





Abstract
Human cancers exhibit genomic instability and an increased mutation rate due to underlying defects in DNA repair genes. Hypermethylation of CpG islands in gene promoter regions is an important mechanism of gene inactivation in cancer. Many cellular pathways, including DNA repair, are inactivated by this type of epigenetic lesion, resulting in mutator pathways. In this review, we discuss the adverse consequences suffered by a cell when DNA repair genes such as the DNA mismatch repair gene hMLH1, the DNA alkyl-repair gene O6-methylguanine-DNA methyltransferase, the familial breast cancer gene BRCA1 and the Werner syndrome gene WRN become epigenetically silenced in human cancer.
Introduction
The primary structure of DNA is constantly subjected to alteration by cellular metabolites and exogenous DNA-damaging agents. These alterations may cause complex genetic changes, including deletions, fusions, translocations and aneuploidy, or, alternatively, single-base changes, which can be either transversions (change of purine to pyrimidine or vice versa) or transitions (change of purine to another purine or pyrimidine to another pyrimidine) (1). Such alterations may ultimately lead to cellular death of unicellular organisms or degenerative changes and aging of multicellular organisms.
There are many kinds of DNA lesions occurring in vivo, which are repaired by different DNA repair pathways. These pathways include (i) direct repair of alkyl adducts by O6-alkylguanine DNA alkyltransferase (AGT); (ii) repair of base damage and single-strand breaks (SSBs) by base excision repair (BER); (iii) repair of double-strand breaks (DSBs) by homologous recombination (HR), non-homologous end joining (NHEJ) and single-strand annealing (SSA); (iv) repair of bulky DNA adducts by nucleotide excision repair; (v) repair of cross-links by DNA interstrand cross-link repair and (vi) repair of mismatches and insertion/deletion loops by DNA mismatch repair (MMR) (2).
The critical role played by DNA repair in the maintenance of genome stability is underpinned by the fact that many enzymes involved have been conserved through evolution. Germ line mutations in several of the DNA repair genes are the cause of cancer-predisposing syndromes, and are associated with an increased rate of chromosome breakage and mutagenesis (3).
The inheritance of information based on gene expression levels is known as epigenetics, as opposed to genetics, which refers to information transmitted on the basis of gene sequence. The main epigenetic modification in mammals, and in particular in humans, is the methylation of the cytosine nucleotide residue in CG dinucleotide sequences. Methylation of promoter CpG islands, CG-rich regions that coincide with the promoter of protein-coding genes, correlates with transcriptional silencing. In a normal cell, the DNA methylation patterns are maintained through cell divisions allowing the expression of the particular set of cellular genes necessary for that cell type and blocking the expression of exogenous-inserted sequences (4). In addition, cells also store epigenetic information in the post-transcriptional modification profile of histones. Different amino acid residues from histones are targets for a variety of modifications, including lysine acetylation, lysine and arginine methylation and serine phosphorylation, with specific functional significance. The hypermethylation of CpG islands in gene promoter regions is associated with specific histone modifications, including dimethylation of histone H3 at lysine 9, deacetylation at this residue, trimethylation of histone H3 at lysine 27 and loss of the transcriptional activating mark H3K4me2 (5,6,7,8). The interconnection between DNA methylation and the presence/absence of specific histone modifications in gene promoter regions suggests that DNA methylation is part of an epigenetic program that leads to transcriptional silencing. In cancer, the epigenetic equilibrium characteristic of a normal cell undergoes dramatic transformations that can be summarized as follows: (i) transcriptional silencing of tumour suppressor genes by CpG island promoter hypermethylation, (ii) global genomic hypomethylation, (iii) loss of imprinting and (iv) epigenetic lack of the repression of exogenous-inserted sequences (4). The epigenetic inactivation of tumour suppressor genes by DNA hypermethylation described in Figure 1 seems to be tumour-type specific (9) and affects all cellular pathways. Examples of genes suffering this aberrant DNA methylation include genes involved in cell cycle (p16INK4a, p15INK4b, Rb, p14ARF), carcinogen metabolism (glutathione s-transferase p1), cell adherence (e-cadherin 1[CDH], CDH13) and apoptosis (death associated protein kinase-1, target of methylation induced silencing-1) (10).
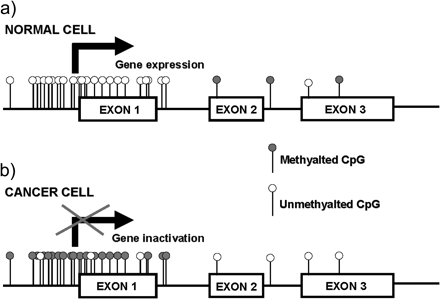
Graphical representation of a typical gene with a CpG island. (a) In a normal cell, the CpG island is devoid of methylation allowing gene expression. (b) In a cancer cell, the CpG island becomes hypermethylated preventing gene transcription.
However, we still do not clearly understand why certain CpG islands are hypermethylated in cancer cells while others remain methylation free. We can hypothesize, as has been done in the case of genetic mutations, that a particular gene is preferentially methylated with respect to others in certain tumour types because its inactivation confers a selective clonal advantage. Another possibility that can explain this local hypermethylation is the role played by the environment and nutrition, since the most hypermethylated tumour types are those of the gastrointestinal tract that are more exposed to external carcinogen agents. If we take in consideration the study from Fraga et al. (11) that reports that patterns of epigenetic modifications of MZ twin pairs diverge as they become older, it is not surprising that external factors like smoking habits, physical activity or diet, among others, together with internal factors can influence the hypermethylation status of specific tumour suppressor genes.
More recently, it was suggested that the tumour-specific targeting of de novo methylation is preprogrammed by an enhancer of zeste homolog 2 (EZH2)-containing polycomb complex that normally has a role in marking embryonic genes for repression (12,13,14). This de novo methylation of EZH2-target promoters packaged with nucleosomes containing histone H3 trimethylated on lys27 is established through direct physical contact between the protein EZH2 and DNA methyltransferases (15), However, the EZH2 system does not contribute to de novo methylation of all tumour suppressor genes in cancer, since several tumour suppressor genes tested were not enriched with trimethylated H3K27 when tested in normal cell types. It thus seems that both gene targeting and adaptive mechanisms are involved in de novo methylation that occurs in cancer (12).
One of the most compelling examples of the role of epigenetic gene silencing in the development of human cancer is the inactivation of DNA repair genes by promoter CpG island hypermethylation. In this review, we will discuss the importance of the epigenetic silencing of a set of genes that are involved in DNA repair, namely, the familial breast cancer gene BRCA1, the DNA MMR gene hMLH1, the DNA alkyl-repair gene O6-methylguanine-DNA methyltransferase (MGMT) and the Werner syndrome (WS) gene WRN in the uncover of new mutator pathways in cancer.
Epigenetic inactivation of BRCA1
One of the earliest indications that BRCA1 was involved in DNA repair was the observation that BRCA1 associates and co-localizes with RAD51 in nuclear foci in mitotic cells (16). These foci were also observed to contain BRCA2 and the BRCA1-binding protein BARD1, both before and after DNA damage (17). Further evidence came from the observation that murine BRCA1 was responsible for genomic integrity as BRCA1−/− embryos exhibited hypersensitivity to γ-irradiation and chromosomal abnormalities, which may be a direct consequence of unrepaired DNA damage (18). Whereas initial studies in BRCA1 and BRCA2 mouse mutants have highlighted similarities between both proteins, other research efforts have also revealed clear differences (19). BRCA1 appears to be more of a signal integrator, linking together sensors and response mechanisms of several types of DNA damage. In contrast, BRCA2 is thought to be more directly involved in homology-directed DSB repair, as it mediates the formation of a RAD51-DNA nucleoprotein filament that catalyses strand invasion during HR (20).
Since the cloning of the BRCA1 gene (21), germ line mutations have been found in the hereditary cases of breast and ovarian cancers (22,23). In fact, germ line alterations in BRCA1 have been estimated to be responsible for ∼50% of familial breast cancer (22,24). However, despite an extensive search, the BRCA1 gene had not been shown to be mutated in any cases of truly sporadic breast cancer and in only an extreme minority of sporadic ovarian tumours (25). These findings challenge the role of BRCA1 as a tumour suppressor gene in the non-hereditary forms of breast and ovarian neoplasia that constitutes 90–95% of these tumour types. However, BRCA1 transcripts (26) and protein (27) are often decreased or lost in sporadic breast carcinomas. Consequently, it was not surprising when different groups reported that BRCA1 was epigenetically silenced by promoter hypermethylation in breast and ovarian primary tumours and cell lines (28,29,30).
Since BRCA1 is important for the repair of DSBs by the potentially error-free pathway of HR and since cells that lack this protein repair these lesions by alternative more error-prone mechanism, we can assume that the epigenetic silencing of this protein in non-hereditary forms of breast cancer creates a new mutator pathway that generates mutations and gross chromosomal rearrangements (Figure 2). One pathway affected by the BRCA1 epigenetic silencing is the p53 pathway; the DNA damage that accumulates in sporadic breast cancer cells would trigger a p53-mediated cell cycle checkpoint that could be alleviated by, for example, mutation of p53 or its downstream target p21. This concept is supported by the observation that inactivation of p53 or p21 results in a prolonged survival of BRCA1-deficient embryos (31,32). In fact, the association between BRCA1-deficient cells and p53 mutations was reported by several groups (33,34,35). Greenblatt et al. reported that p53 mutations are more common in breast cancer associated with BRCA1 or BRCA2 germ line mutations than in sporadic breast cancers; however, these observations do not take into consideration the sporadic breast cancer cases where BRCA1 is epigenetically silenced by promoter hypermethylation. Besides p53, also other cell cycle checkpoint and DNA damage response factors can become mutated as a consequence of the epigenetic silencing of BRCA1. If we consider that deficiency in CHK2, a checkpoint kinase that functions in a DNA damage response pathway that result in p53 activation, mimics the loss of p53 and rescues the defective development, growth and cellular demise of BRCA1-deficient T cells at the expense of genomic instability and increased tumorigenicity (36), it will be of great interest to study a possible correlation between the epigenetic silencing of BRCA1 in sporadic breast cancers and mutations in CHK2.
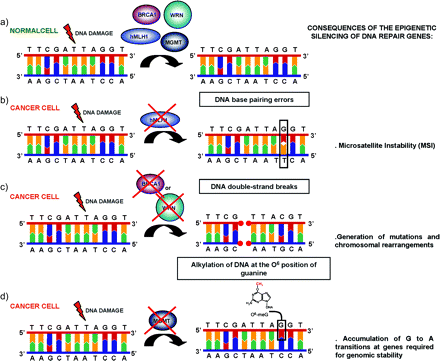
The epigenetic silencing of DNA repair genes uncovers new mutator pathways in human cancer. (a) In a normal cell, DNA lesions occurring in vivo are normally repaired by different DNA repair pathways. (b) The promoter CpG island methylation-associated gene silencing of hMLH1 in sporadic cases of colorectal, endometrial and gastric tumours causes the unusual MSI phenotype. (c) The epigenetic inactivation of BRCA1 in breast and ovarian tumours or the epigenetic inactivation of the WRN protein in various tumour types of both mesenchymal and epithelial cause the accumulation of chromosomal rearrangements and somatic mutations since these two proteins have an important role in the repair of DSBs. (d) The promoter hypermethylation of MGMT in a wide spectrum of human tumours uncovers a new mutator pathway because the O6-methylguanine adducts resulting from alkylating agents are not removed and this consequently causes G:C to A:T transitions that affect genes required for genomic stability.
Epigenetic inactivation of the DNA MMR gene hMLH1
The DNA MMR system corrects DNA base-pairing errors in newly replicated DNA (37). Deficiencies of this system result in mutation rates 100-fold greater than those observed in normal cells (38,39). These mutations are particularly evident in microsatellite sequences consisting of repeats of 1–4 bp. Microsatellite instability (MSI) is thereby a hallmark of MMR gene-deficient cancers. MSI has been observed in ∼13% of sporadic colorectal cancers (CRC) and in virtually all CRC arising in patients with hereditary non-polyposis colorectal cancer (HNPCC) (40,41). In patients with HNPCC, the defect is attributed to germ line mutations in DNA MMR genes, mainly hMLH1 and hMSH2 (42), while other components of the MMR pathway such as hMSH3, hMSH6, hPMS1 and hPMS2 seem to play a minor role in the disease. In MSI+ cancers from patients without HNPCC, these same genes are often mutated and consequently inactivated. However, in a significant subset of sporadic tumours with MSI+, no mutations of MMR genes could be identified (43). The reason for this lack of MMR mutations is that the main cause of MSI in the sporadic cases is the epigenetic inactivation of hMLH1 by promoter hypermethylation. This relationship was reported in colorectal (44,45), endometrial (46) and gastric tumours (47), the three tumour types common in HNPCC patients, while hMLH1 promoter hypermethylation is absent in other tumour types (46). In fact, among the studies described, 90% of MSI+ tumours are methylated at hMLH1 (the remaining 10% probably have somatic mutations in either hMLH1 or hMSH2), while the MSI− tumours are unmethylated in 95% of cases (45,46,47). Further proofs of causality between hMLH1 epigenetic silencing and the MSI+ phenotype came from studies in cancer cell lines, where demethylating agents are not only able to reactivate the hMLH1 gene but also capable of restoring the MMR activity (45).
Abnormalities in the DNA MMR system discussed above were the first well-characterized cause behind the uncover of mutator pathways in human cancer (Figure 2). Since microsatellites and repetitive sequences are frequently present in coding regions, the MSI exhibited by MMR-deficient tumours can be manifested as frameshift mutations that inactivate a variety of genes, including genes that suppress tumour formation, e.g. adenomatovs polyposis coli, transforming growth factor-βRII, hMSH2 and caspase-5 (48,49). Taking this into consideration, we can look to the inactivation of tumour suppressor genes as a final step of a malignant progression characterized by an increase of the genomic instability that is triggered by the early epigenetic inactivation of hMLH1.
Epigenetic inactivation of MGMT
MGMT, also known as AGT, is a DNA repair protein that removes mutagenic and cytotoxic adducts from O6-guanine in DNA (50,51).
Alkylation of DNA at the O6 position of guanine is an important step in the appearance of mutations in cancer, primarily due to the tendency of the O6-methylguanine to pair with thymine during replication, resulting in the conversion of guanine−cytosine to adenine−thymine pairs in DNA (52). Furthermore, the O6-alkylguanine–DNA adduct (especially, the O6-chloroethylguanine) may cross-link with the complementary cytosine residues, blocking DNA replication (53). MGMT protects cells against these lesions, transferring the alkyl group from the O6-guanine in DNA to an active cysteine within its own sequence in a reaction that inactivates one MGMT molecule for each lesion repaired (50). In vitro mutational assays using the adenine phosphoribosyltransferase gene, and in vivo experiments using transgenic mice overexpressing MGMT or knockout mice defective in MGMT function, also demonstrate this genoprotective effect of MGMT (51).
The epigenetic silencing of MGMT by promoter hypermethylation in cancer cell lines and primary human tumours has been reported by several groups (54,55,56) and has been correlated with the loss of MGMT protein (57), lack of mRNA expression (56) and loss of enzymatic activity (58). Furthermore, the CpG island hypermethylation-associated silencing of MGMT occurs very early in human tumorigenesis, such as in small colon adenomas (56), strongly supporting its relevant role in carcinogenesis.
The transcriptional silencing of MGMT by promoter hypermethylation causes an important mutator pathway in human cancer because the O6-methylguanine adducts, resulting from alkylating agents, are not removed and this consequently cause G:C to A:T transitions (Figure 2).
The first gene described to have G:C to A:T transitions as a consequence of MGMT inactivation in human tumours was K-ras (59). Although ras mutation is the most common oncogenic alteration in human cancer (60), the incidence of K-ras activation varies widely among carcinomas. K-ras mutation is rare in human primary breast carcinomas, but occurs in approximately half of colorectal carcinomas. This mutation distribution strongly resembles the pattern of MGMT promoter hypermethylation. While MGMT aberrant methylation is not present in breast carcinomas where K-ras mutations are extremely rare, it occurs in ∼40% of cases of colorectal carcinomas (56) where K-ras mutations are frequent. The association between MGMT promoter hypermethylation and K-ras mutations has been reported not only in colon cancer (61) but also in gastric and gallbladder cancers (62,63).
Other gene that was reported to have G:C to A:T transitions caused by the epigenetic silencing of MGMT in human cancer was the tumour suppressor gene p53 (64). The tumour suppressor gene p53 is the most commonly mutated gene in human cancer, and transition mutations constitute the most common p53 mutations (65). Approximately 52% of the mutational events are missense transitional changes, and, of this subset, ∼72% are G:C to A:T transitions (65). Since the profile of the mutational spectrum varies according to tumour type, there is a very useful p53 mutation database (http://www-p53.iarc.fr/) at the International Agency for Research on Cancer that compiles all p53 gene mutations identified in human cancers and cell lines that have been reported in the peer-reviewed literature since 1989 (66).
Lung and head and neck tumours of smokers have a higher number of transversions, whereas colorectal tumours have the highest rate of transition mutations, reaching 70% of the total number of p53 mutations (65). These last mutations occur frequently in CpG dinucleotides, which are normally methylated (67) through increased rates of spontaneous deamination at methylcytosine, although other mechanisms are also conceivable. However, 17% of p53 mutations are transition mutations in non-CpG dinucleotides, where this causality cannot be invoked (65). Thus, G:C to A:T changes in p53 in non-CpG and CpG dinucleotides could be attributable, in part, to a defect in MGMT that allows the persistence of O6-methylguanine and its reading as an adenine. The link between MGMT promoter hypermethylation and the presence of G:C to A:T transition mutations in p53, particularly in non-CpG dinucleotides, has also been found in glioma (68), liver (69) and non-small-cell lung carcinomas (70).
Epigenetic inactivation of the premature aging WS gene
WS is a rare autosomal recessive disorder characterized by premature onset of age-related pathologies, including hair greying, alopecia, cataracts, osteoporosis, type II diabetes, cardiovascular disorders and mesenchymal neoplasms (71). Mutations in the WS gene (WRN) are found in patients exhibiting the clinical symptoms of WS (72,73). The vast majority of WRN mutations result in loss of function of the WRN protein (74).
The WRN gene product defective in WS belongs to the RecQ family of DNA helicases (72). Mutations in RecQ family members BLM and RecQ4 result in two other disorders associated with elevated chromosomal instability and cancer, Bloom syndrome and Rothmund–Thomson syndrome, respectively (75). RecQ helicase mutants display defects in DNA replication, recombination and repair, suggesting a role for RecQ helicases in maintaining genomic integrity.
The WRN gene encodes a 1432 amino acid protein that has several catalytic activities (76). WRN is a DNA-dependent ATPase and utilizes the energy from adenosine triphosphate hydrolysis to unwind double-stranded DNA. However, unlike other known members of the human RecQ family, WRN is also a 3′ to 5′ exonuclease, consistent with the presence of three conserved exonuclease motifs homologous to the exonuclease domain of Escherichia coli DNA polymerase I and RNase D (76).
Consistent with their ability to act on multiple intermediates in DNA processing, RecQ helicases interact with many proteins involved in DNA metabolism (77). Some are significant functional interactions. For example, the Ku heterodimer strongly stimulates WRN exonuclease (78) and the telomere-binding protein, TRF2, stimulates WRN helicase activity (79). Other proteins, such as p53 and BLM, inhibit WRN exonuclease activity (80). Based on the biochemistry described above and on the known protein partners of WRN, it is likely that the main functions of WRN are in DNA repair and in telomere maintenance (77). In addition, WRN have multiple roles in DNA repair, since is important for the repair of chromosomal DSB by HR, NHEJ and SSA and the repair of SSB by BER (77,81). These multiple roles of WRN protein in DNA repair highlight its importance in aging and cancer.
Because patients with WRN germ line mutations develop a broad spectrum of epithelial and mesenchymal tumours, which is one of the main causes of their death before the age of 50 years, a tumour suppressor function for WRN has been proposed. This putative role is also supported by a very high rate of loss of heterozygosity at the chromosomal WRN loci at 8p11.2–p12 in many tumour types, including colorectal and breast cancer (82,83). However, somatic mutations of WRN have not been described in sporadic neoplasms. More recently, Agrelo et al. (84) reported that the WRN gene undergoes CpG island promoter methylation-associated gene silencing in various tumour types of both mesenchymal and epithelial origin, including those commonly observed in WRN patients (such as osteosarcoma, thyroid and gastric tumours), in a similar fashion to what has been observed with other familial tumour suppressor genes with DNA repair function discussed above, such as hMLH1 or BRCA1 (85).
Since WS cells accumulate chromosomal rearrangements and somatic mutations at an increased rate in an age-dependent manner, and WRN protein has been shown to interact with repair proteins, such as DNA Polβ, Ku and its associated DNA–PKcs, PARP-1 and APE1 (86), we can speculate that the epigenetic inactivation of the WRN gene creates a new mutator pathway in human cancer (Figure 2). This idea is supported by the fact that cancer cells where WRN is silenced by promoter hypermethylation are very sensitive to the action of DNA-damaging agents (84). However, further studies must be done to confirm this hypothesis.
Conclusions
In this review, we have outlined the importance of the epigenetic inactivation of DNA repair genes as a key event in human cancer that uncovers new mutator pathways.
The promoter CpG island methylation-associated gene silencing of hMLH1 in sporadic cases of colorectal, endometrial and gastric tumours that causes the unusual MSI; the promoter hypermethylation of MGMT that prevents the removal of mutagenic and cytotoxic adducts at the O6 position of the guanine and leads to the appearance of K-ras and p53 mutations; the epigenetic inactivation of BRCA1 in breast and ovarian tumours, which generates mutations and gross chromosomal rearrangements; the promoter hypermethylation of the WRN protein in various tumour types of both mesenchymal and epithelial origin, that leads to genomic instability and an increased mutation rate, were the four mutator pathways discussed in this review. However, the list of DNA repair genes that become epigenetically silenced in human cancer is probably uncompleted and future studies will be needed to fill the list that will give us a more global view of the epigenetic events that are capable of modulating the whole environment of a cell.

{kind=link}
{kind=link}