
Abstract
Of huge importance now is to provide a fast, cost-effective, safe, and immediately available pharmaceutical solution to curb the rapid global spread of SARS-CoV-2. Recent publications on SARS-CoV-2 have brought attention to the possible benefit of chloroquine in the treatment of patients infected by SARS-CoV-2. Whether chloroquine can treat SARS-CoV-2 alone and also work as a prophylactic is doubtful. An effective prophylactic medication to prevent viral entry has to contain, at least, either a protease inhibitor or a competitive virus ACE2-binding inhibitor. Using bromhexine at a dosage that selectively inhibits TMPRSS2 and, in so doing, inhibits TMPRSS2-specific viral entry is likely to be effective against SARS-CoV-2. We propose the use of bromhexine as a prophylactic and treatment. We encourage the scientific community to assess bromhexine clinically as a prophylactic and curative treatment. If proven to be effective, this would allow a rapid, accessible, and cost-effective application worldwide.
Similar content being viewed by others


Background
“Pandemic is not a word to use lightly or carelessly.” stated Dr Tedros Adhanom Ghebreyesus, World Health Organization (WHO) Director-General, at a media briefing in March 2020 [1]. According to the WHO, as of April 18, 2020, coronavirus (CoV) disease 2019 (COVID-19) has caused more than 2,000,000 confirmed cases globally, and close to 150,000 deaths [2]. As the world witnesses the alarming levels of spread and severity of atypical pneumonia COVID-19, strategies to combat this outbreak are in dire need. The ripple effect of the COVID-19 outbreak is already starting to come to fruition, having devastating effects on human lives, globally impacting health systems and economies. The flourishing COVID-19 outbreak is reminiscent of epidemics that have caused panic in recent memory. These include SARS, MERS, the Zika virus, swine flu, and Ebola. This is the third documented coronavirus (CoV) transmission from animals to humans (zoonosis) in 2 decades. Previous outbreaks were severe acute respiratory syndrome coronavirus (SARS-CoV) [3, 4] and Middle-East respiratory syndrome coronavirus (MERS-CoV) [5]. We are now dealing with the novel CoV causing severe acute respiratory syndrome (SARS), originally termed 2019-nCoV, and now designated SARS-CoV-2 [6, 7].
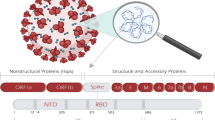
CoVs are spherical enveloped single-stranded positive-sense RNA viruses, characterized by club-like spikes that project from their surface giving them their solar corona or crown-like appearance (hence the name “Corona”). CoVs have an unusually large genome for RNA viruses, containing approximately 30 kilobases (kb). SARS-CoV-2, like the other CoVs, is also spherical and enveloped with a diameter of approximately 60–140 nm [8]. The SARS-CoV-2 virion genome is a single-stranded positive-sense RNA comprising 29,903 nucleotides, and encodes 12 putative, functional open-reading frames (ORFs) responsible for the synthesis of viral structural and nonstructural proteins [9, 10]. The genome encodes the structural [spike (S) protein, envelope (E) protein, membrane (M) protein, and nucleocapsid (N) phosphoprotein] and nonstructural [3-chymotrypsin-like protease (3CLpro), papain-like protease (PLpro), helicase (Hel), and RNA-dependent RNA polymerase (RdRp)] proteins as well as accessory proteins [11] (Fig. 1 below).

Schematic of the SARS-CoV-2 genome organization and virion. This CoV genome comprises a 5′-untranslated region (5′-UTR), open-reading frames (ORFs) 1a and 1b encoding nonstructural proteins 3-chymotrypsin-like protease (3CLpro), papain-like protease (PLpro), helicase (Hel), and RNA-dependent RNA polymerase (RdRp) as well as accessory proteins, and the structural S protein (S), E protein (E), M protein (M), and N phosphoprotein (N)
McLellan and colleagues [12] produced samples of the SARS-CoV-2 S protein and analyzed the structure via the imaging tool cryo-electron microscopy. They confirmed that the S protein on SARS-CoV-2 is similar to that of its close relative, SARS-CoV, with sequence identity of 76% for this protein between the two origins [12]. The SARS-CoV-2 S protein is known as a class I fusion protein. It is a large transmembrane protein of approximately 150 kDa and is highly glycosylated. S glycoproteins assemble into trimers on the virion surface (Fig. 1). The S protein consists of two subunits: the S1 subunit contains a signal peptide, followed by an N-terminal domain (NTD) and receptor-binding domain (RBD). The S1 subunit mediates attachment of the virion to host cell surface receptors and rests above the other subunit, the C-Terminal S2, containing conserved fusion peptide (FP), heptad repeat (HR) 1 and 2, transmembrane domain (TM), and cytoplasmic domain (CP), responsible for fusion of viral and cellular membranes [10] (Fig. 2). The S2 subunit mediates subsequent fusion between viral and host cellular membranes, facilitating entry into the host cell [13]. The SARS-CoV-2 virus appears to bind human cells more tightly than the SARS virus, which may explain why COVID-19 appears to spread more easily, mainly by respiratory transmission, from person to person.

SARS-CoV-2 S protein primary structure. The S protein consists of two subunits: The S1 subunit contains a signal peptide, followed by an N-terminal domain (NTD) and receptor-binding domain (RBD). The S1 domain is responsible for receptor binding and rests above the other subunit, the C-Terminal S2, containing conserved fusion peptide (FP), heptad repeat (HR) 1 and 2, transmembrane domain (TM), and cytoplasmic domain (CP), responsible for fusion of viral and cellular membranes [10]
An important question is why the lungs seem to be the most vulnerable target organ. One possible reason could be due to the large alveolar epithelial surface of the lungs being incredibly susceptible to inhaled noxious agents and pathogens such as viruses. Another reason could be that the lungs and especially the alveolar epithelium have a high density of cells expressing angiotensin converting enzyme 2 (ACE2). SARS-CoV-2 uses ACE2 as the receptor-binding domain for its S protein [14, 15]. Zhao and colleagues describe that approximately 85% of ACE2-expressing cells are epithelial type II alveolar cells (AECII, or AT2). This particularly suggests that these cells are a preference for viral invasion. In addition, genetic analysis indicates that ACE2-expressing AECII have a high level of multiple proviral genes, including regulatory genes for viral processes, viral life cycle, viral reproduction, and genome replication [16].
This infers that ACE2-expressing AECII cells favor the replication of the CoVs in the lungs and are, therefore, essentially responsible for pulmonary complications. The expression of the ACE2 receptor was also found in many extrapulmonary tissues including pharynx, heart, kidney, endothelium, and intestine [17, 18]. Theoretically, there could be viral invasion and replication in any of these tissues. However, currently, it appears that almost all of the fulminant trends of the virus are primarily pulmonary [16]. This organ seems to be the main replication and distribution center for the CoVs. A potential treatment should, therefore, first target the lungs to prevent a generalized infection or at least mitigate the symptoms. ACE2 is also strongly expressed and used on the lumen surface of intestinal epithelial cells as a co-receptor for the absorption of nutrients, especially for amino acids from ingested food [19]. It can be assumed that the intestine could also be an important entry point for SARS-CoV-2 and that the first infection with SARS-CoV-2 may have resulted from eating food from the seafood and wet animal wholesale market in Wuhan, the suspected site of the outbreak [7]. If SARS-CoV-2 does infect the human intestinal epithelium, it has important effects on the fecal–oral route of transmission, the containment of the virus spread, and treatment for it [20]. The distribution of ACE2 tissue in other organs other than the lungs could also explain expansion of dysfunction observed in patients.
Potential treatment strategies for COVID-19
The topic of clinical management of SARS-CoV-2 infections is too vast to be adequately covered in the current review. Consequently, we would like to focus our effort on lessons learned and practical considerations important to frontline clinical personnel. Although patients with COVID-19 pneumonia and respiratory distress share many clinical similarities with patients suffering from other types of severe viral pneumonia and often meet the Berlin definition of acute respiratory distress syndrome (ARDS) [21], accumulating clinical evidence suggests that there are important phenotypic differences in their presentation [22]. While most patients do not require immediate intubation on emergency department (ED) arrival, patients can decompensate quickly depending upon their viral load, comorbidities, and length of clinical illness among other factors. A systematic, escalating, step-wise approach to respiratory support is essential. A patient who arrives to the ED with hypoxia should immediately be placed on nasal cannula (NC) or facemask (FM) supplemental oxygen and their response monitored closely. Currently, the treatment is mainly symptomatic and supportive care, and for patients with severe infection oxygen therapy represents the major treatment intervention. Preventive strategies aimed at reducing transmission are focused on the isolation of patients and careful infection control.
Vaccine development against SARS-CoV-2 is in advanced stages of development in approximately 40 companies [23]. Disadvantages with cutting-edge vaccines are that they take months to years to develop and to approve, and they become obsolete if the virus evolves. Below, we summarize several approaches to targeting SARS-CoV-2, looking at the notable features of the virus and drug molecules with possible antiviral activity that have repurposing potential. Some of the drugs described have activity against other virus families and have already been used for treating other viral infections in a clinical setting, indicating potential broad-spectrum applications.
Interference with viral host cell entry
An S protein-based antiviral
As the CoV S protein is surface-exposed and mediates entry into host cells, it is the main target of neutralizing antibodies upon infection and the focus of therapeutic and vaccine design. The development of a vaccine based on the S1 subunit of SARS-CoV-2 appears to be a promising approach. Of note, in SARS-CoV-2, the S2 subunit, containing a fusion peptide, a transmembrane domain, and cytoplasmic domain, is highly conserved. Thus, it could be a target for antiviral (anti-S2) compounds. Currently, no SARS-CoV-2 S protein antiviral is available.
Inhibition of (serine protease) TMPRSS2 activity
CoVs depend on activation of their S proteins by host cell proteases [24]. Cleavage or priming of the S protein by TMPRSS2 is essential for the fusion of the virus with the ACE2 receptor needed for viral entry and spread of SARS-CoV-2 [25]. Hoffmann et al. [26] confirmed that SARS-CoV-2 enters the host cells mainly via binding and fusion with ACE2. Inhibition of TMPRSS2 activity is thus an excellent target for antiviral intervention. TMPRSS2 plays a crucial role in modulating the metastatic behavior of certain tumors, e.g., in prostate cancer and in modulating inflammation of organs, such as in the context of pancreatitis. Hoffman et al. [26] suggested that TMPRSS2 could be a potential therapeutic target for COVID-19, since entry of the virus into cells was reduced by camostat mesilate, a TMPRSS2 inhibitor. It is currently only approved for treatment of chronic pancreatitis in Japan [27]. Very little data are available on the drug’s risk profile with little or no experience with use in children, pregnant women, multimorbid patients, and other high-risk patients. It is a non-selective TMPRSS2 inhibitor, which means that it has greater, more severe side effects than a selective TMPRSS2. Camostat mesilate is currently undergoing Phase 1/2 trial testing in the USA for chronic pancreatitis [28]. If deemed safe, it could be a potential treatment option of CoV infections [29]. Nafamostat mesilate is also a serine protease inhibitor widely used as an anticoagulant and used to treat pancreatic and kidney disease. It is structurally closely related to camostat mesilate. Researchers previously identified nafamostat mesilate’s potential to inhibit MERS-CoV [30]. Risks of complications (including anaphylaxis) must be considered in non-emergency care.
TMPRSS2 is expressed highly in localized high-grade prostate cancers and in the majority of human prostate cancer metastases. Lucas et al. [31] showed a decrease in the frequency of metastases and a slowdown of the spread of metastases in mice with prostate cancer using TMPRSS2 inhibitors. In particular, they identified bromhexine, an FDA-approved ingredient [31] in mucolytic cough suppressants, as a potential TMPRSS2 inhibitor for their application. Bromhexine is orally readily bioavailable. Endonasal application is also a good alternative option. Bromhexine is an over-the-counter (OTC) drug [32] that is affordable with proven safety. Typically bromide compounds, especially aromatic bromide compounds, show a relatively high binding affinity for serine-containing peptide sequences, proteins, and enzymes [31, 33]. Lucas et al. [31] show that this effect is due to a selective inhibition of TMPRSS2 by bromhexine.
TMPRSS2 is known to harbor an androgen-responsive enhancer and is androgen-regulated, and thus, an alternative strategy, other than selectively inhibiting TMPRSS2, would be to modulate the expression of TMPRSS2 using androgen receptor-targeted therapies. Mikkonen et al. [34] administered androgen (testosterone) to lung adenocarcinoma-derived A549 cells and witnessed approximately a twofold upregulation of the TMPRSS2 transcript, as well as androgen-dependent loading of androgen receptor onto the TMPRSS2 androgen-responsive enhancer. It is likely that androgen antagonists (antiandrogens) can, thus, downregulate TMPRSS2 expression and thereby reduce the severity of COVID-19. Commercially available antiandrogens include, but are not limited to bicalutamide, enzalutamide, apalutamide, flutamide, nilutamide, or darolutamide [35].
Interference with viral fusion and uptake
Viral fusion and release of the genetic components are highly dependent on the endosomal pathway and particularly pH. Chloroquine (N4-(7-chloro-4-quinolinyl)-N1,N1-diethyl-1,4-pentanediamine), an FDA-approved drug, [36] has been used to treat malaria for many years, [37] as well as autoimmune diseases. Chloroquine is known to block virus infection by increasing endosomal pH required for virus/endosome fusion [38] and release of viral DNA or RNA into the cytosol. Moreover, chloroquine has immunomodulatory effects, suppressing the production/release of TNF-α, IL-1β, and IL-6 [39], which may synergistically enhance its antiviral effect in vivo. It also works as a novel class of autophagy inhibitor, [40] further interfering with viral infection and replication. A combination of remdesivir and chloroquine was proven to effectively inhibit the recently emerged SARS-CoV-2 in vitro [41]. Chloroquine and hydroxychloroquine (HCQ) have been found to be efficient against SARS-CoV-2, and reported to be efficient in Chinese COVID-19 patients [42]. Chloroquine is easily available and relatively low in cost. Therefore, it is potentially clinically applicable against COVID-19. However, an overdose of chloroquine can cause acute poisoning and death [43]. HCQ sulfate, a derivative of chloroquine, demonstrated much less (~ 40%) toxicity than chloroquine in animals [44] and is a good alternative. As of March 2020, seven clinical trial registries were found using HCQ sulfate in the Chinese Clinical Trial Registry (https://www.chictr.org.cn) and three were found using HCQ sulfate in ClinicalTrials.gov (https://clinicaltrials.gov/) to treat COVID-19.
Blocking or changing of the ACE2 receptor
The interaction sites between ACE2 and SARS-CoV-2 are interesting therapeutic targets with the aim of blocking or changing the ACE2 receptor. It is important to note that ACE2 has an imperative lung protective function [45] and is, therefore, of limited use as a target as it has to maintain its physiological function. ACE2 inhibitors will go hand-in-hand with partial loss of this lung protection. Chloroquine and the less toxic HCQ are of particular importance here, as it interferes with the glycosylation of ACE2 which is considered one of the cellular receptors of SARS-CoV [46].
Delivery of soluble ACE2
Kuba et al. [47] showed that in mice, SARS-CoV down-regulates the ACE2 protein by binding its S protein, and that this, consequently, can lead to serious lung injury. This also suggests that excessive ACE2 may competitively bind SARS-CoV2. In so doing, it not only neutralizes the virus, but also saves the cellular ACE2 activity that regulates the renin angiotensin system (RAS) [47], additionally protecting the lungs from injuries. Therefore, treatment with a soluble form of ACE2 could slow down and/or even prevent virus entry into cells and reduce virus spread, and also protect the lungs from injury. A recombinant human ACE2 (rhACE2; APN01, GSK2586881) is considered safe and healthy subjects showed no negative hemodynamic effects. This is likely to differ in multimorbids or weakened patients.
Inhibition of cathepsin B/L
Most coronaviruses enter their target cells via plasma membrane fusion, a further entry mechanism is the endosomal acidic pH-dependent endocytosis [48]. Once the virus is internalized, only then does the fusion of virus with lysosomes depend on a low endosomal and lysosomal pH [49]. Following this, the S glycoprotein is cleaved into S1 and S2 subunits by endosomal proteases cathepsin B/L, with the resulting S2 subunit-mediating membrane fusion [50]. To date, numerous protein inhibitors of cathepsin B/L have been described, some of which are of endogenous origin and led to the design and development of several specific peptide- and peptidomimetic inhibitor drugs. One such drug, named E-64d, an epoxysuccinyl cathepsin inhibitor blocked SARS-CoV-2-driven entry into the TMPRSS2- (knockout) cell lines 293T and Vero [26]. However, even if E-64d and analogs are therapeutically effective from a drug perspective, they are not widely accepted or approved. In addition, some exogenous protein inhibitors of cathepsin B/L have been isolated from various natural sources. Amongst such inhibitors are aziridines like miraziridine and aldehydes like tokaramide but also other non-peptidic natural compounds such as flavonoids. Quercetin is a strong cathepsin inhibitor with an IC50 in the low micromolar range [51]. Quercetin is a common and readily available supplement and, therefore, must be considered as an adjuvant to prevent or to weaken the course of such viral infections. Cathepsin B/L activity is inhibited by an elevated endosomal pH and, thus, chloroquine treatment will also work [52].
Interference with viral replication
Interference with RNA-dependent RNA polymerase
Remdesivir (GS-5734) is a 1′-cyano-substituted adenosine nucleotide analog prodrug (RNA-dependent RNA polymerase inhibitor) and shows broad-spectrum antiviral activity against several RNA viruses. According to data collected from in vitro cell lines and mouse model experiments, it could interfere with the nsp12 polymerase even in the setting of intact nsp14 exoribonuclease (ExoN) proofreading activity [53]. It has been reported to improve the clinical condition of the first US case of COVID-19 [54]. A phase III clinical trial of remdesivir against SARS-CoV-2 was launched in Wuhan on February 4, 2020. However, it is still an experimental drug not expected to be largely available for treating a very large number of patients in a timely manner. It has to be requested for each individual patient from the manufacturer.
Inhibition of (cysteine proteases) 3CL-pro and PL-pro activity
The CoV genome encodes more than 20 proteins [55] amongst these are two proteases, papain-like protease (PLpro) and 3-chymotrypsin-like protease (3CLpro), also known as the main protease (Mpro), that are important for virus replication [56, 57]. They cleave two translated polyproteins (PP1A and PP1AB) into individual functional components. These proteases are considered promising drug targets, particularly 3CLpro, as all residues involved in the catalysis, substrate binding, and dimerization are 100% conserved between SARS-CoV and SARS-CoV-2 [58].
The HIV protease inhibitors lopinavir/ritonavir inhibit the major CoV protease 3CLpro. A patient in Korea that contracted SARS-CoV-2 was successfully treated with this antiviral combination [59]. However, this result could not be confirmed in a recent clinical trial as a treatment in adults with SARS-CoV-2. Their results demonstrated no difference from standard care with respect to the time to clinical improvement [60]. A recent drug molecules’ docking study of the 3CLpro pocket indicates that chloroquine and bromhexine have high binding affinity to 3CLpro [61]. Interestingly, quercetin also has a strong binding affinity for 3CLpro, greater than both bromhexine and chloroquine, and thus, all three drugs are inhibitors of 3CLpro. This is an added beneficial mechanism of action for these drug molecules. PLpros, in general, are not as well characterized as 3CLpros and have not generated as much interest as pharmaceutical targets.
Is there an ideal drug for a pandemic like COVID-19?
A prophylaxis strategy and a suitable treatment for the emerging SARS-CoV-2 are crucial for reducing the mortality and morbidity of this disease, but developing and obtaining regulatory approval for new drugs can take years and are discordant with the urgent need for a therapy. Drug repurposing is an attractive alternative drug discovery strategy, because it eliminates many steps usually required at the early phase of drug development. There is the advantage of ease of access, decreased cost of development as they have established manufacturing arrangements, and the possibility to provide a wide array of options for combination studies. The background pharmacological knowledge available for such compounds may also reduce concerns regarding adverse effects in patients as they have gone through rigorous safety and risk testing and are already approved as safe for human use.
It is feasible to address this problem from a theoretical perspective. Theoretically, the ideal drug candidate (or combination) has to be specific, effective, already has regulatory approval (for any indication) ideally globally, but at a minimum in multiple countries. It should be widely available, inexpensive, and most important, has a long history of safe use with minimal side effects. Such a drug candidate could be applied immediately off-label and in so doing break the spreading chain or at least decrease the speed of spreading of the disease. Such a drug candidate, if even only partially effective, could have a huge epidemiological impact. Looking at a global perspective, it could mean the difference between a moderate and severe course of the disease, and/or between life and death. A reduction in the replication of the virus and of the total initial viral load in the host cells, especially in young and healthy individuals, could prevent the critical threshold of becoming infected or transmitting the virus from being reached.
Bromhexine or bromhexine hydrochloride (N-cyclo-N-methyl-(2-amino-3,5-dibromo-benzyl) amine hydrochloride) is derived from Vasicin, a plant-derived ingredient and alkaloid that was developed from the Indian lung herb, Adhatoda vasica, and is a brominated aniline and benzylamine derivative [62]. Bromhexine hydrochloride acts as a mucolytic (breaks down mucus and helps clear chest congestion) and is approved in many countries as an OTC drug. It is characterized by low side effects and a low purchase price. Bromhexine is structurally related to ambroxol, an active demethylated metabolite of bromhexine, that is also a known medicine in the market. The available data suggest further that ambroxol is a potent inducer of surfactant synthesis in AT2 cells [63,64,65]. Its lung protective properties have been discussed in infants and severely ill adult patients as well as the potential as an adjuvant in anti-infective therapy [64]. Thus, bromhexine also provides indirect protective effects. Bromhexine hydrochloride is sold under a few brand names, such as, Bisolvon®, Broncholyte Elixir, Paxirasol, and Bisolex amongst others. According to the package inserts (information for use), the medicinal products containing bromhexine hydrochloride are usually administered three times a day (8–16 mg per dose for adults). It shows a quick and almost complete absorption in the intestine. Lung-tissue concentrations 2 h post dose are 1.5–4.5 times higher in bronchiolo-bronchial tissues and between 2.4 and 5.9 times higher in pulmonary parenchyma compared to plasma concentrations. Unchanged bromhexine is bound to plasma proteins by 95% [66, 67].
Lucas et al. [31], as mentioned above, showed a decrease in the frequency of metastases and a slowdown of the spread of metastases in mice with prostate cancer using TMPRSS2 inhibitors. The extent of metastasis reduction with bromhexine was slightly lower in the wild-type mice than in the genetic TMPRSS2− mice. This is most likely due to an incomplete pharmacological blockage of the protease activity by the applied bromhexine, presumably due to the dosage and the long dosage intervals in the study (72 h) [31]. The terminal half-life of bromhexine in an oral application of 8 mg per single dose is an average of 6.6 h [67]. Bromhexine is orally readily bioavailable, and thus, a more frequent and higher oral dose could have a stronger and longer term inactivation of the TMPRSS2 enzyme. Endonasal, sublingual, buccal application, or inhalation might also be good alternatives as this could circumvent the first pass effect. Laporte and Naesens [68] reported that bromhexine did not show any significant cell entry or replication inhibition effect in vitro in influenza viruses. However, the authors showed that influenza viruses utilize, contrary to SARS-CoV-2, a different extracellular host protease for priming. Thus, these results are not representative for SARS-CoV-2 [68].
In theory, bromhexine is an attractive drug against COVID-19, because it is an OTC drug that is available globally, affordable, with proven safety, and in so doing fulfills all criteria hypothesized above for a global and immediate off-label use. The IC90 (inhibitory concentration) for TMPRSS2 inhibition using bromhexine is favorable at 1 μmol compared to 10 μmol for camostat mesilate. However, the first pass effect for bromhexine is much higher than for camostat mesilate (75–80% versus 33–40%). The currently recommended oral dose for camostat mesilate is 300–600 mg per day which would translate into an oral dose of about 68–136 mg and a parenteral dose of 17–32 mg per day. These are only estimations and dose finding has to be done to identify the ideal dose.
A clinical trial (registration number NCT04273763), carried out by WEPON Pharmaceutical Group Co. Ltd., is the first human body-based preliminary exploratory randomized-controlled clinical study on treating COVID-19 with bromhexine hydrochloride tablets (BHT). The clinical study evaluated the efficacy and safety of oral intake of 96 mg (32 mg tid) BHT combined with standard treatment (experimental group) compared to standard treatment alone (control group) in suspected patients and confirmed patients with mild or common COVID-19. The study results showed the signs of efficacy from multiple angles using BHT (WEPON Pharmaceutical Group Co Ltd. communication. Appendix 3. Exploratory Study on First Use of BHT for Prevention and Cure of COVID-19. 2020). Treatment with BHT alleviated lung injury to a certain extent, as we have proposed, and no severe adverse effects were experienced. The experimental group compared to the control group required less oxygen inhalation days (2 days on average for the experimental group and 4 days on average for the control group), had lower proportion of patients requiring oxygen inhalation (16.67% for the experimental group and 33.33% for the control group), and had smaller incidence of adverse events of liver injury which is expected (25% for the experimental group and 66.67% for the control group). Bromhexine and ambroxol appear to have a liver protective effect. Comparatively, liver toxicity when given chloroquine/HCQ and liver damage in COVID-19 is a high risk. The exploratory results of the study support our proposal that bromhexine hydrochloride may have a good effect on the treatment of COVID-19.
Recent publications on COVID-19 have brought attention to the possible benefit of chloroquine in the treatment of patients infected by SARS-CoV-2. Whether chloroquine can treat COVID-19 alone and also work as a prophylactic is doubtful. This needs to be further investigated before masses of people start to take this relatively toxic drug as a preventive measure. People have been poisoned and one death has occurred due to overdose using chloroquine [69]. The FDA does say that studies are under way to see if chloroquine can be effective in the treatment of COVID-19. Chloroquine had been approved for “compassionate use” where patients are in a life-threatening condition. Past research on chloroquine has shown in vitro activity against many different viruses, but no benefit in animal models [70]. Chloroquine in almost all animal models of different viral infections only partially worked or did not work [71,72,73]. Treatment with chloroquine did not prevent influenza infection in a randomized, double-blind, placebo-controlled clinical trial [74]. Conversely, it worked very well in vitro [75,76,77]. This could indicate that the main mechanism of action of chloroquine, in vivo, is via interference with the non-specific endosomal pathway and not with the ACE2 receptor directly.
The extracellular concentration of the orally applied chloroquine, especially in lung tissue, in vivo, may not be high enough to inhibit virus binding via the discussed glycosylation of the binding pocket [38]. After the viral infection has spread in the body and due to the incredibly high viral loads, the non-specific endocytotic pathway is mainly used for further virus replication. This may explain the recent success reported with chloroquine to assist in the curing of the virus. In already infected individuals, we believe that it is essential to combine HCQ with a TMPRSS2 inhibitor, like bromhexine, to block complete entry of the virus into host cells. In the case of prophylaxis, the inhibition of the TMPRSS2 is essential [26] and the non-specific endosomal entry is negligible. An effective prophylactic medication to prevent viral entry has to contain, at least, either a TMPRSS2 inhibitor, e.g., bromhexine or a competitive virus ACE2-binding inhibitor, e.g., a peptide inhibitor. This will prevent further spreading of the virus through the host’s body. Furthermore, a combination with the lesser toxic chloroquine derivate, HCQ sulfate, that is (amongst other functions) an effective endosomal protease inhibitor, inhibiting cathepsin B/L, could be a favorable combination for the treatment of moderate-to-severe COVID-19 cases. The addition of the 3CLpro inhibitor, quercetin, is also a favorable addition. This combination would block virus-host cell entry completely by blocking the specific receptor-mediated entry (via bromhexine) and non-specific endocytotic virus entry (via HCQ sulfate and quercetin) as well as viral replication (quercetin).
The recommended dose of HCQ sulfate for prophylaxis is 400 mg per week and for a curative treatment a loading dose of 800 mg (twice daily 400 mg) for the first day and 400 mg (twice daily 200 mg) for the following 4 days [78]. The toxic dosage range of chloroquine and HCQ is close to the therapeutic range [79]. Especially, since chloroquine derivatives are quite toxic, a combination with bromhexine and a lower dose of HCQ could be applicable. A combination of airway protease inhibitors with other antiviral drugs is known to obtain a synergistic effect or reduce the risk of resistance. An example shows that a combination of oseltamivir with the serine protease inhibitor BAPA (benzylsulfonyl-d-Arg-Pro-4-amidino-benzylamide) is able to suppress influenza virus replication in human airway epithelial cells at remarkably lower concentrations compared to a treatment with each inhibitor alone [80]. One can deduce that the same could be applicable for the herein proposed drug application. Bromhexine would be a valuable addition in combination with antivirals such as remdesivir. The beneficial role of flavonoid supplements like quercetin to contribute to an inhibition of the viral entry and replication must also be considered as additional support to current and also our proposed treatment scheme [51] (Fig. 3)

Adapted from Simmons et al. [24])
Host–virus interaction: how we can exploit these mechanisms to treat SARS-CoV-2 using bromhexine and/or hydroxychloroquine (HCQ) and/or quercetin. SARS-CoV-2 employs two routes for host cell entry, which are dependent on the localization of the proteases required for activation of the S protein. Binding of SARS-CoV-2 to the cellular receptor, ACE2, can result in uptake of virions into endosomes, where the S protein is activated by the pH-dependent cysteine protease cathepsin B/L. Activation of the S protein by cathepsin B/L can be blocked by HCQ and quercetin. Alternatively, the S protein can be activated by TMPRSS2, resulting in fusion of the viral membrane with the plasma membrane. Activation of the S protein by TMPRSS2 can be blocked by bromhexine. Quercetin also blocks viral replication via inhibition of the viral cysteine protease 3CLpro. (
Summary and perspectives
A rationale was put forward for the repurposing of existing drugs namely bromhexine in combination with HCQ and/or quercetin as an immediately available and affordable treatment option or prophylactic use in response to the COVID-19 pandemic. It appears the fight against COVID-19 is just beginning. Globally, the economic cost of the pandemic has been estimated at $1 trillion in 2020 (UN’s trade and development agency, UNCTAD). There is, at the time of this writing, still no sign of the COVID-19 pandemic slowing down, and with over 2,000,000 recorded infections, it is time to take unprecedented global actions [2]. It is still in the early stages of the outbreak and we have limited knowledge about the transmission and mutation rate of this virus. As the virus continues to spread to more individuals, more mutations may arise which can potentially make the virus even more virulent and difficult to eradicate. As specific functions of the viral proteins and the pathogenicity of the virus become clearer and the sharing of information on variants and clinical information becomes more accessible, we can plan that therapeutic treatments are near in our future.
Neutralizing antibodies and vaccines will play significant roles in controlling this SARS‐CoV‐2 outbreak, but will realistically only become available once it is too late. A fast and immediate prophylactic option to prevent or potent drug to treat the disease progression in these urgent times is the repurposing of an already existing approved drug to be used for off-label usage. Bromhexine or preferably in combination with HCQ could be such an ideal candidate or candidate combination. A further combination with quercetin could be beneficial, as well. First, the combination could be utilized for prophylactic applications, specifically for the more vulnerable individuals such as the elderly, diabetics, those with coronary diseases, and immune-compromised individuals. Here, quercetin could possibly replace HCQ. Second, this combination can be used to lower the viral burden in newly infected individuals, limiting their capacity to transfer the virus and in so doing also prevent them from developing a severe or even critical manifestation of the disease. We encourage the scientific community to test bromhexine and the suggested combinations and to follow our recommended approach to also identify further ideal repurposing candidates according to the herein proposed criteria.
Availability of data and material
Supporting data are referenced and accessible.
References
World Health Organization (2020) Coronavirus disease (COVID-2019) press briefings. https://www.who.int/emergencies/diseases/novel-coronavirus-2019/media-resources/press-briefings. Accessed 22 Mar 2020
World Health Organization (2020) Coronavirus Disease (COVID-2019) Situation Reports 1–89. https://www.who.int/emergencies/diseases/novel-coronavirus-2019/situation-reports. Accessed 18 Apr 2020
Drosten C, Gunther S, Preiser W, van der Werf S, Brodt HR, Becker S et al (2003) Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N Engl J Med 348(20):1967–1976. https://doi.org/10.1056/NEJMoa030747(Epub 2003/04/12, PubMed PMID: 12690091)
Ksiazek TG, Erdman D, Goldsmith CS, Zaki SR, Peret T, Emery S et al (2003) A novel coronavirus associated with severe acute respiratory syndrome. N Engl J Med 348(20):1953–1966. https://doi.org/10.1056/NEJMoa030781(Epub 2003/04/12, PubMed PMID: 12690092)
Zaki AM, van Boheemen S, Bestebroer TM, Osterhaus AD, Fouchier RA (2012) Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N Engl J Med 367(19):1814–1820. https://doi.org/10.1056/NEJMoa1211721(Epub 2012/10/19, PubMed PMID: 2307514)
Gorbalenya AE, Baker SC, Baric RS, de Groot RJ, Drosten C, Gulyaeva AA et al (2020) The species severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nat Microbiol. https://doi.org/10.1038/s41564-020-0695-z
Zhu N, Zhang D, Wang W, Li X, Yang B, Song J et al (2020) A Novel Coronavirus from patients with pneumonia in China, 2019. N Engl J Med 382(8):727–733. https://doi.org/10.1056/NEJMoa2001017(Epub 2020/01/25, PubMed PMID: 31978945)
Cascella M, Rajnik M, Cuomo A, Dulebohn SC, Di Napoli R (2020) Features, evaluation and treatment coronavirus (COVID-19). StatPearls Publishing, St. Petersburg
Wu F, Zhao S, Yu B, Chen YM, Wang W, Song ZG et al (2020) A new coronavirus associated with human respiratory disease in China. Nature 579(7798):265–269. https://doi.org/10.1038/s41586-020-2008-3(Epub 2020/02/06, PubMed PMID: 32015508)
Chan JF, Kok KH, Zhu Z, Chu H, To KK, Yuan S et al (2020) Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg Microbes Infect 9(1):221–236. https://doi.org/10.1080/22221751.2020.1719902(Epub 2020/01/29, PubMed PMID: 31987001)
Li G, De Clercq E (2020) Therapeutic options for the 2019 novel coronavirus (2019-nCoV). Nat Rev Drug Discov 19(3):149–150. https://doi.org/10.1038/d41573-020-00016-0(Epub 2020/03/05, PubMed PMID: 32127666)
Wrapp D, Wang N, Corbett KS, Goldsmith JA, Hsieh CL, Abiona O et al (2020) Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science (New York, NY) 367(6483):1260–1263. https://doi.org/10.1126/science.abb2507(Epub 2020/02/23, PubMed PMID: 32075877)
Kirchdoerfer RN, Cottrell CA, Wang N, Pallesen J, Yassine HM, Turner HL et al (2016) Pre-fusion structure of a human coronavirus spike protein. Nature 531(7592):118–121. https://doi.org/10.1038/nature17200(Epub 2016/03/05, PubMed PMID: 26935699; PubMed Central PMCID: PMCPMC4860016)
Lu R, Zhao X, Li J, Niu P, Yang B, Wu H et al (2020) Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet (London, England) 395(10224):565–574. https://doi.org/10.1016/S0140-6736(20)30251-8(PubMed PMID: 32007145)
Wan Y, Shang J, Graham R, Baric RS, Li F (2020) Receptor recognition by novel coronavirus from Wuhan: an analysis based on decade-long structural studies of SARS. J Virol. https://doi.org/10.1128/jvi.00127-20(Epub 2020/01/31, PubMed PMID: 31996437)
Zhao Y, Zhao Z, Wang Y, Zhou Y, Ma Y, Zuo W (2020) Single-cell RNA expression profiling of ACE2, the putative receptor of Wuhan 2019-nCov. BioRxiv. https://doi.org/10.1101/2020.01.26.919985
Crackower MA, Sarao R, Oudit GY, Yagil C, Kozieradzki I, Scanga SE et al (2002) Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature 417(6891):822–828. https://doi.org/10.1038/nature00786
Gu J, Gong E, Zhang B, Zheng J, Gao Z, Zhong Y et al (2005) Multiple organ infection and the pathogenesis of SARS. J Exp Med 202(3):415–424. https://doi.org/10.1084/jem.20050828(Epub 2005/07/27)
Hashimoto T, Perlot T, Rehman A, Trichereau J, Ishiguro H, Paolino M et al (2012) ACE2 links amino acid malnutrition to microbial ecology and intestinal inflammation. Nature 487(7408):477–481. https://doi.org/10.1038/nature11228(Epub 2012/07/28)
Yeo C, Kaushal S, Yeo D (2020) Enteric involvement of coronaviruses: is faecal–oral transmission of SARS-CoV-2 possible? Lancet Gastroenterol Hepatol 5(4):335–337. https://doi.org/10.1016/S2468-1253(20)30048-0
Ranieri VM, Rubenfeld GD, Thompson BT, Ferguson ND, Caldwell E, Fan E et al (2012) Acute respiratory distress syndrome: the Berlin Definition. JAMA 307(23):2526–2533. https://doi.org/10.1001/jama.2012.5669(Epub 2012/07/17)
Gattinoni L, Chiumello D, Caironi P, Busana M, Romitti F, Brazzi L et al (2020) COVID-19 pneumonia: different respiratory treatment for different phenotypes? Intensive Care Med. https://doi.org/10.1007/s00134-020-06033-2
Craven J (2020) Covid-19 vaccine tracker. Regulatory affairs professionals society. https://www.raps.org/news-and-articles/news-articles/2020/3/covid-19-vaccine-tracker. Accessed 30 Mar 2020
Simmons G, Zmora P, Gierer S, Heurich A, Pöhlmann S (2013) Proteolytic activation of the SARS-coronavirus spike protein: cutting enzymes at the cutting edge of antiviral research. Antiviral Res 100(3):605–614. https://doi.org/10.1016/j.antiviral.2013.09.028
Belouzard S, Millet JK, Licitra BN, Whittaker GR (2012) Mechanisms of coronavirus cell entry mediated by the viral spike protein. Viruses 4(6):1011–1033. https://doi.org/10.3390/v4061011(Epub 2012/07/21)
Hoffmann M, Kleine-Weber H, Schroeder S, Kruger N, Herrler T, Erichsen S et al (2020) SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. https://doi.org/10.1016/j.cell.2020.02.052(Epub 2020/03/07)
Drugs. Camostat. 2020. https://www.drugs.com/international/camostat.html. Accessed 22 Mar 2020
Ramsey ML, Nuttall J, Hart PA, on behalf of the TIT (2019) A phase 1/2 trial to evaluate the pharmacokinetics, safety, and efficacy of NI-03 in patients with chronic pancreatitis: study protocol for a randomized controlled trial on the assessment of camostat treatment in chronic pancreatitis (TACTIC). Trials 20(1):501. https://doi.org/10.1186/s13063-019-3606-y
Zhou Y, Vedantham P, Lu K, Agudelo J, Carrion R, Nunneley JW et al (2015) Protease inhibitors targeting coronavirus and filovirus entry. Antiviral Res 116:76–84. https://doi.org/10.1016/j.antiviral.2015.01.011
Yamamoto M, Matsuyama S, Li X, Takeda M, Kawaguchi Y, Inoue J-I et al (2016) Identification of nafamostat as a potent inhibitor of middle east respiratory syndrome coronavirus S protein-mediated membrane fusion using the split-protein-based cell-cell fusion assay. Antimicrob Agents Chemother 60(11):6532–6539. https://doi.org/10.1128/AAC.01043-16(PubMed PMID: 27550352)
Lucas JM, Heinlein C, Kim T, Hernandez SA, Malik MS, True LD et al (2014) The androgen-regulated protease TMPRSS2 activates a proteolytic cascade involving components of the tumor microenvironment and promotes prostate cancer metastasis. Cancer Discov 4(11):1310–1325. https://doi.org/10.1158/2159-8290.cd-13-1010(Epub 2014/08/15)
Chang CC, Cheng AC, Chang AB (2007) Over-the-counter (OTC) medications to reduce cough as an adjunct to antibiotics for acute pneumonia in children and adults. Cochrane Database Syst Rev 4:Cd006088. https://doi.org/10.1002/14651858.CD006088.pub2(Epub 2007/10/19)
Danelius E, Andersson H, Jarvoll P, Lood K, Gräfenstein J, Erdélyi M (2017) Halogen bonding: a powerful tool for modulation of peptide conformation. Biochemistry 56(25):3265–3272. https://doi.org/10.1021/acs.biochem.7b00429(PubMed PMID: 28581720)
Mikkonen L, Pihlajamaa P, Sahu B, Zhang FP, Janne OA (2010) Androgen receptor and androgen-dependent gene expression in lung. Mol Cell Endocrinol 317(1–2):14–24. https://doi.org/10.1016/j.mce.2009.12.022(Epub 2009/12/29)
Drugs (2020) Antiandrogens. https://www.drugs.com/drug-class/antiandrogens.html. Accessed 14 Apr 2020
Sanofi-Synthelabo Inc. (2001) Chloroquine FDA label. https://s3-us-west-2.amazonaws.com/drugbank/fda_labels/DB00608.pdf?1265922797. Accessed 22 Mar 2020
Aguiar ACC, Murce E, Cortopassi WA, Pimentel AS, Almeida M, Barros DCS et al (2018) Chloroquine analogs as antimalarial candidates with potent in vitro and in vivo activity. Int J Parasitol Drugs Drug Resist 8(3):459–464. https://doi.org/10.1016/j.ijpddr.2018.10.002(Epub 2018/11/06)
Vincent MJ, Bergeron E, Benjannet S, Erickson BR, Rollin PE, Ksiazek TG et al (2005) Chloroquine is a potent inhibitor of SARS coronavirus infection and spread. Virol J 2(1):69. https://doi.org/10.1186/1743-422X-2-69
Jang CH, Choi JH, Byun MS, Jue DM (2006) Chloroquine inhibits production of TNF-alpha, IL-1beta and IL-6 from lipopolysaccharide-stimulated human monocytes/macrophages by different modes. Rheumatology (Oxford, England) 45(6):703–710. https://doi.org/10.1093/rheumatology/kei282(Epub 2006/01/19)
Golden E, Cho H-Y, Hofman F, Louie S, Schonthal A, Chen T (2015) Quinoline-based antimalarial drugs: a novel class of autophagy inhibitors. Neurosurg Focus 38:E12. https://doi.org/10.3171/2014.12.FOCUS14748
Wang M, Cao R, Zhang L, Yang X, Liu J, Xu M et al (2020) Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res 30(3):269–271. https://doi.org/10.1038/s41422-020-0282-0
Gautret P, Lagier JC, Parola P, Hoang VT, Medded L, Mailhe M et al (2020) Hydroxychloroquine and Azithromycin as a treatment of COVID-19: preliminary results of an open-label non-randomized clinical trial. medRxiv. https://doi.org/10.1101/2020.03.16.20037135
Weniger H (1979) Review of side effects and toxicity of chloroquine. World Health Organization, Geneva
McChesney EW (1983) Animal toxicity and pharmacokinetics of hydroxychloroquine sulfate. Am J Med 75(1a):11–18. https://doi.org/10.1016/0002-9343(83)91265-2(Epub 1983/07/18)
Zhang H, Penninger JM, Li Y, Zhong N, Slutsky AS (2020) Angiotensin-converting enzyme 2 (ACE2) as a SARS-CoV-2 receptor: molecular mechanisms and potential therapeutic target. Intensive Care Med. https://doi.org/10.1007/s00134-020-05985-9
Liu J, Cao R, Xu M, Wang X, Zhang H, Hu H et al (2020) Hydroxychloroquine, a less toxic derivative of chloroquine, is effective in inhibiting SARS-CoV-2 infection in vitro. Cell Discov 6(1):16. https://doi.org/10.1038/s41421-020-0156-0
Kuba K, Imai Y, Rao S, Gao H, Guo F, Guan B et al (2005) A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat Med 11(8):875–879. https://doi.org/10.1038/nm1267(Epub 2005/07/12)
Rubio-Aliaga I, Frey I, Boll M, Groneberg DA, Eichinger HM, Balling R et al (2003) Targeted disruption of the peptide transporter Pept2 gene in mice defines its physiological role in the kidney. Mol Cell Biol 23(9):3247–3252
Ding N, Zhao K, Lan Y, Li Z, Lv X, Su J et al (2017) Induction of atypical autophagy by porcine hemagglutinating encephalomyelitis virus contributes to viral replication. Front Cell Infect Microbiol 7:56. https://doi.org/10.3389/fcimb.2017.00056(Epub 2017/03/16)
Turk V, Stoka V, Vasiljeva O, Renko M, Sun T, Turk B et al (2012) Cysteine cathepsins: from structure, function and regulation to new frontiers. Biochem Biophys Acta 1824(1):68–88. https://doi.org/10.1016/j.bbapap.2011.10.002(Epub 2011/10/26)
Frlan R, Gobec S (2006) Inhibitors of cathepsin B. Curr Med Chem 13:2309–2327. https://doi.org/10.2174/092986706777935122
Schrezenmeier E, Dorner T (2020) Mechanisms of action of hydroxychloroquine and chloroquine: implications for rheumatology. Nat Rev Rheumatol 16(3):155–166. https://doi.org/10.1038/s41584-020-0372-x(Epub 2020/02/09)
Agostini ML, Andres EL, Sims AC, Graham RL, Sheahan TP, Lu X et al (2018) Coronavirus susceptibility to the antiviral remdesivir (GS-5734) is mediated by the viral polymerase and the proofreading exoribonuclease. mBio 9(2):e00221–e318. https://doi.org/10.1128/mBio.00221-18
Holshue ML, DeBolt C, Lindquist S, Lofy KH, Wiesman J, Bruce H et al (2020) First case of 2019 novel coronavirus in the United States. N Engl J Med 382(10):929–936. https://doi.org/10.1056/NEJMoa2001191(PubMed PMID: 32004427)
Fehr AR, Perlman S (2015) Coronaviruses: an overview of their replication and pathogenesis. Methods Mol Biol (Clifton, NJ) 1282:1–23. https://doi.org/10.1007/978-1-4939-2438-7_1(Epub 2015/02/28)
Harcourt BH, Jukneliene D, Kanjanahaluethai A, Bechill J, Severson KM, Smith CM et al (2004) Identification of severe acute respiratory syndrome coronavirus replicase products and characterization of papain-like protease activity. J Virol 78(24):13600–13612. https://doi.org/10.1128/jvi.78.24.13600-13612.2004(Epub 2004/11/27)
Báez-Santos YM, St John SE, Mesecar AD (2015) The SARS-coronavirus papain-like protease: structure, function and inhibition by designed antiviral compounds. Antiviral Res 115:21–38. https://doi.org/10.1016/j.antiviral.2014.12.015(Epub 12/29)
Chen YW, Yiu C-PB, Wong K-Y (2020) Prediction of the SARS-CoV-2 (2019-nCoV) 3C-like protease (3CL (pro)) structure: virtual screening reveals velpatasvir, ledipasvir, and other drug repurposing candidates. F1000Res 9:129. https://doi.org/10.1288/f1000research.22457.1
Lim J, Jeon S, Shin HY, Kim MJ, Seong YM, Lee WJ et al (2020) Case of the index patient who caused tertiary transmission of COVID-19 infection in Korea: the application of lopinavir/ritonavir for the treatment of COVID-19 infected pneumonia monitored by quantitative RT-PCR. J Korean Med Sci 35(6):e79. https://doi.org/10.3346/jkms.2020.35.e79(Epub 2020/02/15)
Cao B, Wang Y, Wen D, Liu W, Wang J, Fan G et al (2020) A trial of Lopinavir-Ritonavir in adults hospitalized with severe Covid-19. N Engl J Med. https://doi.org/10.1056/NEJMoa2001282
Huynh T, Wang H, Luan B (2020) In silico exploration of the molecular mechanism of clinically oriented drugs for possibly inhibiting SARS-CoV-2’s main protease. J Phys Chem Lett11:4413–4420. https://doi.org/10.1021/acs.jpclett.0c00994
Gulati K, Rai N, Chaudhary S, Ray A (2016) Nutraceuticals in respiratory disorders. Academic Press, Cambridge, pp 75–86
Han S, Mallampalli RK (2015) The role of surfactant in lung disease and host defense against pulmonary infections. Ann Am Thorac Soc 12(5):765–774. https://doi.org/10.1513/AnnalsATS.201411-507FR
Plomer M, de Zeeuw J (2017) More than expectorant: new scientific data on ambroxol in the context of the treatment of bronchopulmonary diseases. MMW Fortschritte der Medizin 159(Suppl 5):22–33. https://doi.org/10.1007/s15006-017-9805-0(Epub 2017/06/24)
Gao X, Huang Y, Han Y, Bai C-X, Wang G (2011) The protective effects of Ambroxol in Pseudomonas aeruginosa-induced pneumonia in rats. Arch Med Sci 7(3):405–413. https://doi.org/10.5114/aoms.2011.23403(Epub 07/11)
Boehringer Ingelheim Limited (2016) Summary of bisolvon product characteristics. https://www.hpra.ie/img/uploaded/swedocuments/LicenseSPC_PA0007-025-002_29042016142022.pdf. Accessed 20 Mar 2020
Therapeutic Goods Administration (TGA) (2012) Bisolvon chesty product information. https://gp2u.com.au/static/pdf/B/BISOLVON_CHESTY-PI.pdf. Accessed 21 Mar 2020.
Laporte M, Naesens L (2017) Airway proteases: an emerging drug target for influenza and other respiratory virus infections. Curr Opin Virol 24:16–24. https://doi.org/10.1016/j.coviro.2017.03.018(Epub 2017/04/18)
Waldrop T, Alsup D, McLaughlin EC (2020) Fearing coronavirus, Arizona man dies after taking a form of chloroquine used to treat aquariums. CNN Health. https://edition.cnn.com/2020/03/23/health/arizona-coronavirus-chloroquine-death/index.html. Accessed 30 Mar 2020.
Touret F, de Lamballerie X (2020) Of chloroquine and COVID-19. Antiviral Res 177:104762. https://doi.org/10.1016/j.antiviral.2020.104762
Keyaerts E, Li S, Vijgen L, Rysman E, Verbeeck J, Van Ranst M et al (2009) Antiviral activity of chloroquine against human coronavirus OC43 infection in newborn mice. Antimicrob Agents Chemother 53(8):3416–3421. https://doi.org/10.1128/aac.01509-08
Tan YW, Yam WK, Sun J, Chu JJH (2018) An evaluation of chloroquine as a broad-acting antiviral against hand, foot and mouth disease. Antiviral Res 149:143–149. https://doi.org/10.1016/j.antiviral.2017.11.017(Epub 2017/11/28)
Yan Y, Zou Z, Sun Y, Li X, Xu K-F, Wei Y et al (2013) Anti-malaria drug chloroquine is highly effective in treating avian influenza A H5N1 virus infection in an animal model. Cell Res 23(2):300–302. https://doi.org/10.1038/cr.2012.165
Paton NI, Lee L, Xu Y, Ooi EE, Cheung YB, Archuleta S et al (2011) Chloroquine for influenza prevention: a randomised, double-blind, placebo controlled trial. Lancet Infect Dis 11(9):677–683. https://doi.org/10.1016/S1473-3099(11)70065-2
Shimizu Y, Yamamoto S, Homma M, Ishida N (1972) Effect of chloroquine on the growth of animal viruses. Archiv für die gesamte Virusforschung 36(1):93–104. https://doi.org/10.1007/BF01250299
Keyaerts E, Vijgen L, Maes P, Neyts J, Van Ranst M (2004) In vitro inhibition of severe acute respiratory syndrome coronavirus by chloroquine. Biochem Biophys Res Commun 323(1):264–268. https://doi.org/10.1016/j.bbrc.2004.08.085(Epub 2004/09/08)
Inglot AD (1969) Comparison of the antiviral activity in vitro of some non-steroidal anti-inflammatory drugs. J Gen Virol 4(2):203–214. https://doi.org/10.1099/0022-1317-4-2-203
Yao X, Ye F, Zhang M, Cui C, Huang B, Niu P, Liu X, Zhao L, Dong E, Song C, Zhan S, Lu R, Li H, Tan W, Liu D (2020) In vitro antiviral activity and projection of optimized dosing design of hydroxychloroquine for the treatment of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Clin Infect Dis. https://doi.org/10.1093/cid/ciaa237
Good MI, Shader RI (1982) Lethality and behavioral side effects of chloroquine. J Clin Psychopharmacol 2(1):40–47. https://doi.org/10.1097/00004714-198202000-00005(Epub 1982/02/01)
Bottcher-Friebertshauser E, Lu Y, Meyer D, Sielaff F, Steinmetzer T, Klenk HD et al (2012) Hemagglutinin activating host cell proteases provide promising drug targets for the treatment of influenza A and B virus infections. Vaccine 30(51):7374–7380. https://doi.org/10.1016/j.vaccine.2012.10.001(Epub 2012/10/18)
Acknowledgements
The authors would like to thank WEPON Pharmaceutical Group Co. Ltd. for access to their preliminary results on their clinical study project, entitled “Evaluating the Efficacy and Safety of Bromhexine Hydrochloride Tablets Combined with Standard Treatment/ Standard Treatment in Patients with Suspected and Mild Novel Coronavirus Pneumonia (COVID-19)”, officially released on Clinical Trials, with a registration number NCT04273763.
Funding
The author(s) received no specific funding for this work.
Author information
Authors and Affiliations
Contributions
The authors wish it to be known that, in their opinion, the first two authors (MD and DdV) should be regarded as joint first authors. MD contributed to writing the article, literature research, and proposed the drug based on the criteria discussed in the article. DdV contributed to writing the article, literature research, and contributed the illustrations. GL contributed literature research, additional writing, advise, and context. MM contributed literature research, additional writing, advise, and context. SDS contributed literature research, additional writing, advise, and context.
Corresponding author
Ethics declarations
Conflicts of interest
The authors have declared that no competing interests exist.
Ethical approval
No ethics approval is required.
Consent to participate
This article does not contain any study with humans performed by any of the authors.
Consent for publication
No informed consent is required.
Code availability
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Depfenhart, M., de Villiers, D., Lemperle, G. et al. Potential new treatment strategies for COVID-19: is there a role for bromhexine as add-on therapy?. Intern Emerg Med 15, 801–812 (2020). https://doi.org/10.1007/s11739-020-02383-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11739-020-02383-3