





Abstract
DNA double-strand breaks enhance homologous recombination in cells and have been exploited for targeted genome editing through use of engineered endonucleases. Here we report the creation and initial characterization of a group of rare-cutting, site-specific DNA nucleases produced by fusion of the restriction enzyme FokI endonuclease domain (FN) with the high-specificity DNA-binding domains of AvrXa7 and PthXo1. AvrXa7 and PthXo1 are members of the transcription activator-like (TAL) effector family whose central repeat units dictate target DNA recognition and can be modularly constructed to create novel DNA specificity. The hybrid FN-AvrXa7, AvrXa7-FN and PthXo1-FN proteins retain both recognition specificity for their target DNA (a 26 bp sequence for AvrXa7 and 24 bp for PthXo1) and the double-stranded DNA cleaving activity of FokI and, thus, are called TAL nucleases (TALNs). With all three TALNs, DNA is cleaved adjacent to the TAL-binding site under optimal conditions in vitro . When expressed in yeast, the TALNs promote DNA homologous recombination of a LacZ gene containing paired AvrXa7 or asymmetric AvrXa7/PthXo1 target sequences. Our results demonstrate the feasibility of creating a tool box of novel TALNs with potential for targeted genome modification in organisms lacking facile mechanisms for targeted gene knockout and homologous recombination.
INTRODUCTION
Perhaps the most significant application of endonucleases in the post genome era is their coupling with custom-engineered proteins that recognize long stretches of DNA sequences and their application for targeting specific genes for knockout or gene replacement ( 1 ). The key component of these engineered nucleases is the DNA recognition domain that is capable of precisely directing the nuclease to the target site for the purpose of introducing a DNA double-strand break (DSB). These breaks are principally repaired by one of two pathways, non-homologous end-joining (NHEJ) or homologous recombination (HR). Repair by NHEJ often results in mutagenic deletions/insertions in the targeted gene. Moreover, DSBs can stimulate HR between the endogenous target gene locus and an exogenously introduced homologous DNA fragment with desired genetic information, a process called gene replacement or genome editing ( 2–4 ). To date the most promising method involving genome editing is the use of custom-designed zinc finger nucleases (ZFNs) ( 5 ). ZFN technology involves the use of hybrid proteins derived from the DNA-binding domains of zinc finger (ZF) proteins fused with the non-specific DNA-cleavage domain of the endonuclease FokI ( 6 , 7 ).
FokI, a type IIS nuclease, was first isolated from the bacterium Flavobacterium okeanokoites ( 8 ). The nuclease consists of two separate domains, an N-terminal DNA-binding domain and a C-terminal DNA-cleavage domain. The DNA-binding domain recognizes the non-palindromic sequence 5′-GGATG-3′ while the catalytic domain cleaves double-stranded DNA non-specifically at a fixed distance of 9 and 13 nucleotides downstream of the recognition site ( 9 , 10 ). FokI exists as an inactive monomer in solution and becomes an active dimer upon binding to its target DNA and in the presence of specific divalent metals ( 11 ). It has been proposed that to form a functional complex, two molecules of FokI each bind to their recognition sequence in a double stranded DNA molecule and then dimerize in such a way that the two nuclease domains unite to form a functional endonuclease that cleaves both DNA strands at sites downstream of the FokI recognition sequence ( 12 , 13 , 14 ).
ZFNs can be assembled as modules that are custom-designed to recognize selected DNA sequences, typically 18–24 bp with a 4–7 bp spacer between two half sites ( 15–17 ). Following binding at the preselected site, a DSB is produced by the action of the ZFN’s FokI cleavage domain ( 4 ). ZFN technology has been successfully applied to genetic modification in a variety of organisms, including yeast, plants, mammals and even human cell lines [reviewed in ( 18 , 19 )]. Despite the promise of ZFN technology in basic and applied research, widespread adoption of this technology has been hampered by a bottleneck in custom-engineering zinc fingers that possess the requisite high specificity and affinity for preselected DNA target site. Such engineering is labor intensive, time consuming and associated with high rates of failure ( 20 , 21 ). Since the effectiveness of these endonucleases depends almost solely on their DNA-binding specificity, the required specificity theoretically can be supplanted by any high-fidelity DNA-binding domain when fused with a functional endonuclease domain. The TAL effector proteins from a number of bacterial plant pathogens represent a potential source of modularly constructed DNA-binding domains that could supply the needed DNA-binding specificity.
TAL effectors belong to a large group of highly conserved bacterial proteins that exist in various strains of Xanthomonas sp. and are translocated into host cells by a Type III secretion system and, thus, are called Type III effectors [reviewed in ( 22 )]. Once in host cells, some TAL effectors have been found to transcriptionally activate their corresponding host target genes either for strain virulence (ability to cause disease) or avirulence (capacity to trigger host resistance responses) dependent on the host genetic context ( 23–26 ). Each of these bacterial effectors contains a functional nuclear localization motif and a potent transcription activation domain that are characteristic of eukaryotic transcription activators. Each TAL effector also contains a central repetitive region consisting of varying numbers of repeat units of 34 amino acids (aa). It is this repeat region that recognizes a specific DNA sequence and determines the biological specificity of each effector [ Figure 1 A, and reviewed in ( 27 )]. Each repeat is nearly identical except for two variable amino acids at positions 12 and 13, the so called repeat variable di-residues (RVD) ( 28 ). Recent studies have revealed that the recognition of DNA sequences within the promoter regions of specific host target genes is defined by the repeat regions of TAL effectors. The DNA sequence recognition is based on a fairly simple code where one nucleotide of the DNA target site is recognized by the RVD of one repeat (i.e. one repeat/one nucleotide recognition). The sequential tandem array of repeats thus specifies the DNA sequence that will be bound ( 28 , 29 ). The majority of naturally occurring TAL effectors contains repeat units in a range of 13–29 that presumably recognize DNA elements consisting of the same number of nucleotides. Thus, in theory, the so called TAL recognition code can be used as a guide for custom-design of novel TAL effectors with the required specificity to target a single locus in a genome.
![Schematics of TAL effectors AvrXa7 and PthXo1 as well as their fusions with FN. ( A ) A typical TAL effector contains a central region of 34 aa direct repeats (open boxes) and three nuclear localization motifs (NLS, black bars) as well as a transcription activation domain (AD, red solid box) at the C-terminus. The representative 34 aa repeat is shown below with the variable amino acid residues at the positions 12 and 13 in red. ( B ) and ( C ) AvrXa7 and PthXo1 contain central 26 and 24 tandem repeats (shown as boxes) of 34 aa residues, respectively. The repeats are highly conserved, except for residues at positions 12 and 13 (asterisk, missing amino acid residue at position 13). The sequence recognition of AvrXa7 to Os11N3 (and PthXo1 to Os8N3 ) is dictated by the order of the 34 aa repeat units that each contain at positions 12 and 13, a pair of amino acids that exactly recognize one nucleotide at one position in the DNA sequence of the EBE of the Os11N3 gene. ( D–F ) Schematics of TAL effector fusion proteins with FN domain at the N-terminus of AvrXa7 (D) or at the C-termini of AvrXa7 (E) and PthXo1 (F). ( G ) Transient activation by AvrXa7 and FN-AvrXa7 of the Os11N3 promoter (containing the AvrXa7 EBE) driving a GFP reporter gene (GFP-EBE). The plasmid containing the GFP reporter gene was co-injected along with a plasmid containing a gene encoding the AvrXa7 or AvrXa7-FN driven by the CaMV 35S promoter [or with a plasmid containing the 35S promoter but no AvrXa7 coding sequence (Control)]. The dotted line indicates the edge of the inoculation site.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/nar/39/1/10.1093_nar_gkq704/1/m_gkq704f1.jpeg?Expires=1716534452&Signature=Mz~25CEERvfzQxLvyH05KZDKdE27M24Stjcsn7QQOkN-31bh4mNnXb1O1KBFKcAzycl-LF6wXy4pfCjIOlhB2Cod8zB5feHp4X96DbZ9pe8iYOWaXL2pBCSTlRGS~pfTooZt7k2iiZGRLjGmn0vA13xZmUskV7iGwdle2ZFikQ1rgiW7zE-U6GmGaKh2zVMafyVjddSVOfJhhvacgOq5149vN-ucZrIM4hoVu-z3QTJWWAt5DxY4-kViaPMhWmiMOUiSPoSIA-6WUai9G7l-2cDbKV8FCujgnQXxKBk~75feNcwRM-QH~cyC25GtK~vGq2xFlYCHOe0LiipIxFpsWQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Schematics of TAL effectors AvrXa7 and PthXo1 as well as their fusions with FN. ( A ) A typical TAL effector contains a central region of 34 aa direct repeats (open boxes) and three nuclear localization motifs (NLS, black bars) as well as a transcription activation domain (AD, red solid box) at the C-terminus. The representative 34 aa repeat is shown below with the variable amino acid residues at the positions 12 and 13 in red. ( B ) and ( C ) AvrXa7 and PthXo1 contain central 26 and 24 tandem repeats (shown as boxes) of 34 aa residues, respectively. The repeats are highly conserved, except for residues at positions 12 and 13 (asterisk, missing amino acid residue at position 13). The sequence recognition of AvrXa7 to Os11N3 (and PthXo1 to Os8N3 ) is dictated by the order of the 34 aa repeat units that each contain at positions 12 and 13, a pair of amino acids that exactly recognize one nucleotide at one position in the DNA sequence of the EBE of the Os11N3 gene. ( D–F ) Schematics of TAL effector fusion proteins with FN domain at the N-terminus of AvrXa7 (D) or at the C-termini of AvrXa7 (E) and PthXo1 (F). ( G ) Transient activation by AvrXa7 and FN-AvrXa7 of the Os11N3 promoter (containing the AvrXa7 EBE) driving a GFP reporter gene (GFP-EBE). The plasmid containing the GFP reporter gene was co-injected along with a plasmid containing a gene encoding the AvrXa7 or AvrXa7-FN driven by the CaMV 35S promoter [or with a plasmid containing the 35S promoter but no AvrXa7 coding sequence (Control)]. The dotted line indicates the edge of the inoculation site.
AvrXa7 and PthXo1 are TAL Type III effectors from Xanthomonas oryzae pv. oryzae (Xoo), the causal pathogen of bacterial blight of rice. They each contain a unique combination of RVDs in 26 and 24 repeats, respectively ( Figure 1 B and C). For some Xoo strains, AvrXa7 is a key virulence factor in susceptible rice. On the other hand, it is also an avirulence determinant in the otherwise resistant plants containing the cognate resistance gene Xa7 ( 30 , 31 ). As the essential virulence factor, AvrXa7 activates the rice gene Os11N3 to induce a state of disease susceptibility (G.Anthony et al ., manuscript in preparation). The gene induction by AvrXa7 is mediated through its recognition of a specific DNA element within the promoter region of Os11N3 , an element we refer to here as the effector binding element (EBE) (sequence shown in Figure 1 B). Similarly, PthXo1 is also an essential virulence factor for some strains of X. oryzae pv. oryzae and activates the rice gene Os8N3 to promote bacterial multiplication and disease development ( 24 ). PthXo1 recognizes an EBE of 24 nt within the promoter of Os8N3 ( 32 ) (sequence shown in Figure 1 C).
As a proof-of-principle, we have tested the feasibility of generating a new type of DNA sequence-specific endonuclease by utilizing the sequence specificities of AvrXa7 and PthXo1 and the catalytic activity of the endonuclease FokI. Here we report the creation of TAL effector nucleases (TALNs) by fusing the full-length AvrXa7 TAL effector or the full-length PthXo1 TAL effector to the FokI nuclease domain and the characterization of their nuclease activities both in vitro and in vivo using a yeast cell assay.
MATERIALS AND METHODS
Chimeric gene construction
Chimeric genes encoding fusions of TAL effector AvrXa7 or PthXo1 with the FokI nuclease (FN) domain at either the N-terminus or the C-terminus (i.e. FN-AvrXa7, AvrXa7-FN or PthXo1-FN) were constructed using standard Escherichia coli strains and DNA techniques ( 33 ). The full-length AvrXa7 was first modified with PCR primers Tal-F and Tal-R to integrate the restriction sites KpnI and BglII upstream of the start codon at the 5′-end and HindIII, XbaI and a stop codon containing SpeI at the 3′-end based on the plasmid pZWavrXa7 ( 30 ). AvrXa7 without its repetitive central region was PCR amplified using primers Tal-F and Tal-R and cloned into pBluescript KS using KpnI and SpeI. Then the central repeat coding region was cloned back into the plasmid lacking the central repeat element using SphI, resulting in the plasmid pSK/AvrXa7. The SphI fragment for central repeat domain of AvrXa7 in pSK/AvrXa7 was replaced with that of PthXo1 from pZWpthXo1 ( 24 ), resulting in the plasmid pSK/PthXo1. The DNA fragment encoding the DNA-cleavage domain (amino acids 388–583) of FokI (NCBI accession number J04623) was PCR amplified using the primers Fokn-F1 and Fokn-R1 and a plasmid containing the FokI gene as template. Fokn-F1 contained the restriction sites KpnI and BglII, while Fokn-R1 contained a BamHI restriction site. The product was cloned into the A/T cloning vector pGEM-T (Promega, Madison, WI, USA). The KpnI and BamHI digested DNA fragment encoding FN was cloned into KpnI and BglII treated pSK/avrXa7 resulting in pSK/FN-AvrXa7 which contained the chimeric gene with the FN coding region at its 5′ and AvrXa7 at its 3′-end. Similarly, FN coding sequence for C-terminal fusion of AvrXa7 and PthXo1 was PCR amplified with primers Fokn-F2 and Fokn-R2, which contain restriction sites for HindIII and SpeI, respectively. The HindIII and SpeI digested FN fragment was cloned into pSK/AvrXa7 and pSK/PthXo1 individually at their 3′-ends. The accuracy of all PCR products was confirmed by sequencing. Primer sequences are provided in the Supplementary Table S1 .
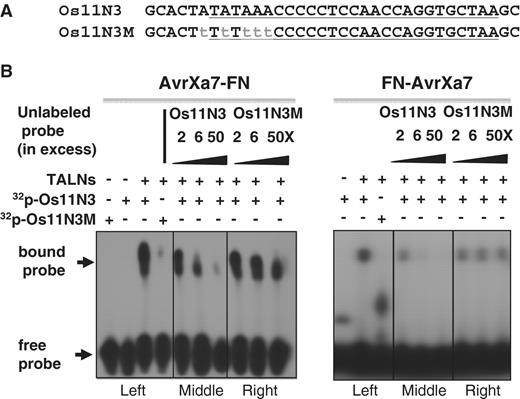
Binding specificity of AvrXa7-FN and FN-AvrXa7 fusion proteins to their target DNA. ( A ) Sense-strand sequences of the wild type (Os11N3) and mutant (Os11N3 M) oligonucleotide duplexes used in the EMS assays. ( B ) EMS assays demonstrating specificities of AvrXa7-FN and FN-AvrXa7 binding to the AvrXa7 EBE sequence. The set of gel images to the left depict results with AvrXa7-FN, while set of gel images to the right depict results with FN-AvrXa7. The left panels of each set show binding of AvrXa7-FN and FN-AvrXa7 to the authentic, 32 P-labeled, Os11N3 DNA target element but not to the mutated target. Competition assays (middle and right panels of each set) showing that the binding of 32 P-labeled Os11N3 EBE target by AvrXa7-FN and FN-AvrXa7 is effectively competed by excess amounts of non-radioactive Os11N3 oligonucleotides (middle panels of each set), but not by the non-radioactive mutated version of Os11N3 DNA (right panels). Positions of the bound and free probes are indicated at the left of the autoradiograph.
Transient expression assay for DNA-binding activity of FN-AvrXa7
A construct containing green fluorescence protein (GFP) reporter gene cloned downstream of Os11N3 promoter, which contains the AvrXa7 EBE sequence was made as follows. The coding region for the GFP gene in plasmid pEGFP (Clontech Laboratories, Mountain View, CA, USA) was PCR amplified using primers GFP-F and GFP-R and cloned into pGEM-T for sequence confirmation. The GFP coding region with added restriction sites was cloned downstream of the promoter region containing the AvrXa7 EBE and upstream of the Os11N3 terminator, resulting in pEBE-GFP. The GFP expression cassette was then cloned into pCAMBIA1300 (CAMBIA) at KpnI and HindIII restriction sites. The resulting construct was transformed into Agrobacterium tumefaciens strain EHA105 to create a ‘reporter’ Agrobacterium strain. DNA encoding FN-AvrXa7 was cloned downstream of the cauliflower mosaic virus (CaMV) 35S promoter in a modified pCAMBIA1300 vector and mobilized into EHA105 to create an ‘effector’ Agrobacterium strain. The effector strain containing AvrXa7 (lacking the FN domain) was similarly constructed to serve as a positive control. The reporter and the effector strains were co-infiltrated into Nicotiana benthamiana leaves. The inoculated leaves were checked for expression of GFP using a Leica M205 FA fluorescent stereomicroscope.
Expression and purification of FN-AvrXa7, AvrXa7-FN and PthXo1-FN
The chimeric gene FN-AvrXa7 was cloned into pPROEX HTb (Invitrogen, Carlsbad, CA, USA) by ligating the BglII and SpeI digested FN-AvrXa7 fragment from pSK/FN-AvrXa7 into the BamHI and SpeI digested expression vector. Similarly, the BglII and SpeI digested AvrXa7-FN DNA fragment was cloned into pPROEX HTb for AvrXa7-FN overexpression. For PthXo1-FN, the expression vector pET28a (Novagen, Madison, WI, USA) was used. The expression constructs were transformed into E. coli strain BL21 (DE3) for overexpression of the recombinant proteins with induction by isopropyl-1-thio-β- d -galactopyranoside (IPTG) following the manufacturer’s instructions (Invitrogen). The 6Xhistidine tagged FN-AvrXa7, AvrXa7-FN and PthXo1-FN were purified with Ni-NTA agarose (Qiagen, Valencia, CA, USA) and the protein concentrations were determined using the BioRad Bradford protein quantification kit (BioRad, Hercules, CA, USA).
Electromobility shift assay
The complementary oligonucleotides of Os11N3-F & Os11N3-R containing AvrXa7 EBE and Os11N3 M-F & Os11N3 M-R containing a mutated AvrXa7 EBE were annealed, respectively, and 5′-end labeled with [γ- 32 P]ATP catalyzed by T4 polynucleotide kinase. The labeled oligonucleotide duplex DNA was mixed individually with AvrXa7-FN and FN-AvrXa7 in a 10 μl reaction volume containing Tris–HCl (15 mM, pH 7.5), KCl (40 mM), DTT (1 mM), glycerol (2.0%), poly(dI.dC) (50 ng/ul), EDTA (0.2 mM), 32 P-labeled DNA (final concentration, 5 fmol) and hybrid protein (final concentration, ∼15 fmol). Unlabeled oligonucleotides were used as competitor probes and were added in increasing amounts (final concentration, 0–250 fmol) to successive reactions. The binding reactions were kept at room temperature for 30 min before loading onto a 6% TBE polyacrylamide gel. After electrophoresis, the gel was exposed to X-ray film for radioactive image capture.
In vitro DNA cleavage
A 406 bp genomic region of the rice Os11N3 gene encompassing the AvrXa7 EBE was PCR amplified with the forward primer Os11N3P-F, and the reverse primer Os11N3P-R, then cloned into the cloning vector pTOPO (Invitrogen), resulting in pTOP/11N3. The plasmid was also used to generate pTOP/Os11N3m2 using linker scanning mutagenesis method with an insertion of 6 nt (5′-cccggg-3′) within the AvrXa7 EBE site. A DNA fragment containing the dual EBEs for PthXo1 and AvrXa7 in a tail-to-tail orientation (TD) was cloned into pPCR Script Amp (Stratagene, Santa Clara, CA, USA), resulting in the plasmid pEBE-TD. For pTOP/Os11N3 and pTOP/Os11N3m2 digestions, the sequenced clone was digested to completion with EcoNI or MluI and purified, 1 μg of uncut plasmid or the digested DNA was incubated individually with FN-AvrXa7 (225 ng or otherwise indicated in the text) and AvrXa7-FN (200 ng) in a volume of 15 μl. The buffer condition was the same as used for the electromobility shift (EMS) assay described earlier, but in the presence of 2.5 mM MgCl 2 .
In vitro digestions of pEBE-TD with TALNs AvrXa7-FN and PthXo1-FN were performed differently. Assays for TALN activities with or without post-digestion with restriction enzyme BglI were conducted in vitro using 1 µg of pEBE-TD supercoiled plasmid DNA in a 30 µl reaction volume containing Tris–HCl (20 mM), pH 8.5, NaCl (150 mM), MgCl 2 (2 mM), glycerol (5%), BSA (0.5 mg/ml), DTT (1 mM) and 5 µg of Ni + -affinity column-purified PthXo1-FN and/or AvrXa7-FN incubated for 30 min at 37°C. For reactions in which BglI was used after TALN digestion, 10 U of BglI were added and digestion continued for an additional 20 min. For reactions in which both PthXo1-FN and AvrXa7-FN were used simultaneously, the reaction conditions were identical except that only 1 µg of each TALN was employed.
Yeast HR assay
The yeast strains YPH499 ( MAT a ura3-52 lys2-801_amber ade2-101_ochre trp1-Δ63 his3-Δ200 leu2-Δ1) and YPH500 ( MAT α ura3-52 lys2-801_amber ade2-101_ochre trp1-Δ63 his3-Δ200 leu2-Δ1 ) as well as expression vectors (pCP3, pCP4 and pCP5) were described and kindly provided by Dr Dan Voytas ( 34 ). The pCP5 derived reporter construct containing a single AvrXa7 EBE was made by inserting the annealed oligonucleotides (EBES-F and EBES-R) into the BglII- and SpeI-digested pCP5. A set of duplexes of oligonucleotides (x7-HDn1-F and x7-HDn1-R, n1 = 6, 10, 14, 19, 24, 30, 35 and 40 bp) containing the dual AvrXa7 EBEs in a head-to-head (corresponding to FN domain to FN domain of FN-AvrXa7) orientation separated by a serial of spacers (n1) were cloned into the BglII and SpeI digested pCP5 plasmid. Similar constructs with two AvrXa7 EBEs in a TD and separated by various lengths (2, 5, 8 and 19 bp) of spacers were also made using duplexes of oligonucleotides (x7-TDn2-F and x7-TDn2-R, n2 = 2, 5, 8 and 19 bp). For the 19 bp spacer configuration, an additional one reporter construct with a mutation in one AvrXa7 EBE and another with a mutation in each of the AvrXa7 EBEs were made. A third set of dual asymmetric EBEs with one corresponding to AvrXa7 and the other to PthXo1 in a TD were also cloned into pCP5 using PstI and SpeI. The duplexes of oligonucleotides are x7/o1-TDn3, n3 = 6, 11, 16, 21, 26, 31 and 36 bp. The expression vectors pCP3 and pCP4 were first modified with a linker sequence containing multiple cloning sites (MCSs) downstream of the translation elongation factor 1α (TEF1) promoter, resulting in pCP3M and pCP4M, respectively. The linker was made by annealing two oligonucleotides (Linker-F and Linker-R) and cloned into the XbaI and XhoI digested pCP3 and pCP4 individually. The chimeric genes FN-AvrXa7 and AvrXa7-FN were digested with BglII and SpeI and cloned into the BamHI and SpeI digested pCP3M vector, while the chimeric gene PthXo1-FN was cloned into pCP4M by BglII and SpeI. The reporter plasmids each were transformed into the yeast mating strain YPH500 (MAT-α) and effector plasmids in a combination of pCP3M-FN-AvrXa7/pCP4M, pCP3M-AvrXa7-FN/pCP4M and pCP3M-AvrXa7-FN/pCP4M-PthXo1-FN into YPH499 (MATa). Yeast cells from a single yeast colony carrying the reporter plasmid with little β-galactosidase background was mixed with cells from single colony with the effector plasmid (in a combination indicated in text) in triplicate on yeast nutrient medium (YPD) overnight, then cultured in synthetic complete medium lacking leucine, histidine and tryptophan to select for mated cells. The cells were then harvested for quantitative measurement of β-galactosidase activity by using the yeast β-galactosidase assay kit from Thermo Fisher Scientific (Rockford, IL, USA) following the manufacture’s manual. Enzyme activity is calculated based on the equation: β-galactosidase activity = (1000 × A 420 )/( t × V × OD 660 ), t = time of incubation in minutes, V = volume of cells used in the assay in ml.
As a positive control to our yeast assay for TALN activity, we also constructed an effector plasmid containing a ZFN and a reporter plasmid containing the corresponding ZF-binding site. A DNA fragment encoding the ‘original’ BCR-ABL three-finger array was obtained by the digestion of pGP-FB-orig BA ( 35 ) with XbaI and BamHI and cloned into pCP3, resulting in a translational fusion of BCR-ABL with a C-terminal FokI nuclease domain. The oligonucleotide duplex containing the dual ZFN target sites in a TD separated by a 6 bp spacer was cloned into pCP5 by BglII and SpeI, resulting in the reporter plasmid. The effector plasmid pCP4-ZFN and pCP3 M were transformed into YPH499. The transformants were mated with YPH500 containing the reporter plasmid pzf-TD6 and measured for β-galactosidase activity as described earlier for TALNs.
Sequence information for oligonucleotide duplexes employed is provided in Supplementary Figure S4 .
RESULTS
Construction of the chimeric genes for FN-AvrXa7, PthXo1-FN and PthXo1-FN
AvrXa7 and PthXo1 are two naturally occurring TAL effectors containing a central region of 26 and 24 repeat units, respectively, with, like their relatives, the last repeat containing only the first 20 aa residues similar to other repeats. The specific arrays of 26 RVDs in AvrXa7 and 24 RVDs in PthXo1 make the target sequences of AvrXa7 and PthoXo1 unique in comparison with all other TAL effectors [ Figure 1 B and C; ( 26 )]. AvrXa7 binds to a specific 26 bp promoter element, EBE, in the rice Os11N3 gene through its DNA-binding repeats [ Figure 1 B; ( 32 ); and G.Anthony et al ., manuscript in preparation], while PthXo1 binds to a specific 24 bp sequence in the promoter region of Os8N3 ( 24 ) . The nucleotide ‘T’ that precedes all known TAL effector target sites and is essential for the target gene activation ( 28 , 29 ) is also present immediately upstream of the first nucleotide of EBEs of Os8N3 and Os11N3 and, therefore, is treated as part of EBE throughout this article. We reasoned that a chimeric protein composed of AvrXa7 or PthXo1 and a DNA cleavage domain of an endonuclease might function in recognizing the Os11N3 and Os8N3 target sequences, respectively, and cleaving DNA adjacent to the recognition site. The DNA-cleavage domain of the endonuclease FokI was chosen due to its well-documented non-specific catalytic activity when linked with other DNA-binding domains, such as zinc finger proteins. Reflecting the configuration of the chimeric protein we designed, FN-AvrXa7 was constructed by fusing the DNA sequence encoding the full-length AvrXa7 TAL effector downstream of the DNA sequence encoding the cleavage domain of FokI. AvrXa7 and PthXo1 constructs were also made that each express hybrid proteins with the FN domain at the C-terminus of each TAL effector. The resulting chimeric genes were predicted to encode proteins of 1645, 1648 and 1574 aa residues, respectively ( Figure 1 D–F). The 196 aa FokI domain is linked by 4 aa residues to the 1459 aa of AvrXa7 in FN-AvrXa7 and by 2 aa in both AvrXa7-FN and PthXo1-FN (also see Supplementary Figure S1 for the complete nucleotide and amino acid sequences of FN-AvrXa7, AvrXa7-FN and PthXo1-FN).
Transcription enhancement by FN-AvrXa7 in vivo
In addition to a DNA-binding domain, the TAL effectors contain a potent C-terminal transcription activation domain ( 27 ) . Thus, we reasoned that by placing the FokI nuclease domain at the N-terminus of TAL effectors, the activation domain would remain functional. We could then take advantage of transcription enhancement as an indirect measure of the DNA-binding ability of the hybrid protein, FN-AvrXa7 in this case, or of any newly synthesized TAL fusion protein, in general.
We adapted a modified A. tumefaciens mediated transient expression assay that has been successfully used for studying the interaction of TAL effectors with their target host genes ( 26 , 29 ). In our case, the ‘reporter’ construct contained the gene encoding a GFP under the control of the Os11N3 promoter that contains the AvrXa7 EBE. Two ‘effector’ constructs were made to express either AvrXa7 or FN-AvrXa7 under control of the strong and constitutive CaMV 35S promoter. Both reporter and effector genes were delivered by injection of Nicotiana benthamiana leaves with A. tumefaciens to allow co-expression of genes from the effector and reporter constructs. Both AvrXa7- and FN-AvrXa7-containing constructs induced the expression of GFP while the construct lacking either AvrXa7 or FN-AvrXa7 did not ( Figure 1 G). These results indicated that the hybrid FN-AvrXa7 retained the DNA-binding ability of AvrXa7, and that the transient expression assay could provide a means to test the DNA-binding ability or specificity of TAL-derived hybrid proteins in vivo .
Overexpression and purification of FN-AvrXa7, AvrXa7-FN and PthXo1-FN
The FN-AvrXa7 and AvrXa7-FN were each cloned into an overexpression vector in frame with an N-terminal 6Xhistidine tag to allow for affinity chromatography purification on a Ni-containing column after overproduction of the TAL hybrid protein in E. coli . The proteins were successfully isolated in a relatively high purity ( Supplementary Figure S2 ). The identity of FN-AvrXa7 and AvrXa7-FN were further confirmed with protein blot analysis using an antibody against the FLAG epitope that was integrated after the repeat regions of AvrXa7 and PthXo1 ( Supplementary Figure S2B and C ). The FLAG-tagged proteins detected were ∼175 KDa, the calculated sizes of FN-AvrXa7 and AvrXa7-FN, respectively. IPTG-induced E. coli cells overexpressing FN-AvrXa7 or AvrXa7-FN did not exhibit any obvious growth defects (data not shown). Overexpression and purification of PthXo1-FN were achieved in a similar fashion to that for FN-AvrXa7 and AvrXa7-FN.
DNA-binding activities of AvrXa7-FN and FN-AvrXa7 in vitro
AvrXa7-FN and FN-AvrXa7 purified from E. coli were tested for their DNA-binding specificities in vitro . The abilities of purified AvrXa7-FN and FN-AvrXa7 to bind DNA targets in vitro were investigated using double-stranded oligonucleotides containing the AvrXa7 EBE of Os11N3 or its mutated version, Os11N3 M, containing 5 nt substitutions near the 5′-end of its EBE sequence ( Figure 2 A). The binding reactions were carried out in the solution lacking divalent metal cations such as Mg 2+ to prevent cleavage of the oligonucleotides (‘Materials and Methods’ section). The EMS assay demonstrated that AvrXa7-FN and FN-AvrXa7 preferentially bound to the labeled double stranded DNA containing the authentic AvrXa7 EBE target sequence but did not effectively bind to the probe containing the mutated target sequence containing five nucleotide changes near the 5′-end of the EBE ( Figure 2 B, left panels). Furthermore, AvrXa7-FN and FN-AvrXa7 binding to the 32 P-labeled AvrXa7 EBE could be competed away using increasing amounts of unlabeled DNA nucleotides of the same sequence ( Figure 2 B, middle panels). However, the binding to the labeled Os11N3 was not competed away with an excess of the variant oligonucleotide, Os11N3 M ( Figure 2 B, right panels).
DNA cleavage activities of FN-AvrXa7, AvrXa7-FN and PthXo1-FN in vitro
We also tested the abilities of FN-AvrXa7, AvrXa7-FN and PthXo1-FN to cleave substrate DNA individually or in combination in vitro . For experiments with individual FN-AvrXa7 and AvrXa7-FN TALNs, we chose a plasmid containing a cloned DNA fragment of the Os11N3 promoter from rice. The plasmid pTOP/Os11N3 was first linearized at a unique restriction site (EcoNI) or digested with MluI ( Figure 3 A and B, lanes 2 and 3). The DNA was then incubated with FN-AvrXa7 or AvrXa7-FN at 37°C for 1 h using the EMS assay buffer with the addition of 2.5 mM MgCl 2 . The FN-AvrXa7 and AvrXa7-FN each cleaved the EcoNI-linearized pTOP/Os11N3 plasmid into two fragments of the expected size (0.8 and 2.1 kb) ( Figure 3 B, lanes 5 and 8). Incubation of the supercoiled pTOP/11N3 ( Figure 3 B, lane 1) with an appropriate amount of FN-AvrXa7 or AvrXa7-FN resulted in partial linearization of the plasmid ( Figure 3 B, lanes 4 and 7, respectively). Individual FN-AvrXa7 and AvrXa7-FN also cleaved the MluI predigested pTOP/11N3 into an expected pattern of DNA fragments ( Figure 3 B, lanes 6 and 9, respectively). Taken together, the results indicate that both FN-AvrXa7 and AvrXa7-FN mediated-cleavages occur at the AvrXa7 EBE site.
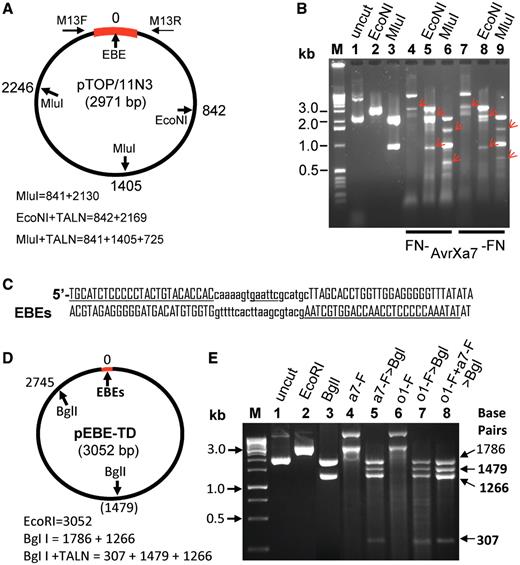
Target DNA digestion by the FN-AvrXa7, AvrXa7-FN and PthXo1-FN TALNs. ( A ) Circular plasmid containing a 400 bp Os11N3 promoter region (thick bar in red) with one AvrXa7 EBE site (position 0). EcoNI cuts once at position 842 and MluI cuts twice at positions 1405 and 2246 relative to EBE of the plasmid. The expected sizes of fragments after cleavages are shown below the plasmid map. ( B ) Gel image of plasmid DNA treated with different enzymes individually or in combinations. M, 1 kb markers with size labeled on the left side. Different treatments are indicated above lanes 1–9 and in combination with TALNs labeled below lanes 4–9. Arrows in red indicate DNA fragment sizes expected if the NF-AvrXa7 or AvrXa7-FN nuclease cleavage takes place correctly at the AvrXa7 EBE target site in pTOP/Os11N3. ( C ) Structure of adjacent PthXo1 and AvrXa7 EBE sites (underlined) in a TD (to allow FN to FN proximity) and separated by 19 spacer nucleotides (lower case letters) which contains a unique EcoRI site (underlined). ( D ) Map of plasmid pEBE-DT with predicted cleavage sites for EcoRI (position 0), PthXo1-FN and/or AvrXa7-FN EBEs and BglI (positions 1479 and 2745). ( E ) Gel image of intact pEBE-DT (lane 1), EcoRI–linearized plasmid ( 2 ), BglI-cleaved plasmid ( 3 ), AvrXa7-FN digested plasmid DNA ( 4 ), AvrXa7-FN digested plasmid followed by BglI digestion ( 5 ), PthXo1-FN digested plasmid ( 6 ), PthXo1-FN digested plasmid followed by BglI digestion ( 7 ) and double digestion with PthXo1-FN and AvrXa7-FN (at one-fifth the concentrations of each protein used individually in reactions for lanes 3 to 6) followed by BglI digestion ( 8 ). The label ‘M’ indicates the DNA marker lane with marker sizes provided in kilo-base (kb). Arrows to the right indicate sizes of DNA fragments expected from complete DNA digestion with BglI and partial digestion with PthXo1-FN (lane 5), AvrXa7-FN (lane 7) or PthXo1-FN combined with AvrXa7-FN (lane 8).
To investigate the specificity of cleaving action by FN-AvrXa7 in more detail, a plasmid containing a mutated binding site of AvrXa7 was generated from pTOP/Os11N3 using a linker scanning mutagenesis method to contain a 6 bp insertion immediately downstream of the initial ‘T’ residue of the Os11N3 EBE ( Supplementary Figure S3A ). This EBE mutation abolished the ability of AvrXa7 to elicit GFP gene expression when placed in the Os11N3 promoter in our Os11N3 promoter assay (data not shown), which is consistent with the finding that the ‘T’ immediately upstream of the first EBE nucleotide is essential for the TAL effector mediated activation of target genes ( 29 ). Incubation of the same amount (1 μg) of EcoNI pretreated DNA of pTOP/Os11N3, the mutant pTOP/Os11N3m2 and pTOP/GFP (a plasmid containing a GFP gene sequence unrelated to the AvrXa7 EBE) with an appropriate amount of FN-AvrXa7 resulted in digestion of pTOP/Os11N3, but not the mutant- and GFP-containing plasmids ( Supplementary Figure S3B ). The specific and expected cleavage pattern observed with digestion of pTOP/Os11N3 DNA was not observed with the mutant pTOP/Os11N3m2 even with increasing amounts of FN-AvrXa7 protein ( Supplementary Figure S3C ). These experiments demonstrate that the FN-AvrXa7 is a highly selective endonuclease, having the ability to cleave double stranded DNA while discerning the preferred target site from a slight variant or unrelated DNA sequence.
Additional evidence for specific cleavage by TALNs in close proximity to EBE sites has come from cleavage patterns of DNA sequences that are targets for PthXo1-FN as well as AvrXa7-FN and PthXo1-FN in combination. Cleavages of the EBE site ( Figure 3 C) of plasmid pEBE-TD ( Figure 3 D) with either of the two TALNs (or EcoRI) cause linearization (or partial linearization) of the plasmid to produce a band of 3052 bp ( Figure 3 E, lanes 2, 4 and 6). To define the site of TALN-mediated DNA cleavage, post treatment of TALN-digested plasmid DNA with BglI was employed. BglI alone produces DNA fragments of 1786 and 1266 bp ( Figure 3 E, lane 3). Incubation with AvrXa7-FN alone results in incomplete digestion of the supercoiled plasmid and produces a band of DNA migrating at ∼3 kb along with another band migrating with an apparent molecular size of >10 kb (lane 4). When BglI is added to the digestion mix 30 min after cutting with AvrXa7-FN has begun, four DNA fragments are observed that correspond to sizes interpreted to be 1786, 1479, 1266 and 307 bp ( Figure 3 E, lane 5). This pattern is fully compatible with the interpretation that the initial cutting by AvrXa7-FN was at the EBE target site, but was only partially complete. Post incubation with BglI resulted in two BglI-BglI DNA fragments of 1786 and 1266 bp and two AvrXa7-FN-BglI bands of 1479 and 307 bp. When pEBE-TD is incubated with PthXo1-FN three DNA fragments are observed ( Figure 3 E, lane 6), a weak band co-migrating with uncut supercoiled plasmid, a band co-migrating with linearized DNA and a band >10 kb. When BglI is subsequently added to the reaction, again four bands of DNA are observed ( Figure 3 E, lane 7). Double digestion of pEBE-TD with AvrXa7-FN and PthXo1-FN using only one-fifth the amount of each TALN used in the reactions whose products are displayed in lanes 4 through 7, results once more in the appearance of four bands, but with somewhat greater representation of bands of molecular sizes of 1479, 1266 and 307 bp—and significantly less non-specific DNA cleavage (compare lanes 5 and 7 with lane 8). The possible nature of the DNA band migrating with an apparent molecular size >10 kb is considered in the ‘Discussion’ section below.
To further identify the major cleavage sites of the sense and antisense strand, the cleaved DNA fragments (expected sizes of ∼840 and 2120 bp) derived from pTOP/Os11N3 treated independently with AvrXa7-FN and FN-AvrXa7 were purified and subjected to sequencing. Each band was expected to contain part of the Os11N3 promoter fragment, but it was unclear which contained the actual target site. Primers that flank the original 0.4 kb Os11N3 promoter fragment were chosen to sequence each of the digested plasmid bands. These primers presumably were able to sequence through the entire EBE site if it was present. For AvrXa7-FN cleavage, the right side primer (M13 R on pTOPO) was used to sequence the sense strand of the 0.8 kb fragment, while M13F was used to sequence the antisense strand of the 2.1 kb fragment ( Figure 4 A). The reverse complementary DNA sequencing trace ( Figure 4 A, the upper chromatograph) from the sense strand of the 0.8 kb fragment ends 2 bp downstream of the last EBE nucleotide. On the other hand, the sequence from the antisense strand of 2.1 kb fragment ends 17 bp downstream of last EBE nucleotide ( Figure 4 A, lower chromatograph), resulting in a 15 bp cutting ‘zone’ of each DNA fragment. These results indicate that the major cleavages of double stranded DNA by the action of AvrXa7-FN occur downstream of the EBE site, a finding that is consistent with the expected locations of the FokI nuclease domain and of the AvrXa7 binding domain given the configuration of the two components. For FN-AvrXa7 cleavage ( Figure 4 B), the sequence trace from the antisense strand of the 1.2 kb fragment ends 14 bp upstream of the first nucleotide of the EBE ( Figure 4 B, lower chromatograph), while the sequencing trace (reverse complementary to the original trace) generated from the sense strand of the 0.8 kb fragment terminated completely after the sixth nucleotide upstream of the last nucleotide of the EBE sequence ( Figure 4 B, upper chromatograph), but within the EBE site. These latter cutting sites were unexpected given the position of the FN domain relative to the FN-AvrXa7 DNA-binding site and the presumed protection of the site by the AvrXa7 binding. Repeated sequencing of DNA fragments from additional experiments yielded similar results and, thus, provide no explanation for the observed, but unexpected, DNA-cleavage pattern with FN-AvrXa7.
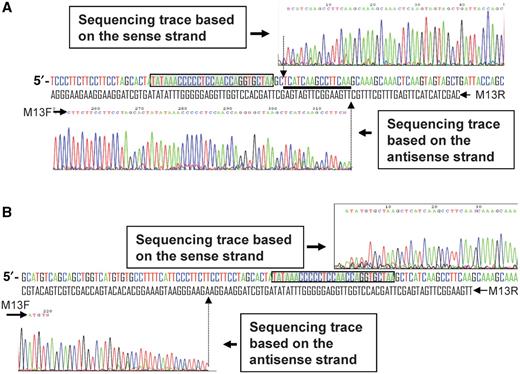
DNA sequencing revealing the major cleavage sites created by the AvrXa7-FN ( A ) and FN-AvrXa7 ( B ). (A) DNA sequencing chromatogram above the original Os11N3 dsDNA sequence (sense strand colored for ease of viewing) is based on the sense strand of the 0.8 kb fragment purified from lane 8 in Figure 3 B. The M13 R primer was used for sequencing the sense strand. The chromatogram, which represents the sense strand sequence around the cleavage site, is in reverse-complement orientation for ease of viewing. The chromatogram below the dsDNA sequence is derived from the antisense strand of the 2.1 kb DNA fragment (also from lane 8 in Figure 3 B) to the left of the predicted AvrXa7-FN cleavage site. The AvrXa7-FN binding site is boxed in shaded gray. The vertical arrows denote the obvious cleavage sites in both strands. The thick underline indicates the 15 bp region downstream of the EBE site in which the vast majority of AvrXa7-FN cleavage occurred. (B) DNA sequencing chromatograms of two DNA fragments derived from pTOP/11N3 treated with FN-AvrXa7 and purified from lane 5 in Figure 3 B. Labeling is similar to that used for (A).
Stimulated HR in yeast by FN-AvrXa7, AvrXa7-FN and AvrXa7-FN/PthXo1-FN
We sought to test the ability of the TALNs to bind and cleave target sequences in vivo by using a previously established yeast single-strand annealing (SSA) assay ( 34 , 36 ). In this assay, a ‘reporter’ construct is coexpressed with an ‘effector’ construct in yeast cells. The reporter construct contains a divided LacZ gene in which a duplicated 125 bp portion of the LacZ coding region has been created. The direct repeat within the LacZ gene is separated by a 1.2 kb sequence containing the URA3 gene ( Figure 5 A) or a shorter 0.2 kb sequence ( Figure 5 B) and a MCS. It is expected that the direct DNA repeats will undergo HR at high efficiency when a DSB is created between the repeats, resulting in a reconstituted and functional LacZ gene. Measurement of β-galactosidase ( LacZ product) enzymatic activity was used to quantify the recombination frequency that, in turn, reflects the activity of TALNs in the presence of various target sequences ( 37–39 ).
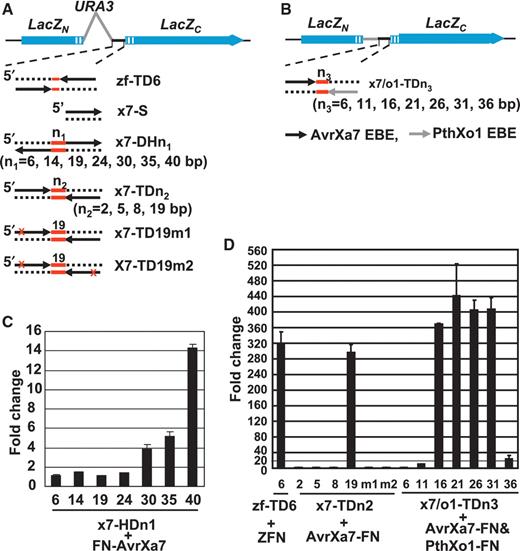
Yeast SSA assay for detection of FN-AvrXa7, AvrXa7-FN and PthXo1-FN TALN-induced HR. ( A ) Schematics of the reporter constructs (not drawn to scale) with dual BCR-ABL target site, AvrXa7 EBE site in different configurations drawn below. Two non-functional LacZ gene fragments ( LacZn and LacZc , blue solid bars) were separated by the yeast URA3 gene (gray line) and a MCS (black line). The 125 bp duplicated LacZ coding sequences are represented as hatched blue boxes. The target sites in the reporter constructs are designated as zf-TD6 (ZFN dual sites), x7-S (single EBE site), x7-DHn1 (dual AvrXa7 EBEs in a HD separated by the red-lined spacers), x7-TDn2 (dual tail-to-tail AvrXa7 EBEs) followed by the number of spacer nucleotides. The constructs x7-TD19m1 and x7-TD19m2 contain a 19 bp spacer and carry a mutation in their EBE site caused by a 4 bp substitution (depicted as a red cross). ( B ) Schematics similar to (A) with a 125 bp fragment (gray line) instead of the URA3 gene between the MCS and LacZn . ( C ) Activity of FN-AvrXa7 on reporters with different lengths of spacer between the AvrXa7 EBE sites (respective spacer lengths are given below each column). Fold change represents the β-galactosidase activity of yeast cells co-expressing the respective reporter and FN-AvrXa7 compared to cells containing the same reporter but lacking the FN-AvrXa7 expression vector. ( D ) Stimulation by the ZFN, AvrXa7-FN and AvrXa7-FN/pthXo1-FN constructs of their respective reporter genes containing various spacers (lengths in base pairs given below each column). Fold change represents the β-galactosidase activity of yeast cells co-expressing each reporter and its respective effector nuclease(s) (indicated below the columns) compared to those containing the same reporter but lacking the presence of genes encoding the respective nuclease(s). Error bars represent the standard deviation of three samples.
For TALN assays, the Saccharomyces cerevisiae YHP500 strain carrying the reporter plasmid with only a single EBE of AvrXa7 EBE (x7-S) was mated with YHP499 harboring pCP3M-FN-AvrXa7/pCP4M or pCP3M-AvrXa7-FN/pCP4 M ( Figure 5 ). Cells carrying the reporter and effector plasmids in either combination did not show increased β-galactosidase activity compared with control cells transformed with two effector plasmids lacking any TALN (data not shown). The results suggested that a single EBE site was insufficient for TALNs FN-AvrXa7 or AvrXa7-FN to effectively cleave double stranded DNA at the target site or at any nearby site within the reporter plasmid in vivo . The results also suggested a possible requirement for binding of two TALNs in an orientation allowing efficient dimerization of their FokI nuclease domains—in a fashion similar to that needed for successful DNA cleavage by ZFNs. We, therefore, reasoned that two TAL EBE sites in a proper orientation and with an appropriate spacing could bring the FokI nuclease domains in sufficiently close vicinity to dimerize and consequentially execute a double strand cleavage. To test the hypothesis, we designed two constructs, one with two identical AvrXa7 EBE sites located in a head-to-head orientation (HD) for FN-AvrXa7 and the other in a TD for AvrXa7-FN. Another construct was made containing two EBE sites, one for AvrXa7 and one for PthXo1, appropriately situated to allow tail-to-tail (TD; FN to FN) interaction between EBE-bound AvrXa7-FN and PthXo1-FN. We also sought to determine the optimal range of spacer lengths between the two EBE sites in each configuration to allow for efficient TALN cleavage ( Figures 5 A and B, Supplementary Figure S4 ). For FN-AvrXa7, the head-to-head oriented dual EBE sites (HD) were separated by a series of spacers of 6, 14, 19, 24, 30, 35 and 40 bp in length ( Figure 5 A, DNA sequences provided in Supplementary Figure 4SC ). Each reporter plasmid was coexpressed with pCP3M-FN-AvrXa7. Yeast cells containing constructs with spacers of 30 nucleotides or more exhibited β-galactosidase activities that increased with the length of the spacer element employed ( Figure 5 C).
To compare the cleavage efficiency of the TALNs containing a C-terminal FN (using a TD of dual target sites) with efficiency obtained with a known ZFN under the similar context, we performed an assay with a ZFN consisting of the ‘original’ BCR-ABL three-finger array and the FokI nuclease domain ( 35 ). For this experiment, YHP499 cells expressing BCR-ABL-FN were mated with YHP500 cells containing the reporter plasmid pzf-TD6 that contained a dual tail-to-tail target site for the BCR-ABL three-finger domain. As expected, the diploid cells exhibited high β-galactosidase enzymatic activity compared to cells lacking the effector plasmid ( Figure 5 D). For experiments with AvrXa7-FN, the tail-to-tail dual EBE sites were separated by spacers of 2, 5, 8 and 19 bp ( Figure 5 A, Supplementary Figure S4D ). Among this collection of reporter plasmids, only the plasmid containing the 19 bp spacer produced increased β-galactosidase in yeast cells when coexpressed with AvrXa7-FN. The magnitude of response was directly comparable to that obtained with the ZFN ( Figure 5 D). Only background levels of β-galactosidase expression were obtained when FN-AvrXa7 was substituted for AvrXa7-FN (data not shown). Furthermore, no stimulation of β-galactosidase activity was observed in cells expressing AvrXa7-FN if either one or both AvrXa7 EBEs was mutated ( Figure 5 D). We further tested the ability of two different species of TALNs, AvrXa7-FN and PthXo1-FN, to act in concert to recognize and cleave an asymmetric target sequence separated by a serial spacer (x7/o1-TDn3, n3 = 6, 11, 16, 21, 26, 31 and 36 bp) ( Figure 5 B; also see sequences in Supplementary Figure S4 E). The yeast cells containing reporter plasmids each with spacer in length of 16, 21, 26 or 31 exhibited significantly increased β-galactosidase activity in the presence of AvrXa7-FN and PthXo1-FN together ( Figure 5 D). Taken together, these results suggest requirements for dual EBE target sites, optimized spacer lengths between the EBEs as well as dimerization of TALN FN domains for efficient cleavage of double stranded DNA target sites by TALNs in living yeast cells.
DISCUSSION
Many years of effort spent in elucidating the interaction between TAL effectors and their modulated host genes has led to a recent breakthrough in deciphering the DNA recognition code of TAL effectors ( 28 , 29 ). The predictability and potential manipulability of the TAL central repeat domain for DNA-binding specificities make TAL an excellent system for exploiting potential biotechnological applications. In the present study, we created chimeric TALNs, FN-AvrXa7, AvrXa7-FN and PthXo1-FN containing the entire AvrXa7 or PthXo1 TAL effectors and the nuclease domain of the FokI restriction enzyme either at the C-terminal end or the N-terminal end of each TAL effector. All three constructs were tested for the ability to bind to the respective EBE recognition site and to cleave adjacent DNA. Binding of FN-AvrXa7 to the AvrXa7 EBE in vivo was demonstrated by its ability to activate transcription of a GFP coding sequence driven by the rice Os11N3 promoter that contains the AvrXa7 EBE binding site ( Figure 1 G). All three TALNs were successfully overproduced and purified from E. coli cells (AvrXa7 TALNs, Supplementary Figure S2 ). FN-AvrXa7 and AvrXa7-FN each were shown to bind specifically to double-stranded oligonucleotides containing the AvrXa7 EBE target site, but not to a slightly modified version of the binding site in an EMS assay ( Figure 2 ). Moreover, the purified AvrXa7-FN and FN-AvrXa7 TALNs exhibited cleavage activity near the expected EBE binding site under optimized reaction conditions in an in vitro assay, the results of which were confirmed by DNA sequencing ( Figures 3 and 4 ). Likewise, PthXo1-FN (alone and together with AvrXa7-FN) was shown to specifically cleave at its specific EBE DNA target site and produce the predicted sized DNA fragments ( Figure 3 ). Finally, expression of the chimeric FN-AvrXa7, AvrXa7-FN and PthXo1-FN TALNs in yeast stimulated HR between internal repeats of a disrupted and non-functional reporter gene ( LacZ ) that contained appropriately paired AvrXa7 or asymmetric AvrXa7/PthXo1 target sites ( Figures 5 ). These observations demonstrate the successful creation of functional TALNs and lead the way to future experimentation directed toward development of a technology for high-specificity gene knockout and HR in organisms that currently lack the ability to support either process in a practical manner for laboratory research.
FokI and its fusion proteins with zinc finger DNA-binding domains have been extensively studied. The endonuclease domain (FN) by itself has no specificity for cleavage, but cuts DNA at a set distance from the binding site specified by the FokI DNA-binding domain when the two domains are linked together ( 13–15 ). In this sense, several types of FN based fusion proteins have been successfully created that combine new DNA sequence binding specificities with the FN cleavage activities, with ZFNs being the most familiar ( 6 , 7 , 40 ). Study has shown that fusion of FN to ZF motif does not change the DNA-binding specificity of the ZF protein although it may cause slight decrease in binding affinity ( 41 ). We chose the FokI cleavage domain to fuse with members of the TAL effector family and, as a proof of principle, demonstrated the feasibility and generality of creating a new class of rare-cutting, site-specific DNA nucleases with sequence specificities attributable to the TAL effectors. The DNA-binding features of TAL effectors make this group of proteins or their repetitive domains desirable as the key component of such chimeric endonucleases for a number of applications, including various sorts of genome editing. For example, the majority of naturally occurring TAL effector proteins contains a large number of repeat units and, correspondingly, recognizes lengthy DNA target sites ( 32 , 42 ). These TAL EBE sites are comparable in length to, or longer than, target sites of rare-cutting meganucleases or homing nucleases (i.e. 14–40 bp) as well as binding sites for artificial zinc finger proteins assembled from multiple single fingers (i.e. 18 or 24 bp) ( 5 , 43 ). All TAL effector proteins investigated thus far exhibit high sequence specificity to the EBEs of their target genes ( 32 , 42 ). The known code of TAL effectors predicts an alignment of a single type of repeat unit to a single nucleotide species (A, G, C or T) based on the specific di-residues at positions 12 and 13 in the repeat unit. This modular nature of the TAL repeat domain for DNA-binding specificity suggests that techniques can be developed to produce an array of repeat units that can precisely recognize a unique, lengthy sequence of nucleotides in any given gene. If so, investigators will be able to create truly gene-specific TALNs for use in organisms with large genomes and lacking robust systems for HR.
Thus far, several TAL effectors have been found to function as transcription activators. Like many other transcription factors, TAL effectors may function as dimers to bind target DNA. However, to date, AvrBs3 is the only TAL effector shown to dimerize in vitro and in the cytoplasm in vivo before entry into nuclei of host cells ( 44 ). The sequence specificity of known TAL effectors that bind DNA can be aligned to only one strand of the target site which is usually asymmetric ( 28 , 29 ). Thus, it is not yet clear if most TAL effector proteins form dimers or multimers in the presence of target DNA (or in the absence of DNA). The results from the present yeast SSA assay imply that TALNs do not form homo- or hetero-dimer at a single TAL EBE site, or at least the dimerization of TAL subdomain does not facilitate the dimerization of FokI nuclease domains for effective double stranded DNA cleavage in yeast cells. More detailed structural studies of TAL effectors or TALNs likely will be needed to resolve this uncertainty, which may or may not negatively influence the ability to easily and successfully design sequence specific TALNs in the future.
It has been established that for efficient double strand cleavage of target DNA dimerization of FokI monomer nuclease domains is required ( 11 ). Therefore, it is conceivable that TALNs need to dimerize for the efficient cleavage of DNA in solution where sufficient concentrations of purified proteins and substrates are present or in vivo where TALN and target substrate are otherwise limited. This could be achieved through various mechanisms, three of which are presented below. In one model, one EBE-bound TALN might form a dimer with another bound or unbound TALN through an as yet uncharacterized dimerization motif of TAL effector. In such a case, the TAL subdomain-mediated dimerization could bring the two FokI nuclease domains in close proximity near the binding site and allow DNA cleavage. Alternatively, one TALN monomer could bind to one EBE target site and, similar to the model proposed for the native FokI or hybrid ZFNs in vitro ( 13 , 14 ), dimerization of two DNA bound-TALNs through the well characterized dimerization motif in the FokI nuclease subdomain could occur in close proximity and, thereby, support DNA cleavage in trans if sufficient concentrations of nucleases were present. Successful in vitro cleavage of DNA carrying a single AvrXa7 EBE by the FN-AvrXa7, AvrXa7-FN and PthXo1-FN TALNs ( Figure 3 ) is consistent with this model. In a third model in which two tandem, head-to-head or tail-to-tail EBE sites are present, TALNs could bind to each of the EBE sites. This would bring the two FN domains of the two TALNs into sufficiently close proximity to allow dimerization and DNA cleavage. Our yeast SSA data ( Figure 5 ) is consistent with this latter model.
The function of native FokI is allosterically regulated through DNA and divalent metal binding. Without DNA binding and in the absence of divalent metal, FN is sequestered through tight interaction with the DNA recognition motifs of FokI and, thus, the FokI monomer maintains an idle state. Following binding of two FokI holoenzymes to the FokI recognition site and in the presence of metals, the two FokI nuclease domains are freed and can dimerize. This dimerization then allows double stranded DNA cleavage ( 14 , 16 , 17 ). It is possible that the interaction between the FN subddomain and the TAL DNA-binding subdomain in hybrid TALN lacks such tight regulatory mechanism and, hence nuclease domains form dimers with less difficulty. That may explain why the apparent stringency of cleavage by the presently studied chimeric TALNs (and also ZFNs) is lower and leads to the non-specific cleavage observed in the presence of excess of TALNs ( Supplementary Figure S3 ). The structure (length and composition) of linker segment between the DNA-binding and -cleaving domains of FokI and its derived nucleases (i.e. ZFNs and, likely, TALNs) also dictate the enzymes’ cleavage pattern, for example, the distance of cleavage sites from the DNA-binding site ( 45–47 ). The linker segment of native FokI is 15 aa (residues 373–387) long and allows the FN to extend and cleave the sense strand 9 bp and antisense strand 13 bp downstream of the binding site ( 12 ). An 18 aa flexible linker of a ZFN accommodates effective cleavage of target spacer in a range of 6–18 bp with 8 bp as an optimum in Xenopus oocytes as determined in a single-strand annealing reporter assay ( 45 ). The latter study also reported that the dependence of efficient cleavage on spacer length in vitro differed from that under in vivo condition. Because the minimum-sized TAL effector fragment required for efficient DNA binding is unknown, the full-length AvrXa7 and PthXo1 were used to construct the TALNs in our study. Therefore, the N-terminal 288 aa in FN-AvrXa7 and the C-terminal 295 aa in AvrXa7-FN and PthXo1-FN function as long inter-domain linkers between FN and the repeat DNA-binding domain in TALNs. Such an extended inter-domain linker may allow significant ‘reach’ for the nuclease domain to cut at a moderate distance away from the ends of the EBE or allow greater flexibility for the nuclease domain to cut within a moderately wide zone as we observed in vitro with our TALN enzyme assays and in vivo with our yeast assays. Future investigations will be required to determine how various combinations of inter-domain sequence and length affects the cleavage efficiency of TALNs [e.g. tests of TALNs consisting of the FN connected directly to the TAL effector repeat domain or through linkers of various lengths and composition].
Other important questions are presently under active investigation. One such question is the nature of the DNA fragment with an apparent molecular size >10 kb when supercoiled plasmid DNA is mixed with a TALN (lanes 4 and 7 in Figure 3 B, lanes 4 and 6 in 3E). Such a band of DNA is not observed if the supercoiled plasmid is cleaved before mixing with the TALN ( Figure 3 B and E). Our working hypothesis is that the uppermost, slow migrating DNA band may be a complex between plasmid DNA and the TALN protein that exists prior to DNA cleavage (by the TALN or a restriction enzyme), but not after cleavage. Future analyses of the component(s) of this DNA band (i.e. ethidium bromide stained band) should resolve this present enigma. Another issue is TALN stability. We have observed that all of the TALNs created for the present study have quite short half-lives (i.e. ∼30–45 min). This currently imposes serious practical constraints on the degree of purity of recombinant TALNs that can be obtained and on the duration and extensiveness of biochemical analyses, including accurate measurements of TALN binding affinities to DNA. Further attempts to discover conditions that stabilize TALNs overproduced in E. coli will be important undertakings along with endeavors to determine if such instability does or does not exist in vivo in various cell types.
The work presented here unambiguously demonstrates several points: the ability to fuse TAL effectors with other proteins to create functional chimeric proteins; the specificity of DNA binding by TAL effectors when fused with a non-specific nuclease domain; and the specific cleavage of DNA target sites by engineered TALNs both in vitro and in vivo . The newly emerging TALN-based approach could be an attractive alternative to the still improving ZFN-based or meganuclease-based ( 48 ) genomic tools for a wide variety of possible applications including targeted genome editing. However, a few basic questions remain unanswered regarding the feasibility of using TALNs for genome modification. First, can novel TALN DNA-binding domains with the requisite specificity and affinity be synthesized based on the actual DNA target sequences? Although arbitrarily assembled, TAL effectors were able to activate promoters containing sequence elements synthesized based on the ‘code’ ( 29 ), this capability is not yet demonstrated for a TALN fusion protein. Second, can two different TALNs work coordinately at preselected adjacent target sites? The reaction involving cleavage of a dual asymmetric PthXo1 EBE/AvrXa7 EBE site with a mixture of PthXo1-FN and AvrXa7-FN ( Figure 3 E, lane 7) hints this may be possible. That is, the two TALNs together produced somewhat better, albeit still incomplete, plasmid cleavage than when each TALN was used independently at five times the concentration employed in the dual cutting reaction—and with less non-specific plasmid DNA cleavage (compare Figure 3 E, lanes 5 and 7 with lane 8). Third, will the DNA recognition and cleavage by the TALNs occur in a chromosomal context in living cells? Data from the yeast experiment described in this paper ( Figure 5 ) provide strong initial evidence that a TALN or sets of TALNs can successfully find, bind and cleave an EBE target site within yeast chromatin. Addressing these issues will assist in achieving the goal of generating a tool box of TALNs for targeted genome editing based on the DNA-binding specificity of custom-designed, synthetic TAL repeat domains and the DNA-cleavage function of FokI or other nucleases.
FUNDING
The Iowa State University (to B.Y. and to M.H.S.); National Science Foundation grants (0820831 to B.Y. and MCB-0952533 to D.P.W.); Department of Energy Advanced Research Projects Agency-Energy Program (DEAR0000010 to M.H.S.). Funding for open access charge: National Science Foundation award (0820831).
Conflict of interest statement . None declared.
ACKNOWLEDGEMENTS
The authors thank Dr Dan Voytas for providing the components of the yeast SSA system and Dr Keith Joung for providing the plasmid pGP-FB-orig BA [obtained through Addgene (Addgene Inc., Cambridge, MA, USA), also designated as Addgene plasmid 13420].
REFERENCES
Author notes
The authors wish it to be known that, in their opinion, the first two authors should be regarded as joint First Authors.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments