





Comparative genomic studies extensively rely on alignments of orthologous sequences. Yet, selecting, gathering, and aligning orthologous exons and protein-coding sequences (CDS) that are relevant for a given evolutionary analysis can be a difficult and time-consuming task. In this context, we developed OrthoMaM, a database of ORTHOlogous MAmmalian Markers describing the evolutionary dynamics of orthologous genes in mammalian genomes using a phylogenetic framework. Since its first release in 2007, OrthoMaM has regularly evolved, not only to include newly available genomes but also to incorporate up-to-date software in its analytic pipeline. This eighth release integrates the 40 complete mammalian genomes available in Ensembl v73 and provides alignments, phylogenies, evolutionary descriptor information, and functional annotations for 13,404 single-copy orthologous CDS and 6,953 long exons. The graphical interface allows to easily explore OrthoMaM to identify markers with specific characteristics (e.g., taxa availability, alignment size, %G+C, evolutionary rate, chromosome location). It hence provides an efficient solution to sample preprocessed markers adapted to user-specific needs. OrthoMaM has proven to be a valuable resource for researchers interested in mammalian phylogenomics, evolutionary genomics, and has served as a source of benchmark empirical data sets in several methodological studies. OrthoMaM is available for browsing, query and complete or filtered downloads at http://www.orthomam.univ-montp2.fr/.
Introduction
Orthologous protein-coding sequences (CDS) are of great interest to study patterns of organismal evolution (species phylogenies) and genomic processes (molecular evolution). The wide use of exons and CDS in phylogenomics and comparative genomics is facilitated by the existence of several independent databases of orthologs (Alexeyenko et al. 2006), each with their pros and cons. Some are generalist—for example, COG/KOG (Tatusov et al. 2003), HOGENOM (Dufayard et al. 2005), and InParanoid (Östlund et al. 2010), some are taxonomically specialized—for example, OPTIC (Heger and Ponting 2008) for vertebrates, INVHOGEN (Paulsen and von Haeseler 2006) for nonvertebrates, EvolMarkers (Li et al. 2012) for metazoans, FUNYBASE for fungi (Marthey et al. 2008), GreenPhylDB (Conte et al. 2008) for plants, HOBACGEN (Perriere et al. 2000) for bacteria, and some are built on functional information, such as OrthoDisease (O’Brien et al. 2004). In particular taxonomic groups, researchers have identified potentially useful phylogenetic DNA markers from complete genomes and have validated their use in nonmodel species such as primates (Horvath et al. 2008), actinopterygian fishes (Li et al. 2007), or rosids (Duarte et al. 2010). However, these databases generally do not provide end-users with key parameters describing the evolutionary pattern of orthologs, and orientating the choice of the molecular markers to be studied from the viewpoint of phylogenomic and molecular evolution. Also, few of them provide high-quality nucleotide and amino acid alignments preserving the key underlying codon structure.
OrthoMaM (Ranwez et al. 2007) is a database of ORTHOlogous MAmmalian coding sequence Markers, which helps filling these gaps. It provides high-quality codon alignments of exon and CDS markers associated with a detailed characterization of their evolutionary dynamics in terms of phylogenetic signal, base composition, substitution rate, and chromosome location. Moreover, OrthoMaM focuses only on one-to-one orthologs identified by Ensembl (Flicek et al. 2014), that is, sequences for which no duplication is detected since the last common ancestor of the corresponding species. Indeed, as one-to-one orthologs are unaffected by complex intragenomic processes such as gene duplication or gene loss, the differences in their sequences are ensured to have occurred through common descent and therefore reflect the divergence between species.
Database Overview and Improvements
Mammalia is among the first animal taxa with many complete genomes available and has been extensively used to define most of the gold-standard methods in phylogenomic and molecular evolution studies. Based on the 12 mammalian genomes available in Ensembl v41, the first version of OrthoMaM was released in July 2007 and contained 3,170 exons (Ranwez et al. 2007).
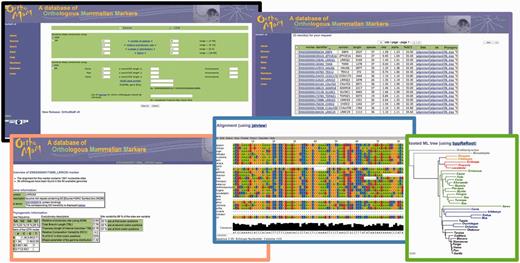
Screenshots from the OrthoMaM website. Here, we searched for CDS with 15–40 mammals, a relative evolutionary rate between 0.5 and 3, an α parameter of the Γ distribution ranging from 1 to 1.5, and a GC3 between 22% and 35%. We got 23 target CDS and focused on the LRRC63 marker. We then visualized the evolutionary dynamics parameters, the first 80 sites of the DNA alignment, and the corresponding phylogenetic tree.
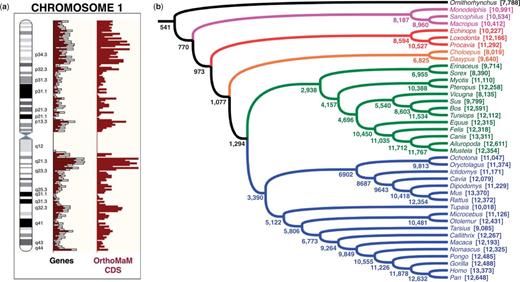
(a) Genomic distribution of OrthoMaM CDS along human chromosome 1. The ideogram for human chromosome 1 is provided together with the distribution of OrthoMaM CDS (dark red bars to the right). The distributions of Ensembl predicted genes (white bars) and database-known genes (red bars) are also indicated (centre). (b) The phylogeny of the 40 species present in OrthoMaM. For each species, we provide the number of available CDS. For each node, we also indicate the number of CDS markers containing all species of the corresponding clade.
The OrthoMaM Pipeline
Identification of Orthologous Sequences
We start by using Ensembl annotations (Flicek et al. 2014) to identify one-to-one orthologous genes among pairs of three high-coverage reference species (Homo–Mus, Homo–Canis, and Mus–Canis). We then enrich each of those clusters of one-to-one orthologs by adding sequences of additional mammals that are annotated as one-to-one orthologs to the human gene (Ranwez et al. 2007). Note that the chromosomal distribution of OrthoMaM human genes basically mirrors the distribution of the full set of Ensembl human genes (fig. 2a), which is to be expected from an unbiased database.
Those clusters of one-to-one orthologous genes are turned into clusters of one-to-one orthologous CDS by selecting the longest transcript of each gene. We choose to consider the longest sequence as this is the one used by Ensembl to define the orthology relationships among genes, and this will maximize the evolutionary information to be analyzed.
The one-to-one orthologous exon clusters are not provided by Ensembl. Their identification is complicated by alternative splicing and by the variability in number and length of exons of a given gene across species. We tackle those problems by relying on the alignments of the one-to-one orthologous CDS to infer one-to-one orthology among their exons. Each human exon annotated by Ensembl initiates a one-to-one orthologous exon cluster. Exons from additional species are added to this cluster if they share a number of identical amino acids greater than half the length of the CDS alignment restricted to the candidate exon and the human one. This similarity threshold ensures that no more than one exon from a given species will be included in the predicted set of orthologs. Initial exon alignments longer than 400 sites are selected as our evolutionary marker descriptors are not meaningful enough for shorter sequences. Clusters with less than four sequences are discarded for the same reason.
Alignments and Trees
CDS and exon sequences are aligned at the codon level in two steps. First, the translated amino acids are aligned using MAFFT (Katoh et al. 2005) and gaps are reported onto the nucleotide sequences. This alignment is then refined using MACSE (Ranwez et al. 2011) to obtain a final codon alignment unaffected by frameshifts, misassemblies, and sequencing errors. Nucleotide and amino acid alignments are then filtered to remove spurious sequences and/or codons using trimAl (Capella-Gutiérrez et al. 2009). The filtering is conducted under the “automated1” option, which has been specifically designed to clean alignments before conducting ML phylogenetic inference. This step can yield final alignments shorter than 400 sites though the average length is far higher for both exons (956 sites) and CDS (1,850 sites). To ensure data traceability, each sequence is linked to the corresponding Ensembl CDS/exon. Moreover, each OrthoMaM alignment is available for download before and after filtering. All previous releases of OrthoMaM also remain available through the website.
The ML tree is identified for each marker by analyzing codon alignments with RAxML (Stamatakis 2006) under the general time reversible (GTR)+Γ model (Yang 1996). We acknowledge that using the proper model of sequence evolution is vital in probabilistic inference. However, we here used the same model for all CDS and exons because 1) it warrants a fair comparison among all markers of the database, 2) it is the one that best fits the majority of the markers (Ranwez et al. 2007), 3) the GTR exchangeability matrix is the only one available at the nucleotide level in RAxML, and 4) the parameter-rich GTR+Γ model is more likely to introduce increased variance rather than bias in the estimates (Lemmon and Moriarty 2004).
All parameters describing the evolutionary dynamics of exons and CDS are gathered by running PAUP* (Swofford 2003) on the ML tree inferred by RAxML. Branch lengths of ML phylograms are also examined, and if some exceed the unrealistic value of two substitutions per site, the corresponding alignment is excluded from OrthoMaM. This phylogenetic-based filter enables to detect and remove markers that likely contain misaligned sequences, misspecified open reading frames, or misannotated paralogs.
Database Updates and Scalability
OrthoMaM is regularly updated and its pipeline is constantly optimized to keep pace with the ever increasing number of available genomes and software developments in the field. Orthology annotation and sequences are now retrieved using the BioMart facilities, which allow massive retrieval of Ensembl data (Flicek et al. 2014). Those data are processed by home made Java tools to identify clusters of one-to-one orthologous CDS/exons. Phylogenetic analyses rely on shell scripts to chain up-to-date software. The website is based on a php/mysql database for query facilities and on XML/XSLT for exchange and graphic representation of marker details. All analyses are performed on the computing cluster of the Montpellier Bioinformatics Biodiversity (MBB) platform.
Query Options
There are three entry points in OrthoMaM. First, exon and CDS sections can be graphically browsed using a clickable phylogeny and ideograms of human chromosomes. Second, markers can be queried according to several of their key characteristics, including: minimal alignment length, number of sequences, mandatory species, base composition (%GC3), relative evolutionary rate of the marker, Ensembl gene identifier or HUGO gene symbol (see fig. 1). Third, a BLAST (Altschul et al. 1990) similarity search can be run to find OrthoMaM markers related to a given request.
Examples of Contributions
OrthoMaM has proven its usefulness in several phylogenomic and comparative genomic studies. We briefly list some of them to illustrate the broad spectrum of analyses facilitated by OrthoMaM. Our database has been used for developing new markers in multigene phylogenetic studies (Zhou et al. 2011; Hassanin et al. 2013) and also as a source of large-scale molecular data in phylogenomic (Parker et al. 2013; Romiguier et al. 2013), molecular dating (Schrago and Voloch 2013), and evolutionary genomic (Galtier et al. 2009; Romiguier et al. 2010; Rorick and Wagner 2011; Lartillot 2013) analyses. The high-quality codon alignments have also been utilized as benchmark empirical data sets for testing new analytical methods (Egan et al. 2008; López-Giráldez and Townsend 2011; Li and Drummond 2012; Wu et al. 2013) and for detecting footprints of purifying or positive selection (Jobson et al. 2010; Laguette et al. 2012). Finally, the inferred ML gene trees have served for assessing the performance of supertree methods (Scornavacca et al. 2008; Ranwez et al. 2010). With the ongoing pace of mammalian genome sequencing, we envision an enhanced potential for the uses of OrthoMaM in comparative genomic studies aiming at understanding the evolutionary dynamics of protein-coding genes.
Future Prospects
The primary aim of OrthoMaM is to provide high-quality genome scale alignments and phylogenetic analysis for one-to-one orthologous exons and CDS among mammals. Its analysis pipeline strategy has been adapted to cope with the increasing number of mammalian genomes that will be released in the upcoming years. This bioinformatic pipeline is constantly improved and we are currently testing the possibility of relying on codon-based phylogenetic inference using codon-phyML (Gil et al. 2013) and including in future releases per branch dN/dS estimations using mapNH (Romiguier et al. 2012). Moreover, we are considering possible solutions to filter only parts of a sequence in order to further improve the quality of our codon alignments with respect to potential exon annotation errors in CDS. We are also evaluating the relevance of expanding the database toward noncoding markers, such as intronic, untranslated, and regulatory regions.
Acknowledgments
This work was supported by the Montpellier Bioinformatics Biodiversity platform, the Agence Nationale de la Recherche “Investissements d’avenir/Bioinformatique” (ANR-10-BINF-01-02 “Ancestrome”), and the European Research Council (“PopPhyl”: Population phylogenomics). The authors thank two reviewers for their comments on the manuscript. This publication is contribution 2014-022 of the Institut des Sciences de l’Evolution de Montpellier (UMR 5554—CNRS-IRD-UM2).
References
Author notes
Associate editor: Xun Gu

{kind=link}
{kind=link}