiol. https://doi.org/10.1038/s41569-020-0360-5
Xiong T-Y, Redwood S, Prendergast B, Chen M (2020) Coronaviruses and the cardiovascular system: acute and long-term implications. Eur Heart J 41:1798–1800. https://doi.org/10.1093/eurheartj/ehaa231
Clerkin KJ, Fried JA, Raikhelkar J et al (2020) COVID-19 and cardiovascular disease. Circulation 141:1648–1655. https://doi.org/10.1161/CIRCULATIONAHA.120.046941
Adiguzel Y (2021) Molecular mimicry between SARS-CoV-2 and human proteins. Autoimmun Rev 20:102791. https://doi.org/10.1016/j.autrev.2021.102791
Yazdanpanah F, Hamblin MR, Rezaei N (2020) The immune system and COVID-19: friend or foe? Life Sci 256:117900. https://doi.org/10.1016/j.lfs.2020.117900
Akhmerov A, Marbán E (2020) COVID-19 and the heart. Circ Res 126:1443–1455. https://doi.org/10.1161/CIRCRESAHA.120.317055
Comini L, Pasini E, Bachetti T et al (2005) Acute haemodynamic effects of IL-6 treatment in vivo: involvement of vagus nerve in NO-mediated negative inotropism. Cytokine 30:236–242. https://doi.org/10.1016/j.cyto.2005.01.009
Mann DL (2015) Innate immunity and the failing heart: the cytokine hypothesis revisited. Circ Res 116:1254–1268. https://doi.org/10.1161/CIRCRESAHA.116.302317
Magadum A, Kishore R (2020) Cardiovascular manifestations of COVID-19 infection. Cells. https://doi.org/10.3390/cells9112508
Unudurthi SD, Luthra P, Bose RJC et al (2020) Cardiac inflammation in COVID-19: lessons from heart failure. Life Sci 260:118482. https://doi.org/10.1016/j.lfs.2020.118482
Seta Y, Shan K, Bozkurt B et al (1996) Basic mechanisms in heart failure: the cytokine hypothesis. J Card Fail 2:243–249. https://doi.org/10.1016/s1071-9164(96)80047-9
Generali E, Folci M, Selmi C, Riboldi P (2017) Immune-mediated heart disease. Adv Exp Med Biol 1003:145–171. https://doi.org/10.1007/978-3-319-57613-8_8
Novelli L, Motta F, De Santis M et al (2021) The JANUS of chronic inflammatory and autoimmune diseases onset during COVID-19 - a systematic review of the literature. J Autoimmun 117:102592. https://doi.org/10.1016/j.jaut.2020.102592
Verdoni L, Mazza A, Gervasoni A et al (2020) An outbreak of severe Kawasaki-like disease at the Italian epicentre of the SARS-CoV-2 epidemic: an observational cohort study. Lancet 395:1771–1778. https://doi.org/10.1016/S0140-6736(20)31103-X
Consiglio CR, Cotugno N, Sardh F et al (2020) The immunology of multisystem inflammatory syndrome in children with COVID-19. Cell 183:968-981.e7. https://doi.org/10.1016/j.cell.2020.09.016
Gruber CN, Patel RS, Trachtman R et al (2020) Mapping systemic inflammation and antibody responses in multisystem inflammatory syndrome in children (MIS-C). Cell 183:982-995.e14. https://doi.org/10.1016/j.cell.2020.09.034
Belhadjer Z, Méot M, Bajolle F et al (2020) Acute heart failure in multisystem inflammatory syndrome in children in the context of global SARS-CoV-2 pandemic. Circulation 142:429–436. https://doi.org/10.1161/CIRCULATIONAHA.120.048360
Bajaj R, Sinclair HC, Patel K et al (2021) Delayed-onset myocarditis following COVID-19. Lancet Respir Med 9:e32–e34. https://doi.org/10.1016/S2213-2600(21)00085-0
Hoffmann M, Kleine-Weber H, Schroeder S et al (2020) SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181:271-280.e8. https://doi.org/10.1016/j.cell.2020.02.052
Siripanthong B, Nazarian S, Muser D et al (2020) Recognizing COVID-19-related myocarditis: the possible pathophysiology and proposed guideline for diagnosis and management. Heart Rhythm 17:1463–1471. https://doi.org/10.1016/j.hrthm.2020.05.001
Goulter AB, Goddard MJ, Allen JC, Clark KL (2004) ACE2 gene expression is up-regulated in the human failing heart. BMC Med 2:19. https://doi.org/10.1186/1741-7015-2-19
Basso C, Leone O, Rizzo S et al (2020) Pathological features of COVID-19-associated myocardial injury: a multicentre cardiovascular pathology study. Eur Heart J 41:3827–3835. https://doi.org/10.1093/eurheartj/ehaa664
Pietsch H, Escher F, Aleshcheva G et al (2021) Proof of SARS-CoV-2 genomes in endomyocardial biopsy with latency after acute infection. Int J Infect Dis 102:70–72. https://doi.org/10.1016/j.ijid.2020.10.012
Jacobs W, Lammens M, Kerckhofs A et al (2020) Fatal lymphocytic cardiac damage in coronavirus disease 2019 (COVID-19): autopsy reveals a ferroptosis signature. ESC Heart Fail. https://doi.org/10.1002/ehf2.12958
Gauchotte G, Venard V, Segondy M et al (2021) SARS-Cov-2 fulminant myocarditis: an autopsy and histopathological case study. Int J Legal Med 135:577–581. https://doi.org/10.1007/s00414-020-02500-z
Kawakami R, Sakamoto A, Kawai K et al (2021) Pathological evidence for SARS-CoV-2 as a cause of myocarditis: JACC review topic of the week. J Am Coll Cardiol 77:314–325. https://doi.org/10.1016/j.jacc.2020.11.031
Puntmann VO, Carerj ML, Wieters I et al (2020) Outcomes of cardiovascular magnetic resonance imaging in patients recently recovered from coronavirus disease 2019 (COVID-19). JAMA Cardiol 5:1265–1273. https://doi.org/10.1001/jamacardio.2020.3557
Huang L, Zhao P, Tang D et al (2020) Cardiac involvement in patients recovered from COVID-2019 identified using magnetic resonance imaging. JACC Cardiovasc Imaging 13:2330–2339. https://doi.org/10.1016/j.jcmg.2020.05.004
Engelmann B, Massberg S (2013) Thrombosis as an intravascular effector of innate immunity. Nat Rev Immunol 13:34–45. https://doi.org/10.1038/nri3345
Borges L, Pithon-Curi TC, Curi R, Hatanaka E (2020) COVID-19 and neutrophils: the relationship between hyperinflammation and neutrophil extracellular traps. Mediators Inflamm 2020:8829674. https://doi.org/10.1155/2020/8829674
Zuo Y, Yalavarthi S, Shi H et al (2020) Neutrophil extracellular traps in COVID-19. JCI Insight
Blasco A, Coronado M-J, Hernández-Terciado F et al (2020) Assessment of neutrophil extracellular traps in coronary thrombus of a case series of patients with COVID-19 and myocardial infarction. JAMA Cardiol. https://doi.org/10.1001/jamacardio.2020.7308
Abdel-Wahab N, Talathi S, Lopez-Olivo MA, Suarez-Almazor ME (2018) Risk of developing antiphospholipid antibodies following viral infection: a systematic review and meta-analysis. Lupus 27:572–583. https://doi.org/10.1177/0961203317731532
Xiao M, Zhang Y, Zhang S et al (2020) Antiphospholipid antibodies in critically ill patients with COVID-19. Arthritis Rheumatol 72:1998–2004. https://doi.org/10.1002/art.41425
Popovic B, Varlot J, Metzdorf PA et al (2021) Changes in characteristics and management among patients with ST-elevation myocardial infarction due to COVID-19 infection. Catheter Cardiovasc Interv 97:E319–E326. https://doi.org/10.1002/ccd.29114
Libby P, Loscalzo J, Ridker PM et al (2018) Inflammation, immunity, and infection in atherothrombosis: JACC review topic of the week. J Am Coll Cardiol 72:2071–2081. https://doi.org/10.1016/j.jacc.2018.08.1043
Tschöpe C, Sherif M, Anker MS et al (2021) COVID-19-convalescence phase unmasks a silent myocardial infarction due to coronary plaque rupture. ESC Heart Fail 8:971–973. https://doi.org/10.1002/ehf2.13186
Magro C, Mulvey JJ, Berlin D et al (2020) Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: a report of five cases. Transl Res 220:1–13. https://doi.org/10.1016/j.trsl.2020.04.007
Blagova O, Varionchik N, Zaidenov V et al (2021) Anti-heart antibodies levels and their correlation with clinical symptoms and outcomes in patients with confirmed or suspected diagnosis COVID-19. Eur J Immunol 51:893–902. https://doi.org/10.1002/eji.202048930
Lucchese G, Flöel A (2020) Molecular mimicry between SARS-CoV-2 and respiratory pacemaker neurons. Autoimmun Rev 19:102556. https://doi.org/10.1016/j.autrev.2020.102556
Angileri F, Legare S, Marino Gammazza A et al (2020) Molecular mimicry may explain multi-organ damage in COVID-19. Autoimmun Rev 19:102591. https://doi.org/10.1016/j.autrev.2020.102591
Gil-Cruz C, Perez-Shibayama C, De Martin A et al (2019) Microbiota-derived peptide mimics drive lethal inflammatory cardiomyopathy. Science 366:881–886. https://doi.org/10.1126/science.aav3487
Şelli ME, Thomas AC, Wraith DC, Newby AC (2017) A humanized HLA-DR4 mouse model for autoimmune myocarditis. J Mol Cell Cardiol 107:22–26. https://doi.org/10.1016/j.yjmcc.2017.04.003
Kaya Z, Katus HA, Rose NR (2010) Cardiac troponins and autoimmunity: their role in the pathogenesis of myocarditis and of heart failure. Clin Immunol 134:80–88. https://doi.org/10.1016/j.clim.2009.04.008
Revzin MV, Raza S, Srivastava NC et al (2020) Multisystem imaging manifestations of COVID-19, part 2: from cardiac complications to pediatric manifestations. Radiographics 40:1866–1892. https://doi.org/10.1148/rg.2020200195
Sandoval Y, Januzzi JL, Jaffe AS (2020) Cardiac troponin for assessment of myocardial injury in COVID-19: JACC review topic of the week. J Am Coll Cardiol 76:1244–1258. https://doi.org/10.1016/j.jacc.2020.06.068
Thygesen K, Alpert JS, Jaffe AS et al (2018) Fourth universal definition of myocardial infarction (2018). J Am Coll Cardiol 72:2231–2264. https://doi.org/10.1016/j.jacc.2018.08.1038
Capaccione KM, Leb JS, D’souza B et al (2020) Acute myocardial infarction secondary to COVID-19 infection: a case report and review of the literature. Clin Imaging 72:178–182. https://doi.org/10.1016/j.clinimag.2020.11.03
Citro R, Pontone G, Bellino M et al (2020) Role of multimodality imaging in evaluation of cardiovascular involvement in COVID-19. Trends Cardiovasc Med. https://doi.org/10.1016/j.tcm.2020.10.001
Galea N, Catapano F, Marchitelli L et al (2020) How to perform a cardio-thoracic magnetic resonance imaging in COVID-19: comprehensive assessment of heart, pulmonary arteries, and lung parenchyma. Eur Heart J Cardiovasc Imaging. https://doi.org/10.1093/ehjci/jeaa335
Galea N, Marchitelli L, Pambianchi G et al (2021) T2-mapping increase is the prevalent imaging biomarker of myocardial involvement in active COVID-19: a cardiovascular magnetic resonance study. J Cardiovasc Magn Reson 23:68. https://doi.org/10.1186/s12968-021-00764-x
Tavazzi G, Pellegrini C, Maurelli M et al (2020) Myocardial localization of coronavirus in COVID-19 cardiogenic shock. Eur J Heart Fail 22:911–915. https://doi.org/10.1002/ejhf.1828
Kelle S, Bucciarelli-Ducci C, Judd RM et al (2020) Society for Cardiovascular Magnetic Resonance (SCMR) recommended CMR protocols for scanning patients with active or convalescent phase COVID-19 infection. J Cardiovasc Magn Reson 22:61. https://doi.org/10.1186/s12968-020-00656-6
Kwee TC, Kwee RM (2020) Chest CT in COVID-19: what the radiologist needs to know. Radiographics 40:1848–1865. https://doi.org/10.1148/rg.2020200159
Pontone G, Baggiano A, Conte E et al (2020) “Quadruple rule-out” with computed tomography in a COVID-19 patient with equivocal acute coronary syndrome presentation. JACC Cardiovasc Imaging 13:1854–1856. https://doi.org/10.1016/j.jcmg.2020.04.012
Ferrante G, Fazzari F, Cozzi O et al (2020) Risk factors for myocardial injury and death in patients with COVID-19: insights from a cohort study with chest computed tomography. Cardiovasc Res 116:2239–2246. https://doi.org/10.1093/cvr/cvaa193
Nai Fovino L, Cademartiri F, Tarantini G (2020) Subclinical coronary artery disease in COVID-19 patients. Eur Heart J Cardiovasc Imaging 21:1055–1056. https://doi.org/10.1093/ehjci/jeaa202
Wu Q, Zhou L, Sun X et al (2017) Altered lipid metabolism in recovered SARS patients twelve years after infection. Sci Rep 7:9110. https://doi.org/10.1038/s41598-017-09536-z
Rudski L, Januzzi JL, Rigolin VH et al (2020) Multimodality imaging in evaluation of cardiovascular complications in patients with COVID-19: JACC Scientific Expert Panel. J Am Coll Cardiol 76:1345–1357. https://doi.org/10.1016/j.jacc.2020.06.080
Liguori C, Farina D, Vaccher F et al (2020) Myocarditis: imaging up to date. Radiol Med 125:1124–1134. https://doi.org/10.1007/s11547-020-01279-8
Murphy WJ, Longo DL (2021) A possible role for anti-idiotype antibodies in SARS-CoV-2 infection and vaccination. N Engl J Med. https://doi.org/10.1056/NEJMcibr2113694
Paque RE, Miller R (1991) Autoanti-idiotypes exhibit mimicry of myocyte antigens in virus-induced myocarditis. J Virol 65:16–22. https://doi.org/10.1128/JVI.65.1.16-22.1991
Ghugre NR, Orbach A, Biswas L et al (2021) Suspected subclinical myocarditis detected by cardiac magnetic resonance imaging late post COVID-19 recovery. J Cardiol Cases 24:203–205. https://doi.org/10.1016/j.jccase.2021.04.014
Salerno M, Kwong RY (2020) CMR in the era of COVID-19: evaluation of myocarditis in the subacute phase. JACC Cardiovasc Imaging 13:2340–2342. https://doi.org/10.1016/j.jcmg.2020.06.013
Musher DM, Abers MS, Corrales-Medina VF (2019) Acute infection and myocardial infarction. N Engl J Med 380:171–176. https://doi.org/10.1056/NEJMra1808137
Smilowitz NR, Kunichoff D, Garshick M et al (2021) C-reactive protein and clinical outcomes in patients with COVID-19. Eur Heart J 42:2270–2279. https://doi.org/10.1093/eurheartj/ehaa1103
Linde JJ, Kelbæk H, Hansen TF et al (2020) Coronary CT angiography in patients with non-ST-segment elevation acute coronary syndrome. J Am Coll Cardiol 75:453–463. https://doi.org/10.1016/j.jacc.2019.12.012
Stefanini GG, Montorfano M, Trabattoni D et al (2020) ST-elevation myocardial infarction in patients with COVID-19: clinical and angiographic outcomes. Circulation 141:2113–2116. https://doi.org/10.1161/CIRCULATIONAHA.120.047525
Pontone G, Scafuri S, Mancini ME et al (2020) Role of computed tomography in COVID-19. J Cardiovasc Comput Tomogr. https://doi.org/10.1016/j.jcct.2020.08.013
Singh V, Choi AD, Leipsic J et al (2020) Use of cardiac CT amidst the COVID-19 pandemic and beyond: North American perspective. J Cardiovasc Comput Tomogr. https://doi.org/10.1016/j.jcct.2020.11.004
Zoghbi WA, DiCarli MF, Blankstein R et al (2020) Multimodality cardiovascular imaging in the midst of the COVID-19 pandemic: ramping up safely to a new normal. JACC Cardiovasc Imaging 13:1615–1626. https://doi.org/10.1016/j.jcmg.2020.06.001
Motwani M, Kidambi A, Greenwood JP, Plein S (2014) Advances in cardiovascular magnetic resonance in ischaemic heart disease and non-ischaemic cardiomyopathies. Heart 100:1722–1733. https://doi.org/10.1136/heartjnl-2013-304680
Zeng J-H, Liu Y-X, Yuan J et al (2020) First case of COVID-19 complicated with fulminant myocarditis: a case report and insights. Infection 48:773–777. https://doi.org/10.1007/s15010-020-01424-5
Alhogbani T (2016) Acute myocarditis associated with novel Middle east respiratory syndrome coronavirus. Ann Saudi Med 36:78–80. https://doi.org/10.5144/0256-4947.2016.78
Inciardi RM, Lupi L, Zaccone G et al (2020) Cardiac involvement in a patient with coronavirus disease 2019 (COVID-19). JAMA Cardiol 5:819–824. https://doi.org/10.1001/jamacardio.2020.1096
Cuomo G, Menozzi M, Carli F et al (2020) Acute myocarditis as the main clinical manifestation of SARS-CoV 2 infection. Infect Dis Rep 12:8609. https://doi.org/10.4081/idr.2020.8609
Pirzada A, Mokhtar AT, Moeller AD (2020) COVID-19 and myocarditis: what do we know so far? CJC Open 2:278–285. https://doi.org/10.1016/j.cjco.2020.05.005
Han Y, Chen T, Bryant J et al (2020) Society for Cardiovascular Magnetic Resonance (SCMR) guidance for the practice of cardiovascular magnetic resonance during the COVID-19 pandemic. J Cardiovasc Magn Reson 22:26. https://doi.org/10.1186/s12968-020-00628-w
Friedrich MG, Sechtem U, Schulz-Menger J et al (2009) Cardiovascular magnetic resonance in myocarditis: a JACC White Paper. J Am Coll Cardiol 53:1475–1487. https://doi.org/10.1016/j.jacc.2009.02.007
Ferreira VM, Schulz-Menger J, Holmvang G et al (2018) Cardiovascular magnetic resonance in nonischemic myocardial inflammation: expert recommendations. J Am Coll Cardiol 72:3158–3176. https://doi.org/10.1016/j.jacc.2018.09.072
Cundari G, Galea N, De Rubeis G et al (2021) Use of the new Lake Louise Criteria improves CMR detection of atypical forms of acute myocarditis. Int J Cardiovasc Imaging 37:1395–1404. https://doi.org/10.1007/s10554-020-02097-9
Turkbey EB, Nacif MS, Guo M et al (2015) Prevalence and correlates of myocardial scar in a US cohort. JAMA 314:1945–1954. https://doi.org/10.1001/jama.2015.14849
Ng M-Y, Ferreira VM, Leung ST et al (2020) Patients recovered from COVID-19 show ongoing subclinical myocarditis as revealed by cardiac magnetic resonance imaging. JACC Cardiovasc Imaging 13:2476–2478. https://doi.org/10.1016/j.jcmg.2020.08.012
Knight DS, Kotecha T, Razvi Y et al (2020) COVID-19: myocardial injury in survivors. Circulation 142:1120–1122. https://doi.org/10.1161/CIRCULATIONAHA.120.049252
Lagan J, Schmitt M, Miller CA (2018) Clinical applications of multi-parametric CMR in myocarditis and systemic inflammatory diseases. Int J Cardiovasc Imaging 34:35–54. https://doi.org/10.1007/s10554-017-1063-9
Cruz Rodriguez JB, Lange RA, Mukherjee D (2020) Gamut of cardiac manifestations and complications of COVID-19: a contemporary review. J Investig Med 68:1334–1340. https://doi.org/10.1136/jim-2020-001592
Revzin MV, Raza S, Warshawsky R et al (2020) Multisystem imaging manifestations of COVID-19, part 1: viral pathogenesis and pulmonary and vascular system complications. Radiographics 40:1574–1599. https://doi.org/10.1148/rg.2020200149
Suh YJ, Hong H, Ohana M et al (2021) Pulmonary embolism and deep vein thrombosis in COVID-19: a systematic review and meta-analysis. Radiology 298:E70–E80. https://doi.org/10.1148/radiol.2020203557
Desborough MJR, Doyle AJ, Griffiths A et al (2020) Image-proven thromboembolism in patients with severe COVID-19 in a tertiary critical care unit in the United Kingdom. Thromb Res 193:1–4. https://doi.org/10.1016/j.thromres.2020.05.049
Contou D, Pajot O, Cally R et al (2020) Pulmonary embolism or thrombosis in ARDS COVID-19 patients: a French monocenter retrospective study. PLoS One 15:e0238413. https://doi.org/10.1371/journal.pone.0238413
Cattaneo M, Bertinato EM, Birocchi S et al (2020) Pulmonary embolism or pulmonary thrombosis in COVID-19? Is the recommendation to use high-dose heparin for thromboprophylaxis justified? Thromb Haemost 120:1230–1232. https://doi.org/10.1055/s-0040-1712097
Kermani-Alghoraishi M, Ghahramani R (2021) A review of venous thromboembolism phenomena in COVID-19 patients. Curr Probl Cardiol 46:100692. https://doi.org/10.1016/j.cpcardiol.2020.100692
Visseren FL, Bouwman JJ, Bouter KP et al (2000) Procoagulant activity of endothelial cells after infection with respiratory viruses. Thromb Haemost 84:319–324
Ciceri F, Beretta L, Scandroglio AM et al (2020) Microvascular COVID-19 lung vessels obstructive thromboinflammatory syndrome (MicroCLOTS): an atypical acute respiratory distress syndrome working hypothesis. Crit Care Resusc 22:95–97
Moore AJE, Wachsmann J, Chamarthy MR et al (2018) Imaging of acute pulmonary embolism: an update. Cardiovasc Diagn Ther 8:225–243. https://doi.org/10.21037/cdt.2017.12.01
van Dam LF, Kroft LJM, van der Wal LI et al (2020) Clinical and computed tomography characteristics of COVID-19 associated acute pulmonary embolism: a different phenotype of thrombotic disease? Thromb Res 193:86–89. https://doi.org/10.1016/j.thromres.2020.06.010
Mueller-Peltzer K, Krauss T, Benndorf M et al (2020) Pulmonary artery thrombi are co-located with opacifications in SARS-CoV2 induced ARDS. Respir Med 172:106135. https://doi.org/10.1016/j.rmed.2020.106135
Vernuccio F, Lombardo FP, Cannella R et al (2020) Thromboembolic complications of COVID-19: the combined effect of a pro-coagulant pattern and an endothelial thrombo-inflammatory syndrome. Clin Radiol 75:804–810. https://doi.org/10.1016/j.crad.2020.07.019
Meinel FG, Nance JW, Schoepf UJ et al (2015) Predictive value of computed tomography in acute pulmonary embolism: systematic review and meta-analysis. Am J Med 128:747–59.e2. https://doi.org/10.1016/j.amjmed.2015.01.023
Ierardi AM, Angileri SA, Arrichiello A et al (2020) Pulmonary embolism in COVID-19: ventilation and perfusion computed tomography. IDCases 21:e00805. https://doi.org/10.1016/j.idcr.2020.e00805
Sharma M, Surani S (2020) Revisiting one of the dreaded outcomes of the current pandemic: pulmonary embolism in COVID-19. Medicina (Kaunas). https://doi.org/10.3390/medicina56120670
Zhang L, Yan X, Fan Q et al (2020) D-dimer levels on admission to predict in-hospital mortality in patients with Covid-19. J Thromb Haemost 18:1324–1329. https://doi.org/10.1111/jth.14859
Obi AT, Barnes GD, Wakefield TW et al (2020) Practical diagnosis and treatment of suspected venous thromboembolism during COVID-19 pandemic. J Vasc Surg Venous Lymphat Disord 8:526–534. https://doi.org/10.1016/j.jvsv.2020.04.009
Mestre-Gómez B, Lorente-Ramos RM, Rogado J et al (2021) Incidence of pulmonary embolism in non-critically ill COVID-19 patients. Predicting factors for a challenging diagnosis. J Thromb Thrombolysis 51:40–46. https://doi.org/10.1007/s11239-020-02190-9
Ventura-Díaz S, Quintana-Pérez JV, Gil-Boronat A et al (2020) A higher D-dimer threshold for predicting pulmonary embolism in patients with COVID-19: a retrospective study. Emerg Radiol 27:679–689. https://doi.org/10.1007/s10140-020-01859-1
Benito N, Filella D, Mateo J et al (2020) Pulmonary thrombosis or embolism in a large cohort of hospitalized patients with Covid-19. Front Med (Lausanne) 7:557. https://doi.org/10.3389/fmed.2020.00557
Taccone FS, Gevenois PA, Peluso L et al (2020) Higher intensity thromboprophylaxis regimens and pulmonary embolism in critically ill coronavirus disease 2019 patients. Crit Care Med 48:e1087–e1090. https://doi.org/10.1097/CCM.0000000000004548
Oudkerk M, Büller HR, Kuijpers D et al (2020) Diagnosis, prevention, and treatment of thromboembolic complications in COVID-19: report of the National Institute for Public Health of the Netherlands. Radiology 297:E216–E222. https://doi.org/10.1148/radiol.2020201629
Revel M-P, Parkar AP, Prosch H et al (2020) COVID-19 patients and the radiology department - advice from the European Society of Radiology (ESR) and the European Society of Thoracic Imaging (ESTI). Eur Radiol 30:4903–4909. https://doi.org/10.1007/s00330-020-06865-y
Whyte MB, Kelly PA, Gonzalez E et al (2020) Pulmonary embolism in hospitalised patients with COVID-19. Thromb Res 195:95–99. https://doi.org/10.1016/j.thromres.2020.07.025
Fang C, Garzillo G, Batohi B et al (2020) Extent of pulmonary thromboembolic disease in patients with COVID-19 on CT: relationship with pulmonary parenchymal disease. Clin Radiol 75:780–788. https://doi.org/10.1016/j.crad.2020.07.002
Meiler S, Hamer OW, Schaible J et al (2020) Computed tomography characterization and outcome evaluation of COVID-19 pneumonia complicated by venous thromboembolism. PLoS One 15:e0242475. https://doi.org/10.1371/journal.pone.0242475
Birk R, Shaw D, Kennedy C et al (2020) Low detection rate of pulmonary embolism in patients presenting to the emergency department with suspected coronavirus disease 2019 (COVID-19): a single-centre UK study. Curr Probl Diagn Radiol. https://doi.org/10.1067/j.cpradiol.2020.09.014
Burger IA, Niemann T, Patriki D et al (2020) Is there a role for lung perfusion [99mTc]-MAA SPECT/CT to rule out pulmonary embolism in COVID-19 patients with contraindications for iodine contrast? Eur J Nucl Med Mol Imaging 47:2062–2063. https://doi.org/10.1007/s00259-020-04837-4
Lu Y, Macapinlac HA (2020) Perfusion SPECT/CT to diagnose pulmonary embolism during COVID-19 pandemic. Eur J Nucl Med Mol Imaging 47:2064–2065. https://doi.org/10.1007/s00259-020-04851-6
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethics Approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Conflict of Interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Laino, M.E., Ammirabile, A., Motta, F. et al. Advanced Imaging Supports the Mechanistic Role of Autoimmunity and Plaque Rupture in COVID-19 Heart Involvement. Clinic Rev Allerg Immunol 64, 75–89 (2023). https://doi.org/10.1007/s12016-022-08925-1
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12016-022-08925-1