





Abstract
Summary: We describe a web server for the automatic prediction of immunoglobulin variable domains based on the canonical structure model. The server is user-friendly and flexible. It allows the user to select the templates for the frameworks and the loops using different strategies. The final output is a full-fledged 3D model of the variable domains of the target immunoglobulin.
Availability: The server is openly accessible to academic users at the address: http://arianna.bio.uniroma1.it/pigs. It does not require registration and there is no limit to the number of sequences that can be submitted.
Contact: anna.tramontano@uniroma1.it
1 INTRODUCTION
Immunoglobulins are key players of the immune response and their overall structure is reasonably well conserved. They are composed of two heavy and two light chains that contain four and two domains with a similar fold, respectively (Chothia et al., 1989). Antibodies bind their cognate antigen using the tip of the first domains of each chain (VL and VH). From a structural point of view, the antigen binding site is formed by six loops, three from the light (L1, L2, L3) and three from the heavy chain (H1, H2, H3) named according to their order of appearance in the amino acid sequence.
The structure of the main chain of five of these loops can be predicted quite accurately by taking into account the position and identity of a few specific key amino acids (Chothia and Lesk, 1987; Chothia et al., 1989; Tramontano et al., 1990; Webster and Rees, 1995). For example, either five or six residues form the L3 loop. The five residue L3 loops all have similar main chain conformation (within 0.2 Å). The six residue L3 loops can only take one of two possible conformations depending upon the position of a proline residue within the loop. The case of the third loop of the heavy chain (H3) is more complex. Extensive analysis of this loop in the many available structures has demonstrated that the conformation of the 10 residues closer to the framework (the torso) can be predicted in a similar fashion as for the other loops, while the remaining do not seem to follow identifiable sequence rules (Morea et al., 1998; Shirai et al., 1996).
The advanced understanding of the sequence to structure relationship in this important class of molecules makes it possible to predict their structure quite accurately and automatically. The crucial steps in the prediction are the correct alignment of the target sequence with those of immunoglobulins of known structure, the identification of the limits of the hypervariable loops (where insertions and deletions occur) and of the key residues determining their conformation. The alignment has to follow immunoglobulin specific rules and cannot by obtained by classical dynamic programming methods, because insertions and deletions can only occur at very specific positions and some important conserved residues, for example, two bonded cysteine and a tryptophan, need to be aligned. Nevertheless, rule-based techniques for the alignment, the identification of the canonical structures and the detection of the appropriate templates for the loops can be implemented and automated.
A server, named WAM (Webster and Rees, 1995), mostly based on the rules described earlier, is already available for immunoglobulin structure prediction. However, its usage by the academic community at large is limited by a number of factors. Users, who need to register via fax, are restricted to five sequences per month, which is a rather low threshold in the genomic era. Furthermore, the server is rather rigid: it does not allow any input for the selection of the template structures and it only works if the sequence spans the precise boundaries of the domain. Finally the alignment often requires manual intervention.
2 THE PIGS SERVER
We have developed PIGS (prediction of immunoglobulin structure), a tool to build the structure of immunoglobulins available to the academic community. The PIGS server is flexible and user-friendly and relies upon a database of known immunoglobulin structures and of their structural alignment that is regularly updated. The user only needs to input the sequence of the variable chains of the antibody of interest and the program will display a list of putative templates for both the loops and the framework for each chain, together with other useful information (Fig. 1).
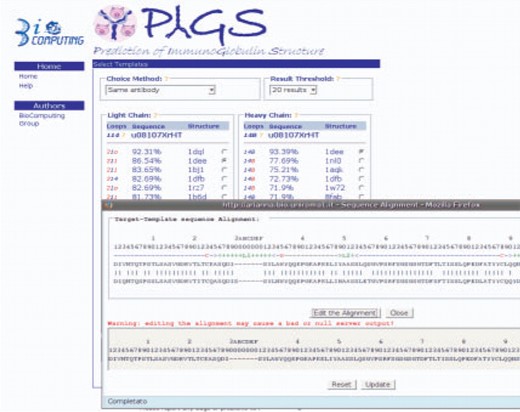
The main page of PIGS, displayed after the user has uploaded the target sequences. The template structures for each chain can be selected manually or according to a predefined strategy. The numbers in the ‘Loop’ column indicate the canonical structure of the loop, with the blue color indicating the same canonical structure as the target immunoglobulin chain. The alignment with each template structure can be viewed and edited in the pop-up window by clicking on the %id figure.
The user can either manually select the templates, or automatically select one of four possible strategies:
Same antibody: select the known structure that can provide a template for both the heavy and light chain, even if a different template with a higher sequence identity exists for one of the chains. If two different antibodies are selected for each chain, the program needs to reconstruct the complete molecule by matching residues known to be conserved at the VL–VH interface and this can introduce more errors than taking the two chains from the same antibody.
Same canonical structure: select the template having loops with the same canonical structure of the target even if a different template with a higher sequence identity exists for one or both chains. If a chain with different canonical structures is selected, the program needs to reconstruct the complete chain by building a ‘chimera’ taking the loops from an antibody with the matching canonical structures and this can introduce more errors than taking the loops from the same chain used to model the framework.
Same antibody and canonical structure: select an antibody structure that can be used as a template for both Vl and Vh and where the canonical structures of the loops are the same as those of the target even if a different template with a slightly better sequence identity exists for one or both chains.
Best L and H chain: select the two chains with highest sequence identity with the corresponding chains of the target and, if needed, pack the two chains together and take the loops from a different structure.
The user can also select whether he/she prefers to obtain just the main chain coordinates (‘Backbone only’), the coordinates of the main chain and of all the side chains conserved with respect to the template (‘Transfer conserved residues’) or the complete structure. In the latter case (‘Transfer conserved+SCWRL’), the conformation of the conserved side chains is retained and the remaining ones are reconstructed using SCWRL (Bower et al., 1997).
No extensive benchmarking has ever been performed to assess which of the strategies described earlier works best. In our own experience, the ‘Same antibody’ and ‘Transfer conserved+SCWRL’ are preferable, and these are the default parameters of the server. We are presently performing a large scale benchmarking whose results will be made available via the PIGS server.
Once the choice has been made, the user can select the ‘build’ option and obtain the predicted structure of the antibody in PDB format. The model can be visualized via JMol, where a few options allow the user to identify key parts of the molecule.
The reliability of the canonical structure method has been proven over and over again, so there is no need to further demonstrate it. We just show as an example, in Table 1 the r.m.s.d. deviation of the main chain atoms between the models obtained by the completely automatic procedure on three antibody structure recently deposited in the protein structure database and their experimental structures.
Comparison between the predicted and experimental main chain structures of four recently solved immunoglobulin structures
| r.m.s.d. | Region | 2HWZ | 2ADF | 2R29 | 2ZCH |
|---|---|---|---|---|---|
| All | All | 1.11 | 1.08 | 1.42 | 1.16 |
| FW | FW | 0.94 | 0.88 | 1.22 | 0.95 |
| Loops | FW | 1.75 | 1.75 | 2.12 | 1.81 |
| L | L | 1.17 | 0.48 | 1.14 | 0.6 |
| FW L | FW L | 1.03 | 0.48 | 0.94 | 0.47 |
| L loops | FW L | 1.90 | 0.48 | 1.90 | 1.07 |
| L1 (length) | FW L | 1.63 (6) | 0.31 (7) | 2.29 (11) | 1.24 (11) |
| L2 (length) | FW L | 0.91 (3) | 0.32 (3) | 1.05 (3) | 0.44 (3) |
| L3 (length) | FW L | 2.43 (6) | 0.68 (5) | 1.39 (6) | 0.94 (6) |
| H | H | 0.74 | 1.01 | 1.50 | 1.21 |
| FW H | FW H | 0.52 | 0.61 | 1.44 | 0.73 |
| H loops | FW H | 1.38 | 1.97 | 1.90 | 2.44 |
| H1 (length) | FW H | 0.8 (9) | 0.85 (7) | 0.81 (7) | 0.56 (7) |
| H2 (length) | FW H | 1.17 (3) | 0.88 (4) | 1.14 (4) | 0.49 (4) |
| H3 (length) | FW H | 1.66 (10) | 2.52 (9) | 2.47 (7) | 3.06 (13) |
| r.m.s.d. | Region | 2HWZ | 2ADF | 2R29 | 2ZCH |
|---|---|---|---|---|---|
| All | All | 1.11 | 1.08 | 1.42 | 1.16 |
| FW | FW | 0.94 | 0.88 | 1.22 | 0.95 |
| Loops | FW | 1.75 | 1.75 | 2.12 | 1.81 |
| L | L | 1.17 | 0.48 | 1.14 | 0.6 |
| FW L | FW L | 1.03 | 0.48 | 0.94 | 0.47 |
| L loops | FW L | 1.90 | 0.48 | 1.90 | 1.07 |
| L1 (length) | FW L | 1.63 (6) | 0.31 (7) | 2.29 (11) | 1.24 (11) |
| L2 (length) | FW L | 0.91 (3) | 0.32 (3) | 1.05 (3) | 0.44 (3) |
| L3 (length) | FW L | 2.43 (6) | 0.68 (5) | 1.39 (6) | 0.94 (6) |
| H | H | 0.74 | 1.01 | 1.50 | 1.21 |
| FW H | FW H | 0.52 | 0.61 | 1.44 | 0.73 |
| H loops | FW H | 1.38 | 1.97 | 1.90 | 2.44 |
| H1 (length) | FW H | 0.8 (9) | 0.85 (7) | 0.81 (7) | 0.56 (7) |
| H2 (length) | FW H | 1.17 (3) | 0.88 (4) | 1.14 (4) | 0.49 (4) |
| H3 (length) | FW H | 1.66 (10) | 2.52 (9) | 2.47 (7) | 3.06 (13) |
No refinement protocol has been applied. Values are the r.s.m.d. in A of the region in the first column after optimal superposition of the region indicated in the second. FW,Framework; L, Light chain; H, Heavy chain
Comparison between the predicted and experimental main chain structures of four recently solved immunoglobulin structures
| r.m.s.d. | Region | 2HWZ | 2ADF | 2R29 | 2ZCH |
|---|---|---|---|---|---|
| All | All | 1.11 | 1.08 | 1.42 | 1.16 |
| FW | FW | 0.94 | 0.88 | 1.22 | 0.95 |
| Loops | FW | 1.75 | 1.75 | 2.12 | 1.81 |
| L | L | 1.17 | 0.48 | 1.14 | 0.6 |
| FW L | FW L | 1.03 | 0.48 | 0.94 | 0.47 |
| L loops | FW L | 1.90 | 0.48 | 1.90 | 1.07 |
| L1 (length) | FW L | 1.63 (6) | 0.31 (7) | 2.29 (11) | 1.24 (11) |
| L2 (length) | FW L | 0.91 (3) | 0.32 (3) | 1.05 (3) | 0.44 (3) |
| L3 (length) | FW L | 2.43 (6) | 0.68 (5) | 1.39 (6) | 0.94 (6) |
| H | H | 0.74 | 1.01 | 1.50 | 1.21 |
| FW H | FW H | 0.52 | 0.61 | 1.44 | 0.73 |
| H loops | FW H | 1.38 | 1.97 | 1.90 | 2.44 |
| H1 (length) | FW H | 0.8 (9) | 0.85 (7) | 0.81 (7) | 0.56 (7) |
| H2 (length) | FW H | 1.17 (3) | 0.88 (4) | 1.14 (4) | 0.49 (4) |
| H3 (length) | FW H | 1.66 (10) | 2.52 (9) | 2.47 (7) | 3.06 (13) |
| r.m.s.d. | Region | 2HWZ | 2ADF | 2R29 | 2ZCH |
|---|---|---|---|---|---|
| All | All | 1.11 | 1.08 | 1.42 | 1.16 |
| FW | FW | 0.94 | 0.88 | 1.22 | 0.95 |
| Loops | FW | 1.75 | 1.75 | 2.12 | 1.81 |
| L | L | 1.17 | 0.48 | 1.14 | 0.6 |
| FW L | FW L | 1.03 | 0.48 | 0.94 | 0.47 |
| L loops | FW L | 1.90 | 0.48 | 1.90 | 1.07 |
| L1 (length) | FW L | 1.63 (6) | 0.31 (7) | 2.29 (11) | 1.24 (11) |
| L2 (length) | FW L | 0.91 (3) | 0.32 (3) | 1.05 (3) | 0.44 (3) |
| L3 (length) | FW L | 2.43 (6) | 0.68 (5) | 1.39 (6) | 0.94 (6) |
| H | H | 0.74 | 1.01 | 1.50 | 1.21 |
| FW H | FW H | 0.52 | 0.61 | 1.44 | 0.73 |
| H loops | FW H | 1.38 | 1.97 | 1.90 | 2.44 |
| H1 (length) | FW H | 0.8 (9) | 0.85 (7) | 0.81 (7) | 0.56 (7) |
| H2 (length) | FW H | 1.17 (3) | 0.88 (4) | 1.14 (4) | 0.49 (4) |
| H3 (length) | FW H | 1.66 (10) | 2.52 (9) | 2.47 (7) | 3.06 (13) |
No refinement protocol has been applied. Values are the r.s.m.d. in A of the region in the first column after optimal superposition of the region indicated in the second. FW,Framework; L, Light chain; H, Heavy chain
3 CONCLUSIONS
The prediction of antibody structures is not only important, but feasible at a level of accuracy much higher than for any other protein type. The level of understanding of the sequence structure relationship in this class of molecules is sufficiently advanced so that automatic easy to use methods can be developed and employed for the molecular analysis of the ‘immunome’. The purpose of the PIGS server described here is to enable scientists to tackle the open problem of the molecular basis of the specificity of antibodies at large and/or to obtain data useful in the context of specific biological problems.
ACKNOWLEDGEMENTS
Funding: PIGS is supported by the EU contract number LHSG-CT-2003-503265, and AIRC BICG Project.
Conflict of Interest: none declared.
REFERENCES
Author notes
Associate Editor: Alfonso Valencia


{kind=link}