




Abstract
The detection of remote homolog pairs of proteins using computational methods is a pivotal problem in structural bioinformatics, aiming to compute protein folds on the basis of information in the database of known structures. In the last 25 years, several methods have been developed to tackle this problem, based on different approaches including sequence–sequence alignments and/or structure comparison. In this article, we will briefly discuss When, Why, Where and How (WWWH) to perform remote homology search, reviewing some of the most widely adopted computational approaches. The specific aim is highlighting the basic criteria implemented by different research groups and commenting on the status of the art as well as on still-open questions.
SYNOPSIS
The protein-folding problem is traditionally the problem where different expertise from different fields was integrated along with the goal of finding solutions to the long-standing issue of computing the three-dimensional (3D) structure of proteins starting from their residue sequence. This problem was never solved with analytical approaches for several reasons, including the fact we are still lacking an exhaustive description of all the subtle atomic interactions inside the protein world and those among the protein and its solvent environment. Luckily, structural bioinformatics in the last decade was able to show that, after all, protein structure is rather conserved within families of proteins performing the same functions in different organisms belonging to different kingdoms. Based on this notion several approaches have been developed and described in the literature to address the problem of protein structure prediction [1]. From this, it is evident that structural bioinformaticians are willing to provide solutions to protein crystallographers for integrating the information flow in an efficient way. In order to cope with the increasing interest in computational solutions, critical assessments of the different heuristic methods were launched as international experiments through the years (Critical Assessment of Techniques for Protein Structure Prediction, or CASPs) starting some 14 years ago (http://predictioncenter.org/casp6). What did we learn? Very simply, as it can be found on the tens of specialised textbooks on bioinformatics (www.iscb.org/bioinformaticsBooks.shtml), our present knowledge on protein structure comparison can be summarised into the following basic rules:
Highly homologous sequences are endowed with very similar structures.
Distantly related sequences (with low homology, with a threshold that is routinely set in the range of 30% sequence identity) may be endowed with the same 3D structure or may have even partially overlapping structures when they perform similar functions.
Based on these considerations, the first and only reliable method that we may think to adopt when trying to compute a 3D structure of a given, not-yet-structurally determined protein is building by homology, or better building by comparison. What do we need to compare? First of all, sequence-to-sequence comparison must be done. If we are lucky and the sequence identity is above 30% (a remarkable suggestion: the higher the identity value is, the better the computed model will be) and our target sequence can be compared to a sequence already endowed with an experimentally known 3D structure, we may think to adopt this folding as a reliable folding also for our protein.
Unfortunately, when this is not the case, we are forced to search for distantly related proteins, since we know that, through evolution, protein modules performing a given functions have been conserved, also independently of sequence identity. Furthermore, real life is always full of complications: we now know from all the structures in the Protein Database (PDB, about 40.000 according to August 2006 release) that similar functions may also correspond to different structures, and this obviously complicates the general picture.
This last set of difficult problems is generally addressed adopting the so-called ab initio methods [2] and by a broad spectrum of different methods that basically try to take advantage of a search in the protein sequence and structure space with different perspectives. This is what we call searching for distantly related homologs.
THE WWWH OF REMOTE HOMOLOG SEARCH
We may simplify our problem by considering the three questions and their answers, as in the following.
When to search for distantly related homologs? When we are unlucky and after sequence alignment we do not find any sequence with some reliable similarity to our pet protein with an already known 3D structure.
Why to search for distantly related homologs? Because we hope to overcome all the difficulties that gene evolution apparently produced so far to complicate matters.
Where to search for distantly related homologs? In the sequence and structure protein space that we know thanks to the effort of all the experimentalists, namely in the data bases of known protein sequences and structures.
The problem now is how to search for distantly related homologs, and this is still an open problem, since new paradigms are always welcome to improve the results. Before entering into the realm of all the methods that have been developed so far, one should take into consideration that the procedure, whatever implementation is realised later on, is based on the assumption that what we presently know is sufficient to give us an answer. This may be a severe limitation and also an explanation of why we are not always successful.
GLOSSARY OF HOMOLOGY
When we define homologous proteins, we imply that they descend from a common ancestor. Remote homologs are pairs of proteins that have similar structures and functions but lack easily detectable sequence similarity. Many remote homologs have been discovered by a systematic structural neighbouring procedure [1]. Literature dating back to the 1970s reports semantic distinctions of homology into ortholog, paralog or xenolog depending on different evolutionary events, which have been debated and described at length in all textbooks on molecular genetics [3]. Speciation events led to orthologs, gene duplication events produced parologs and lateral (horizontal) gene transfer originated xenologs. This distinction is maintained for genes, especially if one is a genetist/molecular biologist. However if one is a bioinformatician, it is sufficient to remember that homologs have similar protein structures, but that, unfortunately, protein structure similarity does not imply homology [4, 5]. Here, problems start, as well as all the computational methods that were invented to find approximate solutions by taking advantage of the progressive amount of data stored in the databases.
STRUCTURAL CLASSIFICATION
Correct evolutionary classification of proteins is subjective. Structural domains are considered as evolutionary units. When delving into substructures, it is often debated when structural divergence ends and convergence starts [6, 7].
The database of Structural Classification of Proteins (SCOP) [8] has become the gold standard in evolutionary classification [9]. SCOP describes in a hierarchical way four different levels of protein classification, namely: class, fold, superfamily and family (Figure 1). Proteins with the same fold but collected into different superfamilies lack evidence of a common ancestor; those with the same fold and in the same superfamily show some evidence of a common ancestor. This evidence is deduced from characteristics such as conservation of rare structural features, clusters of conserved residues, sequence similarity through intermediate sequences or functional similarities. Proteins with clear sequence similarity are grouped into families. Protein pairs listed in the same superfamily but belonging to different families are typical benchmarks for testing the ability of computational methods to identify remote homology [10].
![A scheme of the SCOP classification procedure [5].](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/bib/8/2/10.1093/bib/bbl032/2/m_bbl032f1.jpeg?Expires=1716321423&Signature=y7KodzZwRfAVi2N9abHeIhYOINqnMjtnA2tKIyG~rxOVAfZeji~rNNOmYz-72Kp5DDuDj2KB2tC9FpjTp0Xylc1Y0nU-6ANUEdE8PhYQHG02RhbduwJh5QhINQSntJlZbCnZ-W5EbARxdz6NvPSk5zrU0HE~m-JDAaT-Q3TQHpJQn8rLGFC~ukzG336F2k2k9S06zfe9Z0FzOSW-xKdBkPJzcnLvL2pa3ien5RxxJPcQDuM2pQ0OhxuEISIxFsLlV5tImRHhrQaX1xhMPoQ2ZgFHcGcRsP3IcewQQBFMBNnkQGcnUdSC63Tx-GEZpDzmiVW5dCfv4xeaa2a3tmf6pw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Over the years, researchers have tried to tackle the problem of remote homolog identification by developing a wealth of different computational methods to detect protein sequence similarities. The computational methods can be divided into two major classes: (A) methods that compare proteins on the basis of their sequence information; (B) methods that compare protein structures (i.e. using their three-dimensional structures). In this article, we review the most general aspect of these different approaches, focusing on their underlying concepts (for a comprehensive list of sources, see also [11]).
COMPUTATIONAL METHODS
The computational methods aiming at detecting remote homolog sequences face two different but connected problems: (i) finding the best true homolog among the proteins in the database; (ii) building the correct alignment. While in the last years there have been a significant improvement for the first task, the latter one has been proved to be more problematic [5, 11].
For sake of clarity, we can further split the classification of the computational methods as follows:
methods that compare proteins on the basis of their sequence information:
(A1) Based only on protein sequences comparison
(A2) Based on protein sequence profiles
(A3) Based on information derived from protein structures
(A4) Based on machine learning predictors
(A5) Based on consensus
Methods that compare protein structures (i.e. using their 3D structures) based on structure-versus-structure alignment.
This classification reflects also what was published through the years and details on the different methods can be found on a recent review article [11]. However, more recent developments, and methods based on machine learning and consensus, were not included. Therefore, we will focus specifically on what is presently considered by the scientific community to be the state of art when homology search is necessary.
Methods based on pairwise sequence comparisons (A1)
Sequence alignment methods are at the basis of most of the tools that are routinely described in the literature, and first of all, pairwise sequence comparison is historically the oldest solution to the problem of determining how distant two sequences are. Such approaches routinely implement dynamic programming, and their similarity search can be either local (Smith–Waterman) [12] or global (Needleman–Wunsch) [13]. Generally, the optimal match between two query sequences is evaluated. For local similarity search, only portions of one sequence are matched against portions of the other. This procedure is routinely more effective. The reason why this is so stems from considering that proteins may have undergone different evolutionary events, such as duplication and fusion. As a consequence, only certain sequence domains match each other [12].
In order to speed up database search heuristic approaches to identify the similarity between two query sequences have been introduced. Among them, BLAST [14] and FASTA [15] are the most well known. Even though neither BLAST nor FASTA can guarantee the optimal alignment, in many cases they are quite fast and very effective and as accurate as the complete Smith–Waterman [9] algorithm. For further details on similarity matrices, gap penalties and other related technical details when producing alignment see any textbook of bioinformatics, or the Web sites where the programs are available [1, 11].
Methods based on profile comparisons (A2)
In order to improve the sensitivity of the searching algorithms, a step forward was made when the profile representation of the sequence was introduced [16]. A sequence profile accounts for position-specific information which in many cases is obtained by pairwise-aligning similar sequences against that of interest, using local or global algorithms. At the end of the process, for each position of the sequence of interest it is possible to compute the frequency of each residue type in that position (Figure 2). A profile can therefore be considered as a matrix whose rows are as many as are the features adopted (in sequence profile the feature number is routinely twenty as the number of residues) and whose columns are the residue positions in the sequence. The most interesting result when using sequence profiles is that the score of the match is position specific, although it takes into consideration the same scoring matrix adopted for sequence–sequence methods. This is due to the fact that aligned sequences contribute differently to the score depending on the residue positions (for instance, two alanines in different chain positions can be represented by different column profiles). Profile methods are more sensitive than single-sequence comparison approaches since they summarise the evolutionary history of a family, identifying more and less conserved positions along the protein chain [5].
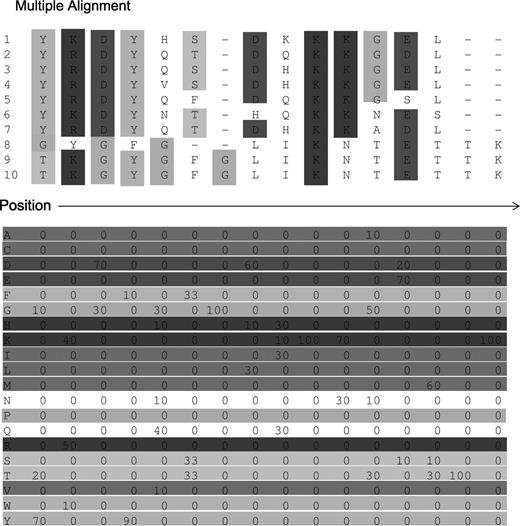
An example of sequence profile.
We can distinguish three different sequence–profile-based approaches: (i) a query profile versus a database of sequences; (ii) a query sequence versus a database of profiles and (iii) a query profile versus a database of profiles. In the first case, one of the most popular and effective approaches is the Position Specific Iterated-BLAST (PSI-BLAST [17]). PSI-BLAST starts with a simple sequence query versus a database of sequences, and iteratively builds a query profile using similar sequences found during the previous run. It stops when no more sequences are retrieved or after a preset number of iterations. PSI-BLAST is an extremely sensitive comparison tool that highlights homologies between sequences that can be retrieved only with structure comparison [18]. PSI-BLAST provides accurate statistical estimates for computed similarities; it occasionally gives good scores to unrelated sequences since the inclusion of a non-homologous sequence in the Position Specific Scoring Matrix (PSSM) may occur (this occurrence is, however, very difficult to detect).
The second way of exploiting sequence profiles is to build a library of sequence profiles and use single-sequence queries. This is done by Integrating Matrix Profiles And Local Alignments (IMPALA), which have been shown to be good and complementary to PSI-BLAST [19].
Finally, the third way is to exploit profile information with a query profile versus a database of profiles, by means of a profile–profile alignment. Recently, several methods have been extended to provide profile–profile based comparisons [20–25]. It has been reported that these methods can give numerous examples of unrecognised sequence similarities reflecting structural similarity and homology [26]. A further improvement of profile–profile alignment accuracy can be obtained using a score based on the information theory, both during the alignment procedure [24], or as a reliability filter [25]. Evaluation of profile–profile comparison methods suggests that profile-profile methods can identify about 20–30% more homologs than PSI-BLAST [2, 26].
Methods exploiting structure information (A3)
In this group of methods, we can include approaches that build 3D structural profiles [27], methods that use secondary structures together with the primary sequence [28–31] and those that thread a protein sequence into the structures of known proteins [32–34]. Most of these methods were originally developed to predict ab initio the protein structures; however, they proved to be quite valuable in detecting remote relationships.
And, 3D profiles [27] extend the notion of sequence profile using information about the spatial environments of the protein residues. The most common environments are described by: (i) the area of the residue buried in the protein and inaccessible to the solvent, (ii) the fraction of side-chain area that is covered by polar atoms (O and N); and (iii) the local secondary structure [27]. As in the case of the protein sequence profiles, each position of the chain can be represented by a vector of values and by this, it is possible to score how effective are the different environments for the different aligned residues.
The methods that incorporate secondary structure information take advantage of the fact that secondary structures of proteins are more likely to be conserved than their sequences. Hence, after predicting secondary structure, two proteins can be aligned, and the alignment of the predicted secondary structures is an additional scoring function [30]. Threading One-dimensional Predictions Into Three-dimensional Structures (TOPITS) first implemented this approach, by defining a scoring matrix that was a linear combination of two different matrices. The first was the classical Point Accepted Mutation (PAM) (or BLOcks SUbstitution Matrix (BLOSUM)), and the second was based on secondary structures and relative solvent accessibility features, both predicted for the two sequences to be aligned. This method performed with significant improvement over methods based on single-sequence alignment [28]. Secondary structure prediction combined with PSI-BLAST was found to improve remote homology detection [29].
Alternatively, when protein sequences have very low sequence identity, secondary structure information can help to improve the alignment quality (you may try YAP at gpcr.biocomp.unibo.it/cgi/predictors/aligns/aligns.cgi). It is also possible to consider the scoring systems that include residue solvation/burial values [35, 36], and in this case profile–profile alignment methods have been accordingly extended to take into account also secondary structure alignment [37].
Several threading methods have been developed with the main purpose of protein structure prediction at low sequence identity [32–34, 38, 39]. These methods are all based on the notions that protein folds are limited in nature and that a sequence can fit to a set of representative folds with different scores. Optimising the procedure gives also the possibility of finding distantly related homologs among the less scoring templates [38].
Machine-learning approaches (A4)
There is an entire group of methods, based on tools first developed within the artificial intelligence/machine-learning community, including Hidden Markov Models (HMMs), Neural Networks (NNs) and Support Vector Machines (SVMs). These methods have been successfully applied to the remote-homolog detection problem. HMMs and NNs can be considered non-linear statistical data-modelling tools that can be ‘trained’ (parameterised) to model complex relationships between inputs and outputs or to find patterns in data; SVMs in turn excel at classification tasks [40].
An alternative way of building sequence profiles is by means of HMMs [41–45]. The main advantage is then that HMMs rely on a solid probabilistic framework and that they can derive from examples not only position specific scores, but also position specific gap penalties. One of the most widely used resources that apply HMMs to find remote homologs is the Protein Families (PFAM) database [46].
The ability of HMM and NN to model complex and unclear relationship between data even of heterogeneous nature, is at the basis of several methods that exploit both sequence- and structure-related information. These approaches use a machine-learning tool either as the core of the scoring system that evaluates the quality of alignments generated using different methods [47, 48] or as generative models that both map and score the alignments between the target and template sequences [49].
In the previous years, SVMs became one of the most widely adopted approach to address several problems of computational biology, including remote homology search [50–57]. The main difference between different implementations consists in the kernel function that measures the similarity between any pair of examples. Different kernels correspond to different notions of similarity and can lead to discriminative functions with different performance.
The basic idea of the kernel-based approaches is to use the sequence comparison methods (sequence-sequence alignments, profile-based, HMMs and others) in order to compute a vector of values representing each sequence. Then, exploiting the SVM learning feature, it is possible to improve the classification and the detection of remote homologs. In practice, different from other approaches that only rely on positive examples, SVMs also add to different methods the ability of learning from negative examples and of discriminating among the positive and negative class [53]. Despite their efficacy in detecting remote sequence relationships, these methods, however, were not constructed to improve the alignment between the query and the target proteins, and so far the problem is still unsolved [5, 11].
Consensus-based approaches (A5)
This category comprises the so-called ‘metaservers’ that have proved to be highly successful in the more recent editions of the CASP experiment [58, 59] where it was shown that the accuracy of remote-homologs detection is improved when different methods are combined to generate a consensus model. Many systems are currently available and they differ both in the number and type of approaches selected as jury components, as well as in the jury implementation [60–63]. The underlying philosophy is, however, the same: these methods leverage on the results provided by other methods, often obtained from remote web servers (thus the name of ‘metaserver’ [64]), such as the one described in the preceding sections. These results are then fed as input to the metaserver scoring system, such as a NN, that selects which of the input prediction is the most reliable for the case under examination.
Structure-Structure comparison (B)
Even though modern sequence-based methods significantly improved the single-sequence search algorithms, they still fail to correctly identify similarities that can be identified through 3D structure alignment. Programs such as SARF [65], Combinatorial Extension (CE) [66], DALI [67], Structural [68], VAST [69], FAST [70] and MAMMOTH [71] have the great advantage of using atomic coordinates for both query and target proteins. Considering that a clear agreement about what is the best structural alignment is still lacking, methods based on structural alignments adopt different rules. For example, some of them require that the matching protein portions have to be topologically connected in the same way [66, 68, 69, 72]; instead, others search for matching regions that are not necessary topologically connected in the same way [65, 67]. The extent of the difference in performance, however, largely depends on both the level of specified selectivity and how the overall performance is computed.
It is worth noticing that the performance of methods comparing protein structure cannot be totally discriminated from that of sequence-based methods [5, 11, 25]: as a general consideration, the first ones perform better, but sometimes the last ones are similarly performing, and the solution is therefore problem-dependent.
To summarise, and according to the data presently available in the literature, structure comparison is better than profile–profile comparison, which is, in turn, better than profile–sequence comparison, which is in turn better than sequence-sequence comparison. However, in some cases, the profile–profile performance was higher than that of structure comparison [25]. An example of this rule of thumb is depicted in Figures 3A–D (evaluated also using MaxSub [72]). For the specific case at hand, it is shown that the alignment obtained using sequence against sequence (Figure 3A) is worse that that obtained using profile versus sequence (Figure 3B); in turn, this is worse than that obtained with profile against profile (Figure 3C); finally, this last alignment is comparable to that obtained with the CE structural comparison program (3D).
![(A) Alignment obtained using sequence against sequence for the yeast transcription regulator MAT A1 and its counterpart ALPHA2 (1AKH chains A and B, PDB code) following the procedure previously described [25]. RMSD = route mean square deviation between the CA coordinates. COV = coverage of the alignment (i.e. the percentage of aligned residues). MS = MaxSub score as described in Reference [71]. ID = percentage of identical aligned residues. (B) Alignment obtained using sequence against profile. For legend, see Figure 3A. (C) Alignment obtained using profile–profile alignment. For legend see Figure 3A. (D) Alignment for the same protein pair of Figure 3A and C obtained with CE [65]. For legend see Figure 3A.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/bib/8/2/10.1093/bib/bbl032/2/m_bbl032f3.jpeg?Expires=1716321423&Signature=4CWzZI~tV0UmTMfrO836NUsM4UgmAhZqHorMZGvAyKQAC-DnnpAJ3tbRy4s-lCf2gOQ2JIVm59EEL~8AFOtQf261dTr3LhCCKnPLx-OH2cEO2COsXIRAlBOmNr4fWJ-7SeQffdaoaWyt5Po1nJaD6d51oUOqVA13jreOn1AJidhG9AS5rWuCklghsehU8cdFyHwL9~-CbGIoBEh0T8PboERnSZCTYed2sRJM1Pwu88uZU8fSgoiiOWeTmvzokeSw52JShYLPsMaur1LRBFWDecKM-mXP0RNnltbcQExB-WTZ8K1FxHF5Z6AbzRd1FM9ygex~aJJH4boTTLKsGTvLLw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
(A) Alignment obtained using sequence against sequence for the yeast transcription regulator MAT A1 and its counterpart ALPHA2 (1AKH chains A and B, PDB code) following the procedure previously described [25]. RMSD = route mean square deviation between the CA coordinates. COV = coverage of the alignment (i.e. the percentage of aligned residues). MS = MaxSub score as described in Reference [71]. ID = percentage of identical aligned residues. (B) Alignment obtained using sequence against profile. For legend, see Figure 3A. (C) Alignment obtained using profile–profile alignment. For legend see Figure 3A. (D) Alignment for the same protein pair of Figure 3A and C obtained with CE [65]. For legend see Figure 3A.
MEASURING THE METHOD RELIABILITY
In order to assign statistical meaning to the scores obtained with the different methods it is also mandatory to estimate what is the chance that a given score could have been obtained also with unrelated pairs of proteins (baseline predictors). One of the most widely adopted ways is to compute the alignment scores with the extreme value distribution [73]. The assumption is that unrelated sequences have local alignment similarity scores that are very accurately described by mathematical models of random sequences. Since unrelated sequences have similarity scores that are indistinguishable from the scores of random sequences, statistically significant similarities derive from homologous sequences. A good estimation of reliability can also reduce the number of false positive hits and increase the quality of searching methods. It is interesting to consider that at the last CASP experiment (CASP6) [58], all the groups that adopted a combination of the methods described earlier (about 10 different groups) with the aid of large computational resources were well performing on the 38 targets that were predicted. This obviously does not guarantee that for specific problem at hand the solution will be successful simply by considering a submission to any of the best performing servers at CASP6 (for a detailed descriptions of the methods and their performance, see [58]).
CONCLUSIONS AND PERSPECTIVES
In the previous years an increase of accuracy in the detection of the remote homologs was reported. This is particularly true for the detection of the closest homologous proteins (if any) in a given data set. However, it is still necessary to improve the results of algorithms capable of aligning remote homolog protein sequences starting from a correctly selected protein pair [5]. Recently divergent evolution within protein superfolds was inferred with sequence profile-based phylogenetic techniques [74]. This suggests that profile-based phylogenetic methods, often adopted for protein function prediction can also be applied for remote homolog identification.
Another relevant, somewhat related problem, that deserves a special attention ‘per se’, is the detection of remote homologs among membrane proteins. This is due to the fact that these proteins are very difficult to solve with atomic resolution, due to technical problems, so that they are under-represented into the database of protein structures [75]. In recent years, several methods have been proposed to predict the topology of membrane proteins, i.e. the position of the transmembrane domains that span the bilayer (either α helices or β strands) along the protein chain and the position of the N and C terminus with respect to the membrane plane [76]. Also new types of web products have been described in order to include diaries and protocols into the computational web experiments [77]. Even though these methods have been built to assign membrane protein topology, they can be also applied to detect remote homologs in prokaryotic genomes with quite high reliability [78]. This suggests that with the increase of membrane protein examples in the near future, a specific class of membrane protein remote homolog detectors can be generated and compared to already available large scale predictions [79].
When to search for distantly related homologs? When we are unlucky and after sequence alignment we do not find any sequence with some reliable similarity (25% identity) to our pet protein with an already known 3D structure.
Why to search for distantly related homologs? Because we want to infer structures, functions of phylogenetics relationships.
Where to search for distantly related homologs? In the sequence and structure protein space that we know, thanks to the effort of all the experimentalists, namely in the databases of known protein sequences and structures.
How to search? This is still an open question and an active research field. Several methods based on improved alignment procedures, machine learning approaches, chemicophysical principles and combinations of them are routinely used and developed.
Protein structures are more conserved than protein sequences. In detecting remote homologs, structure comparison is better than profile–profile comparison, which is in turn better than profile–sequence comparison, which is, in turn, better than sequence–sequence comparison.
Acknowledgments
We thank MIUR for the following grants: PNR-2003 grant delivered to PF, a PNR 2001-2003 (FIRB art.8) and PNR 2003 projects (FIRB art.8) on Bioinformatics for Genomics and Proteomics and LIBI-Laboratorio Internazionale di BioInformatica, both delivered to RC. This work was also supported by the Biosapiens Network of Excellence project (a grant of the European Unions VI Framework Programme, contract number LSHG-CT-2003-503265).

{kind=link}
{kind=link}
{kind=link}