




Abstract
The Protein Data Bank Japan (PDBj) curates, edits and distributes protein structural data as a member of the worldwide Protein Data Bank (wwPDB) and currently processes ∼25–30% of all deposited data in the world. Structural information is enhanced by the addition of biological and biochemical functional data as well as experimental details extracted from the literature and other databases. Several applications have been developed at PDBj for structural biology and biomedical studies: (i) a Java-based molecular graphics viewer, jV; (ii) display of electron density maps for the evaluation of structure quality; (iii) an extensive database of molecular surfaces for functional sites, eF-site, as well as a search service for similar molecular surfaces, eF-seek; (iv) identification of sequence and structural neighbors; (v) a graphical user interface to all known protein folds with links to the above applications, Protein Globe. Recent examples are shown that highlight the utility of these tools in recognizing remote homologies between pairs of protein structures and in assigning putative biochemical functions to newly determined targets from structural genomics projects.
INTRODUCTION
Over the last several years, PDBj [1] has expanded its role from that of a database of macromolecular structures to a provider of structure-derived information and services. To this end, PDBj offers a set of integrated structural bioinformatics tools that enable a variety of queries to be performed on the text, sequence and structural content of PDB data. In this article, we cover the current features of our major web-based services and their underlying data content.
EXPANDED DATA CONTENT
PDBML
A significant step forward in the scope of PDBj's activities was made possible by the development of an XML-based data standard, PDBML [2]. PDBML was developed in collaboration with the Research Collaboratory for Structural Bioinformatics (RCSB) in the United States, and the Macromolecular Structure Database (MSD) in the European Bioinformatics Institute (EBI), which, along with the Biological Magnetic Resonance Data Bank (BMRB), form the worldwide Protein Data Bank (wwPDB) [3–5]. The advantage of an XML description is that it allows structural information to be dynamically integrated with ever-growing evolutionary and functional annotations relating to particular proteins or protein families. For example, Gene Ontology (GO) identifiers and functional information from UniProt, including SwissProt and Prosite, are automatically added on a weekly basis. In addition to such automated annotations, we manually extract experimental and other details from literature sources and include them in the XML description as well.
DATA VISUALIZATION
j V
Since the first macromolecular structures were solved, data visualization tools have been essential to our understanding of biological and biochemical function. Now, with biological data growing more rapidly than ever, the need for integrated visualization software is even more important. For this purpose, PDBj has developed a Java-based molecular viewer, jV [6, 7], that is available both as a stand-alone program and as a web-based applet. To use jV as an applet, only the Java Runtime Environment (JRE) is required, while the stand-alone program also requires the Java bindings for OpenGL (JOGL). What is unique about jV, is that, in addition to displaying the familiar molecular coordinates in a variety of representations, it can also display any three-dimensional (3D) data that is described as a set of polygons or polylines. This currently includes the electrostatic potentials of proteins and nucleic acids mapped onto their molecular surfaces, electron density maps derived from X-ray crystallographic measurements and even the entire known Protein Universe represented as points on a ‘Protein Globe’.
Viewing molecular structures and surfaces
An extensive database of molecular surfaces, with detailed information on functional sites, is maintained at eF-site [7–9]. The electrostatic potentials mapped onto the molecular surfaces along with the atomic coordinates of active sites can both be viewed interactively and downloaded in a variety of formats for local use. Figure 1A shows the active site of L-2-haloacid dehalogenase from Pseudomonas sp. (PDB ID: 1qh9, chain-A) viewed through jV. The surface files with electrostatic potentials computed by solving the Poisson–Boltzmann equations can also be generated on demand for an arbitrary PDB-formatted file provided by the user, such as a 3D structural model, at the eF-surf [10] site.

Viewing structures with jV. (A) The electrostatic potential mapped onto the molecular surface of L-2-haloacid dehalogenase from Pseudomonas sp. (PDB ID: 1qh9, chain-A) viewed through jV at eF-site web site. When viewed on the eF-site web site, the potentials are indicated by colors. The blue and red colors correspond to the positive and negative electrostatic potentials, respectively, from +0.1 V to −0.1 V. The yellow color indicates the hydrophobic side chains. (B) The electron density isosurfaces of the iron–sulfur cluster in human glutaredoxin-2 (PDB ID 2ht9).
Viewing electron density maps
Electron densities are available for a large number of entries, for which the corresponding structure factors are registered. From February 2008, all crystal structures will have to be deposited to the wwPDB with their structure factors. Their electron densities are calculated from the structure factors, and can be displayed either as contour plots or as density isosurfaces. The electron densities and molecular coordinates can be viewed simultaneously, as in Figure 1B, which depicts the iron–sulfur cluster in human glutaredoxin-2 (PDB ID 2ht9).
Viewing the protein universe
Protein Globe [11] is a unique graphical interface to the PDBj services that runs though jV (Figure 2A). All the known protein folds in the PDB are represented as points on a globe. The distance between points indicates their structural similarity as defined by the Alignment of Structural Homologs (ASH) score [11]; that is, structurally similar folds are placed close together on the globe. To facilitate navigation and visual inspection, cartoon representations are provided for some super folds with many family members (Figure 2B). Using the jV-based interface, a user can interactively explore the Protein Globe by rotating and zooming in on the sphere. A point can be picked by clicking it, causing its 3D structure to be shown on the right-hand side of the page for closer examination. Once a point has been picked, the corresponding PDB entry can be sent to other services provided by PDBj including xPSSS (XML-based Protein Structure Search Service), Sequence Navigator, Structure Navigator and eF-site (Figure 2C), each of which will be introduced in the next section, as well as to a few external databases such as SCOP [12] and CATH [13].

Protein Globe. (A) A screenshot of Protein Globe. Each point (brown-colored when viewed on the PDBj web site) in the Globe represents a representative of a protein fold. Some super folds are also represented as polygon cartoons. The 3D structures of a picked domain and of the corresponding PDB entry are shown in the right-hand side of the page. (B) Close-up views of the Globe, showing cartoon polygons of several super folds (helices and strands are colored in red and yellow, respectively, when viewed on the PDBj web site). (C) PDBj services can be sent to a point picked on the Globe, which include xPSSS, Sequence Navigator, Structure Navigator and eF-site (from top to bottom), as well as the SCOP and CATH databases (data not shown).
PERFORMING QUERIES
Text-based queries
Our native XML search engine xPSSS allows the entire PDBML data content to be searched by text-based queries. Simple PDB ID or keyword searches as well as sophisticated compound queries, are possible. xPSSS provides the familiar forms for basic and advanced searches (i.e. compound name, release date, ligands and prosthetic groups, etc.) as well as XQuery and XPath windows for constructing customized queries. Since it takes some experience to become familiar with the XQuery syntax, we provide an XQuery advisor service (XQuad) that allows a query to be constructed from a combination of keyword and category search forms. Figure 3 illustrates two example queries. More detailed tutorials and online help are also available.

Examples queries using XQuery. (A) A search based on secondary structure content. (B) A search based on resolution.
Sequence-based queries
One of the most common queries performed at PDBj is a search for homologous entries. For such queries, PDBj provides Sequence Navigator [14], a BLAST-based sequence query engine. There are a variety of ways of performing Sequence Navigator queries. For example, it is possible to enter Sequence Navigator directly using an amino acid sequence or PDB ID and chain ID to identify the PDB entries that represent a particular sequence family. There are also options available for clustering the output, which is helpful when the number of family members is large. Alternatively, from within xPSSS, a ‘Sequence Neighbor’ button is available for any PDB ID, which automatically fills out the Sequence Navigator form using the PDB ID as a query. Finally, Sequence Navigator is available as a SOAP service so that it can be incorporated in workflows constructed from various Web Services. Figure 4 shows the top five hits to the structural genomics target 2czl, chain A. Note that the hits with BLAST E-values <0.01 are themselves functionally uncharacterized, indicating that 2czl represents a new protein sequence family. PDB entries for new families are important as they can serve as structural templates for homology modeling, thereby defining the structure for an entire family of sequences.

The top five hits to the structural genomics target 2czl, chain A. Note that the hits are all themselves functionally uncharacterized, so we cannot learn the function of 2czl from a Sequence Navigator query.
Structure-based queries
PDBj has a range of tools for structure-based queries. At the tertiary structure level, one can search for structures with a similar fold using Structure Navigator [15]. As with Sequence Navigator, Structure Navigator may be accessed by entering a PDB ID or by uploading an external file that need not be a registered PDB entry. PDBj maintains a large database of structure alignments that have been computed using the program ASH [11, 16]. Using this database, Structure Navigator first tries to find a close match to the query, and retrieve its structural neighbors. The time required for a query depends on whether the query (or a close sequence homolog) has been registered in our structure alignment database or not. If the query (or a sequence homolog) has been registered, the stored result is quickly returned; if not, the query is processed in real time, which will take several minutes or more, depending on the query. If the query was entered as a PDB ID the results are registered in the database, so that subsequent queries can be retrieved more rapidly; if the query was entered as an external file, the results are not saved.
Figure 5A shows the top 10 hits to the structural genomics target 2czl, chain A. Note that in contrast to the Sequence Navigator results, all but one of the hits are functionally characterized and have a common amino acid binding function. In Figure 5B and C the alignment and superposition to a glutamate-binding protein (1ii5, chain A) are shown, respectively. Several conserved ligand-binding residues (S57, T105 and T106) are indicated, as well as G82, which acts as a hinge for the binding site. This example illustrates the potential for using remote evolutionary relationships as manifested in structural homology to functionally annotate structural genomics targets.

Structure Navigator output from query 2czl, chain A. (A) In contrast to the Sequence Navigator results, all but one of the top 10 hits are functionally characterized and have a common amino acid binding function. (B) The alignment to a glutamate-binding protein (1ii5, chain A) indicating several conserved ligand-binding residues (S57, T105 and T106), as well as a hinge for the binding site (G82). (C) The structural superposition indicates the proximity of the conserved residues to the ligands tartaric acid and glutamate in 2czl and 1ii5, respectively.
At a more detailed level, it is possible to search for similar molecular surfaces to a query protein using eF-seek [17–19]. Figure 6 shows an eF-seek search using the query 2czl, chain A. A template found is 1qh9, chain A, L-2-haloacid dehalogenase from Pseudomonas sp., whose molecular surface is shown in Figure 1A. The query and template have only 5% sequence identity, and even belong to different SCOP folds (Periplasmic binding protein II-like and HAD-like, respectively); nonetheless, their active sites are similar enough to correctly identify the ligand-binding site in 2czlA. As this example illustrates, eF-seek can be a very sensitive tool for functional annotation in cases where a close sequence or structural homolog is not available.
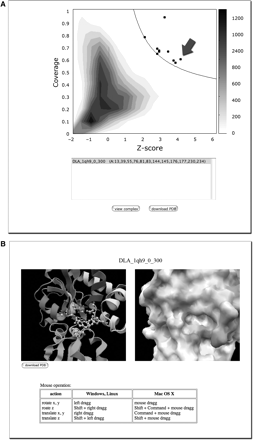
An eF-seek example using 2czl, chain A as the query. (A) The points that appear above and to the right of the solid black line represent templates that are considered significant. The arrow indicates the point for 1qh9. (B) By selecting a point (1qh9 is selected in this example), the predicted binding site in both a molecular and surface representation can be viewed. The bound ligand, a lactic acid, to the template 1qh9 is superimposed on the putative active site of 2czl. The lactic acid and the surrounding side chains of 2czl are displayed with ball-and-stick models, together with the ribbon model of 2czl backbone in the left panel.
CONCLUSION
PDBj offers a range of tools that will assist in the analysis and interpretation of macromolecular structural data. In addition to the tools discussed above, there are services for depositing structural data, checking the status of a deposition and downloading entries from PDBj. Each of the PDBj services is summarized in Table 1. Since the number of PDB depositions has recently been growing rapidly, as a result of structural genomics efforts, the importance of these tools for biomedical research will become even greater in the future. Of particular importance are the structure-based comparison tools such as eF-seek and Structure Navigator, as they are more sensitive to distant evolutionary relationships, and thus putative functional relationships than purely sequence-based techniques. Our recent investigation of such remote functional relationships in a large number of hypothetical proteins illustrates their application [20].
A summary of services available at PDBj
| Data deposition | |
| ADIT | Deposit data to PDB |
| ADIT-NMR | Simultaneously deposit NMR data to BMRB and PDB |
| Search engines | |
| xPSSS | Text-based searches, XQuery and XPath |
| Sequence Navigator | Sequence homology searches |
| Structure Navigator | Structure-based searches |
| EM Navigator | Electron microscopy data searches |
| BMRB | NMR data searches |
| Status Search | Find the status of a PDB entry |
| eF-seek | Functional surface searches |
| Services and software | |
| Protein Globe | Graphical interface to PDB data and services |
| ASH | Pairwise structural alignment |
| jV | Graphical viewer for structures and polygons |
| eF-surf | Server to generate electrostatic molecular surfaces |
| Download | FTP and rsync services |
| Secondary databases | |
| eF-site | Electrostatic surfaces |
| eProtS | Encyclopedia of protein structures for nonexperts |
| ProMode | Protein dynamics based on normal mode analysis |
| Data deposition | |
| ADIT | Deposit data to PDB |
| ADIT-NMR | Simultaneously deposit NMR data to BMRB and PDB |
| Search engines | |
| xPSSS | Text-based searches, XQuery and XPath |
| Sequence Navigator | Sequence homology searches |
| Structure Navigator | Structure-based searches |
| EM Navigator | Electron microscopy data searches |
| BMRB | NMR data searches |
| Status Search | Find the status of a PDB entry |
| eF-seek | Functional surface searches |
| Services and software | |
| Protein Globe | Graphical interface to PDB data and services |
| ASH | Pairwise structural alignment |
| jV | Graphical viewer for structures and polygons |
| eF-surf | Server to generate electrostatic molecular surfaces |
| Download | FTP and rsync services |
| Secondary databases | |
| eF-site | Electrostatic surfaces |
| eProtS | Encyclopedia of protein structures for nonexperts |
| ProMode | Protein dynamics based on normal mode analysis |
A summary of services available at PDBj
| Data deposition | |
| ADIT | Deposit data to PDB |
| ADIT-NMR | Simultaneously deposit NMR data to BMRB and PDB |
| Search engines | |
| xPSSS | Text-based searches, XQuery and XPath |
| Sequence Navigator | Sequence homology searches |
| Structure Navigator | Structure-based searches |
| EM Navigator | Electron microscopy data searches |
| BMRB | NMR data searches |
| Status Search | Find the status of a PDB entry |
| eF-seek | Functional surface searches |
| Services and software | |
| Protein Globe | Graphical interface to PDB data and services |
| ASH | Pairwise structural alignment |
| jV | Graphical viewer for structures and polygons |
| eF-surf | Server to generate electrostatic molecular surfaces |
| Download | FTP and rsync services |
| Secondary databases | |
| eF-site | Electrostatic surfaces |
| eProtS | Encyclopedia of protein structures for nonexperts |
| ProMode | Protein dynamics based on normal mode analysis |
| Data deposition | |
| ADIT | Deposit data to PDB |
| ADIT-NMR | Simultaneously deposit NMR data to BMRB and PDB |
| Search engines | |
| xPSSS | Text-based searches, XQuery and XPath |
| Sequence Navigator | Sequence homology searches |
| Structure Navigator | Structure-based searches |
| EM Navigator | Electron microscopy data searches |
| BMRB | NMR data searches |
| Status Search | Find the status of a PDB entry |
| eF-seek | Functional surface searches |
| Services and software | |
| Protein Globe | Graphical interface to PDB data and services |
| ASH | Pairwise structural alignment |
| jV | Graphical viewer for structures and polygons |
| eF-surf | Server to generate electrostatic molecular surfaces |
| Download | FTP and rsync services |
| Secondary databases | |
| eF-site | Electrostatic surfaces |
| eProtS | Encyclopedia of protein structures for nonexperts |
| ProMode | Protein dynamics based on normal mode analysis |
As a member of the wwPDB, PDBj curates, edits and processes about 25 to 30% of the deposited biomolecular structure data in the world.
Several viewers and derivative databases are developed for bioscience and biomedical researchers. In particular, text-based query services and “analog” query services, for similarities of folds and molecular surfaces of proteins, provide analyses for structure-function relationships.
Protein Globe represents the known protein universe at a glance and enables interaction with PDBj's search, analysis, and visualization tools.
Acknowledgements
The authors would like to thank the PDBj staff for technical support, in particular, Mr Atsuro Yoshihara and Ms Reiko Yamashita. PDBj is financially supported by the Japan Science and Technology Agency, Institute for Bioinformatics Research and Development (JST-BIRD).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}