





Abstract
We have sequenced the genome of the emerging human pathogen Babesia microti and compared it with that of other protozoa. B. microti has the smallest nuclear genome among all Apicomplexan parasites sequenced to date with three chromosomes encoding ∼3500 polypeptides, several of which are species specific. Genome-wide phylogenetic analyses indicate that B. microti is significantly distant from all species of Babesidae and Theileridae and defines a new clade in the phylum Apicomplexa. Furthermore, unlike all other Apicomplexa, its mitochondrial genome is circular. Genome-scale reconstruction of functional networks revealed that B. microti has the minimal metabolic requirement for intraerythrocytic protozoan parasitism. B. microti multigene families differ from those of other protozoa in both the copy number and organization. Two lateral transfer events with significant metabolic implications occurred during the evolution of this parasite. The genomic sequencing of B. microti identified several targets suitable for the development of diagnostic assays and novel therapies for human babesiosis.
INTRODUCTION
Babesia microti is the principal cause of human babesiosis and one of the most common transfusion-transmitted pathogens in the United States ( 1–3 ). The parasite has a worldwide distribution and has been cited as an emerging health threat in the United States by the National Academy of Sciences ( 4 ). B. microti is primarily transmitted to humans by the tick vector, Ixodes scapularis , but also perinatally and through blood transfusion. The mortality rate associated with human babesiosis is estimated to be between 3 and 28%. Most severe cases occur in people over the age of 50 years or those who are asplenic, have cancer or HIV or who are on an immunosuppressive therapy. The majority of patients experience mild to moderate malaria-like symptoms; however, in severe cases, the disease may be associated with respiratory failure, multi-organ system dysfunction or coma ( 1–4 ). Both host and parasite factors contribute to these symptoms, but the exact pathogenic mechanisms remain unknown ( 5 , 6 ). No evidence of extraerythrocytic cellular parasitism exists for B. microti . Although the parasite has been classified as a member of the Babesia genus, the accuracy of this classification has been debated for >50 years with some experts suggesting that it belongs to the Theileria genus .
We describe here the first sequence of the B. microti genome from a patient isolate propagated in gerbils. Sequence analysis revealed important information about the genome organization, gene content and metabolic capacities of this parasite and provides new insights into its pathophysiology. Furthermore, the study identified new targets for the development of diagnostic assays and novel therapies for this important human pathogen. Phylogenetic analysis using a large pool of coding sequences (CDS) strongly suggest that B. microti defines a new taxonomic genus among Apicomplexa distinct from Babesia and Theileria species.
MATERIALS AND METHODS
Strains and genome sequencing
The B. microti R1 isolate was obtained from an adult male patient who experienced severe B. microti infection that required hospital admission. Although the patient was hospitalized in France, he lived in the United States in a Babesia -endemic area. Prior to the onset of his illness, the patient was in general good health, non-splenectomized and with no other evidence of immune suppression. Because the patient had fever, purpura, laboratory evidence of hemolytic anemia and kidney failure, B. microti infection was suspected and subsequently confirmed by blood smear, serology, rodent inoculation and polymerase chain reaction (PCR) among other molecular methods as outlined in the Results and Discussion section. The patient was treated with clindamycin and quinine after which he fully recovered.
The R1 and the Gray strain control were propagated in immune-compromised gerbils or hamsters. B. microti R1 DNA was extracted from agarose gel plugs and fragmented by mechanical shearing producing 3 and 10 kb inserts that were subsequently cloned into the pcDNA2.1 ( www.invitrogen.com ) and pCNS (pSU18 derived) plasmids, respectively. In addition, a large insert (30 kb) bacterial artificial chromosome library was constructed by cloning Sau 3A partially digested genomic fragments into the pBelo-BAC11 vector. Vector DNAs were purified and end-sequenced using dye-terminator chemistry on ABI3730 sequencers. We collected 71 882, 59 193 and 3343 sequences, respectively, from each DNA library. A first assembly was performed using Arachne ( http://www.broadinstitute.org ) resulting in 522 contigs of >500 nt (N50 = 38 216) and 104 scaffolds of >2 kbp (N50 = 951 194). We retained only the clones containing contigs with >4 kb because the B. microti R1 DNA was contaminated with Gerbil DNA. The corresponding reads were assembled using the Phred/Phrap/Consed software package ( www.phrap.com ) as described by Vallenet et al. ( 7 ). We then obtained 139 contigs (N50 = 183 364) with a cumulated size of 6 425 753bp. Three main scaffolds were determined and attributed to the chromosomes. Contigs corresponding to mitochondrial DNAs were identified by Basic Local Alignment Search Tool (BLAST). Primer walks, PCRs and in vitro transposition technology (Template Generation System ™ II Kit; Finnzyme, Espoo, Finland) were used to obtain complete chromosome sequences. A total of 10 328 sequences were used for gap closure and quality assessment. The mitochondrial genome organization was confirmed by PCR and by sequencing full clone inserts. PFGE analyses were performed in 0.5 × Tris-HCl, Borate, EDTA (TBE) at 10°C at 4 V/cm using Gene Navigator ™ (Pharmacia). 2D Pulsed Field Gel Electrophoresis (PFGE) analyses were performed as previously described ( 8 ).
Genome annotation
Integration of resources using GAZE
The automatic gene prediction pipeline ( Supplementary Figure S1 ) was modified from a standard annotation pipeline ( 9 ). Programs were used with default options following standard procedures. A semi-automatic procedure was used to generate a first training set of 690 gene models from chromosome I sequence. A similar approach was previously used to analyze the Encephalitozoon cuniculi genome ( 10 ). To use these curated annotations in the data integration step, transcript sequences of the genes were mapped on the final genomic assembly using BLAST-like alignment tool (BLAT) ( 11 ), best match (best BLAT score) per gene models were selected, and each best match was realigned using Est2Genome ( 12 ) in order to identify exon/intron boundaries. The mapping was used to calibrate the ab initio SNAP ( 13 ) gene prediction software and as an entry for GAZE ( 14 ). The same approach was previously used with Apicomplexan EST data. A collection of 426 440 public messenger RNAs (mRNAs) from the clade of Apicomplexa (downloaded from the EMBL database, release 98) were first aligned with the B. microti genome assembly using BLAT. To refine BLAT alignment, we used Est2Genome ( 12 ). BLAT alignments were made using default parameters between translated genomic and translated ESTs ( 11 ).
For protein sequences, the UniProt database ( 15 ) was used to detect conserved proteins between B. microti and other species. Apicomplexan proteins in the UniProt database were first aligned with the B. microti sequences using BLAT ( 11 ), and alignments with a BLAT score over 20 were selected. Subsequently, we extracted the genomic regions where no protein hit had been found by BLAT and then realigned the UniProt protein with more permissive parameters. Alignments larger than 50 residues and with >20 identity matches were selected. Each match was then refined using GeneWise ( 16 ) in order to identify exon/intron boundaries.
Most of the genome comparisons were performed with repeat-masked sequences. For this purpose, we searched and sequentially masked several kinds of repeats including (i) Apicomplexan-known repeats available in Repbase (instead of the human data) using the RepeatMasker program ( 17 ), (ii) tandem repeats using the TRF program ( 18 ) and (iii) ab initio repeat detection using RepeatScout ( 19 ). From this pipeline, 1.2% of the assembled bases were masked. No further analysis of the repeated sequences has been performed.
All the resources described here were used to automatically build B. microti gene models using GAZE ( 14 ). Individual predictions from each of the programs (SNAP, GeneWise and Est2genome) were broken down into segments (coding, intron and intergenic) and signals (start codon, stop codon, splice acceptor, splice donor, transcript start and transcript stop). Exons predicted by SNAP, GeneWise and Est2genome were used as coding segments. Introns predicted by GeneWise and Est2genome were used as intron segments. Intergenic segments were created from the span of each mRNA with a negative score (to prevent genes splitting by GAZE). Predicted repeats were used as intron and intergenic segments in order to avoid prediction of genes encoding proteins in such regions. A weight was assigned to each resource. More importance was given to alignments than to ab initio predictions. Chromosome I curated gene models were associated with stronger parameters to enable their consideration by GAZE. This weight acts as a multiplier for the score of each information source prior to processing by GAZE automaton. All signals were given a fixed score, but segment scores were context sensitive: coding segment scores were linked to the percentage identity (%ID) of the alignment, while intronic segment scores were linked to the %ID of the flanking exons. Finally, gene predictions created by GAZE were filtered following their scores and lengths. When applied to the entire assembled sequence, GAZE predicted 1426 gene models.
Annotation procedure
GlimmerHMM gene prediction software was used in parallel with GAZE annotation. Parameters and training were performed following published recommendations ( 20 ). The training was performed on each chromosome independently. GlimmerHMM predicts 3923 putative CDS. The gene models have been curated according to a semi-automatic procedure ( Supplementary Figure S2 ). BLASTX homology search ( 21 ) covering the whole genome was performed as described by Katinka et al. ( 10 ) and stored in an AceDB v4.9.38 database ( http://www.acedb.org/index.shtml ). Artemis software ( 22 ) was used as a graphic interface ( Supplementary Figure S3 ).
Subtelomeric sequence organization
The miropeat v2.01 software ( http://www.genome.ou.edu/miropeats.html ) and icatools v2.5 package ( http://www.littlest.co.uk/software/pub/bioinf/freeold ) were compiled on an Ubuntu Linux platform. The coordinates were recovered from postscript output file. Ghostview was used for the graphic representation of the miropeat output ( http://pages.cs.wisc.edu/∼ghost/ ). The genome of B. microti was aligned to itself using BLASTN ( 21 ) to confirm and to refine the structure of the subtelomere-specific duplicated sequences (expect value threshold at 0.0001).
Functional annotation
The functional annotation of the B. microti CDS was performed by BLAST sequence comparison ( 21 ) against UniProt and by searching for orthologues in defined species. Orthologous genes were identified using the bidirectional best hit method (BBH). The analysis was performed with three Apicomplexan parasites Plasmodium falciparum , Babesia bovis and Theileria annulata as well as with the yeast Saccharomyces cerevisiae and the human pathogen Leishmania major . Homology searches were performed at the protein level using BLASTP with default options ( 21 ). Sequence data were obtained from the Kyoto Encyclopedia of Genes and Genomes database (KEGG) ( 23 ), PiroplasmDB and PlasmoDB ( 24 ). BBH scores were collected in a relational database and combined with the metabolic information provided by KEGG ( 23 ) and the Malaria Parasite Metabolic Pathway (MPMP, http://sites.huji.ac.il/malaria/ ) databases ( Supplementary Table S1 ).
The functional annotation was scored using different parameters combining the results of different BBH analyses. A first score was given by the number of organisms having orthologs for each B. microti CDS (parameter o∈[0,5]). The reliability of the annotation was also assessed by the count of KEGG orthology group identification numbers (ko) associated with each ortholog (release 58.1, 1 June 2011). Two parameters were used in this case: the number of KEGG ko number per B. microti CDS (k) and the number of different ko (d). Three levels of annotation were used, each associated with a different validation procedure ( Supplementary Table S2 ): (i) the ko number was directly inferred when o and k values were high; (ii) for intermediate values of d and k, the annotation was the only information stored (the protein was declared as ‘valid’ and annotation was deduced from BLAST best homologues’ description) and (iii) for low o or k scores and also for high d values, we considered that the annotation of a B. microti protein could not be inferred from those of its orthologous sequences (‘No Annotation’ denoted by ‘NA’).
Several metabolic charts of the KEGG and MPMP databases were analyzed for this study. B. microti genes encoding key enzymes and ‘missing’ genes in a given metabolic pathway were further analyzed by gene-specific bioinformatics approaches. Central metabolism has been reconstructed by taking into account the carbon and redox balance.
Genome-wide phylogenetic analyses
Taxon sampling
The Apicomplexan phylogeny was analyzed based on a taxon sampling including nine species representing different families in the Apicomplexa phylum ( 25 ) and one outgroup species, Tetrahymena thermophila. The annotation of B. microti CDS ( Supplementary Table S1 ) was initiated to generate the complete set of data. Additional sequences were directly obtained from KEGG (release 58.1, 1 June 2011).
Dataset assembly
To perform phylogenetic analyses, we assigned CDS of B. microti to existing KEGG gene families ( 23 ). We retained CDS having one orthologous sequence in each tree of the three Apicomplexan parasites (described in Supplementary Table S1 ) and associated with only one KEGG ko identification number ( d = 1). Using this analysis, 1002 CDS were obtained. We filtered those ko families to retain only those containing one, and only one, sequence for: (i) each of the eight Apicomplexan genomes in the KEGG database, (ii) B. microti genome and (iii) T. thermophila . This filtration step reduced the B. microti sequence set to 316 CDS. For each of the 316 orthology groups, the sequences of these 10 organisms were collected and aligned using MUSCLE v3.8.31 ( 26 ) (with default options). Spurious alignment sites were then removed using the ‘automated’ option of TrimAL v1.3 ( 27 ) that establishes optimal thresholds based on the alignment characteristics.
Tree inferences
Phylogenetic analyses were conducted on each of the 316 genes to infer the evolutionary history of each KEGG ko family. The resulting trees were inferred by maximum likelihood, using RAxML v7.2.8 ( 28 ). Inferences were performed starting from 10 distinct maximum parsimony trees randomly chosen. The WAG protein model ( 29 ) using empirical base frequencies and a discrete Gamma law with four categories to model heterogeneity of evolutionary rate among sites were chosen. Branch supports were estimated through full bootstrap analyses, as opposed to the faster RAxML bootstrap approximations. Both supermatrix and supertree analyses were computed using SeqCat.pl and SeqConverter.pl ( http://www.molekularesystematik.uni-oldenburg.de/33997.html#Sequences ) scripts to concatenate the 316 trimmed alignments into a nexus ‘Supermatrix’ of 129 571 protein sites. The Supermatrix was analyzed by RAxML, with the same parameters mentioned above. To obtain the supertree with the Matrix Representation with Parsimony (MRP) method ( 30 ), a matrix for the 316 protein trees was computed with MRTOOLS ( 31 ), weighting each clade of the protein trees according to its bootstrap support. Accordingly, clades that may have arisen from a low signal to noise ratio (low bootstrap) had less influence on the inferred supertree than more reliable clades (high bootstrap). The matrix was then bootstrapped and analyzed with a parsimony criterion in the PAUP* software ( 32 ). Supertree analysis was also conducted by resorting to phySIC_IST ( 33 ), a more conservative supertree method that allows congruent study among clades of individual gene histories with respect to their bootstrap supports.
Supertree inferred by the PhySIC_IST method
The PhySIC_IST method ( 33 ) first establishes a congruence analysis of the gene trees and subsequently modifies these trees to remove pieces of topological signal in each gene tree that conflict with the majority of other gene trees. The 316 gene trees were curated by discarding clades supported by a boostrap value below a threshold were discarded, merging their two delimiting nodes in the gene tree where they belong. Then for each triplet of taxa, the frequencies of occurrence of the three possible topologies for the triplet were compared on the basis of the clades remaining after the bootstrap elimination step. The P value of a χ 2 test was used to determine whether a topology for a triplet observed in a gene tree was an anomaly. In our study, a threshold value (STC = 1-p) was used to eliminate abnormal triplet topologies. STC = 1 means that no triplet topology will be rejected, even if it appears in one gene tree and is in contradiction with all other gene trees. The usual values are STC = 0.95 or STC = 0.8. Then clades of each gene tree inducing rejected triplets were eliminated. Once the gene trees were curated with this procedure, a deduced supertree in total agreement with the modified gene trees was obtained. More information on this method is available on http://www.atgc-montpellier.fr/physic_ist/ .
RESULTS AND DISCUSSION
B. microti genome sequence and analysis
Using a whole-genome shotgun strategy approach, we sequenced the DNA content of the B. microti isolate R1 that was purified from infected human blood. The parasite was positively identified as a B. microti using light microscopy, serological tests, PCR analyses, rDNA sequencing and PFGE-based caryotyping and chromosomal restriction profiling ( Figure 1 ). The karyotype of the R1 strain was similar to that of the standard laboratory Gray strain. Not I restriction digestion revealed small differences on chromosomes I and II between the two isolates ( Figure 1 D). An approximate 140 Gbp of raw sequence data were generated using conventional Sanger sequencing technologies. Three nuclear, one mitochondrial and one apicoplast chromosomes comprise the DNA material of the parasite. The overall size of the B. microti nuclear genome is ∼6.5 Mbp, which is 20% smaller than that of other piroplasms such as B. bovis and T. annulata and 72% smaller than that of the human malaria parasite P. falciparum , thus making it the smallest Apicomplexan genome ever sequenced ( Table 1 ). This small size is most likely the result of a regressive evolution from an ancestral organism with a larger genome. The genome size and structure of Apicomplexan parasites are as diverse as their host range and life-style. Coccidian genomes are between 60 and 80 Mbp and Plasmodium genomes are between 20 and 25 Mbp. Interestingly, piroplasms have some of the smallest Apicomplexan genomes ( Table 1 ). The genome size and chromosome number of B. microti are in the range of what has been described in other piroplasms ( 34–36 ). B. microti belongs to the so-called small Babesia and infects only the erythrocyte of its mammalian host ( 37 ) where it undergoes one or two divisions prior to parasite egress and invasion by merozoites of new red blood cells. The ability of the parasite to invade and replicate in lymphocytes of the vertebrate host has been suggested but was never confirmed ( 38 ). Therefore, the limited cellular host range of B. microti may account for the dramatic reduction its genome size and structure.
Genomic features of B. microti and five other Apicomplexa
| Feature | B. microti | B. bovis | T. parva | T. annulata | P. falciparum | C. parvum |
|---|---|---|---|---|---|---|
| Genome | ||||||
| Size (Mbp) a | 6.5 | 8.2 | 8.3 | 8.4 | 23.3 | 9.1 |
| Number of chromosomes | 3 | 4 | 4 | 4 | 14 | 8 |
| G + C content (%) | 36 | 41.5 | 34.1 | 32.5 | 19.3 | 30.8 |
| Genes | ||||||
| Number of genes b | 3513 | 3706 | 3796 | 4082 | 5324 | 3805 |
| Mean gene length (bp) | 1327 | 1503 | 1407 | 1602 | 2326 | 1844 |
| Mean gene length including introns (bp) | 1471 | 1609 | 1654 | 1802 | 2590 | 1851 |
| Gene density (bp per gene) | 1816 | 2194 | 2059 | 2199 | 4374 | 2411 |
| Coding regions (%) | 73 | 68 | 68 | 73 | 53 | 76 |
| Coding regions including introns (%) | 81 | 73 | 80 | 82 | 59 | 77 |
| Number of genes with introns (%) | 70 | 60 | 75 | 71 | 54 | 4 |
| Exons | ||||||
| Number per gene | 3.3 | 2.8 | 2.7 | 3.9 | 2.6 | 1.1 |
| Mean length (bp) | 397 | 547 | 514 | 416 | 904 | 1748 |
| Total length (%) | 73 | 68 | 68 | 73 | 53 | 77 |
| Introns | ||||||
| Number per gene | 2.3 | 1.7 | 2.6 | 2.9 | 1.6 | 0.1 |
| Number per gene presenting intron | 3.4 | 2.9 | 3.5 | 4 | 2.9 | 1.3 |
| Mean length (bp) | 61 | 60 | 94 | 70 | 167 | 96 |
| Total length (%) | 8 | 5 | 12 | 9 | 6 | 0.02 |
| Intergenic regions | ||||||
| Mean length (bp) | 346 | 585 | 405 | 398 | 1784 | 561 |
| Total length (%) | 19 | 27 | 20 | 18 | 41 | 23 |
| RNAs | ||||||
| Number of tRNA genes c | 44 | 70 | 71 | 47 | 72 | 45 |
| Number of 5S rRNA genes | 2 | ND | 1 | 3 | 3 | 6 |
| Number of 5.8S/18S/28S rRNA units | 2 | 5 | 8 | 1 | 13 | 9 |
| Feature | B. microti | B. bovis | T. parva | T. annulata | P. falciparum | C. parvum |
|---|---|---|---|---|---|---|
| Genome | ||||||
| Size (Mbp) a | 6.5 | 8.2 | 8.3 | 8.4 | 23.3 | 9.1 |
| Number of chromosomes | 3 | 4 | 4 | 4 | 14 | 8 |
| G + C content (%) | 36 | 41.5 | 34.1 | 32.5 | 19.3 | 30.8 |
| Genes | ||||||
| Number of genes b | 3513 | 3706 | 3796 | 4082 | 5324 | 3805 |
| Mean gene length (bp) | 1327 | 1503 | 1407 | 1602 | 2326 | 1844 |
| Mean gene length including introns (bp) | 1471 | 1609 | 1654 | 1802 | 2590 | 1851 |
| Gene density (bp per gene) | 1816 | 2194 | 2059 | 2199 | 4374 | 2411 |
| Coding regions (%) | 73 | 68 | 68 | 73 | 53 | 76 |
| Coding regions including introns (%) | 81 | 73 | 80 | 82 | 59 | 77 |
| Number of genes with introns (%) | 70 | 60 | 75 | 71 | 54 | 4 |
| Exons | ||||||
| Number per gene | 3.3 | 2.8 | 2.7 | 3.9 | 2.6 | 1.1 |
| Mean length (bp) | 397 | 547 | 514 | 416 | 904 | 1748 |
| Total length (%) | 73 | 68 | 68 | 73 | 53 | 77 |
| Introns | ||||||
| Number per gene | 2.3 | 1.7 | 2.6 | 2.9 | 1.6 | 0.1 |
| Number per gene presenting intron | 3.4 | 2.9 | 3.5 | 4 | 2.9 | 1.3 |
| Mean length (bp) | 61 | 60 | 94 | 70 | 167 | 96 |
| Total length (%) | 8 | 5 | 12 | 9 | 6 | 0.02 |
| Intergenic regions | ||||||
| Mean length (bp) | 346 | 585 | 405 | 398 | 1784 | 561 |
| Total length (%) | 19 | 27 | 20 | 18 | 41 | 23 |
| RNAs | ||||||
| Number of tRNA genes c | 44 | 70 | 71 | 47 | 72 | 45 |
| Number of 5S rRNA genes | 2 | ND | 1 | 3 | 3 | 6 |
| Number of 5.8S/18S/28S rRNA units | 2 | 5 | 8 | 1 | 13 | 9 |
Quantitative data have been calculated using Artemis genome browser.
a Estimated size including gaps.
b Data from Plasmodb 8.1 data for Apicomplexa. B. microti : this work.
c Data from piroplasmoDB v1.1 and plasmoDB v8.1.
Genomic features of B. microti and five other Apicomplexa
| Feature | B. microti | B. bovis | T. parva | T. annulata | P. falciparum | C. parvum |
|---|---|---|---|---|---|---|
| Genome | ||||||
| Size (Mbp) a | 6.5 | 8.2 | 8.3 | 8.4 | 23.3 | 9.1 |
| Number of chromosomes | 3 | 4 | 4 | 4 | 14 | 8 |
| G + C content (%) | 36 | 41.5 | 34.1 | 32.5 | 19.3 | 30.8 |
| Genes | ||||||
| Number of genes b | 3513 | 3706 | 3796 | 4082 | 5324 | 3805 |
| Mean gene length (bp) | 1327 | 1503 | 1407 | 1602 | 2326 | 1844 |
| Mean gene length including introns (bp) | 1471 | 1609 | 1654 | 1802 | 2590 | 1851 |
| Gene density (bp per gene) | 1816 | 2194 | 2059 | 2199 | 4374 | 2411 |
| Coding regions (%) | 73 | 68 | 68 | 73 | 53 | 76 |
| Coding regions including introns (%) | 81 | 73 | 80 | 82 | 59 | 77 |
| Number of genes with introns (%) | 70 | 60 | 75 | 71 | 54 | 4 |
| Exons | ||||||
| Number per gene | 3.3 | 2.8 | 2.7 | 3.9 | 2.6 | 1.1 |
| Mean length (bp) | 397 | 547 | 514 | 416 | 904 | 1748 |
| Total length (%) | 73 | 68 | 68 | 73 | 53 | 77 |
| Introns | ||||||
| Number per gene | 2.3 | 1.7 | 2.6 | 2.9 | 1.6 | 0.1 |
| Number per gene presenting intron | 3.4 | 2.9 | 3.5 | 4 | 2.9 | 1.3 |
| Mean length (bp) | 61 | 60 | 94 | 70 | 167 | 96 |
| Total length (%) | 8 | 5 | 12 | 9 | 6 | 0.02 |
| Intergenic regions | ||||||
| Mean length (bp) | 346 | 585 | 405 | 398 | 1784 | 561 |
| Total length (%) | 19 | 27 | 20 | 18 | 41 | 23 |
| RNAs | ||||||
| Number of tRNA genes c | 44 | 70 | 71 | 47 | 72 | 45 |
| Number of 5S rRNA genes | 2 | ND | 1 | 3 | 3 | 6 |
| Number of 5.8S/18S/28S rRNA units | 2 | 5 | 8 | 1 | 13 | 9 |
| Feature | B. microti | B. bovis | T. parva | T. annulata | P. falciparum | C. parvum |
|---|---|---|---|---|---|---|
| Genome | ||||||
| Size (Mbp) a | 6.5 | 8.2 | 8.3 | 8.4 | 23.3 | 9.1 |
| Number of chromosomes | 3 | 4 | 4 | 4 | 14 | 8 |
| G + C content (%) | 36 | 41.5 | 34.1 | 32.5 | 19.3 | 30.8 |
| Genes | ||||||
| Number of genes b | 3513 | 3706 | 3796 | 4082 | 5324 | 3805 |
| Mean gene length (bp) | 1327 | 1503 | 1407 | 1602 | 2326 | 1844 |
| Mean gene length including introns (bp) | 1471 | 1609 | 1654 | 1802 | 2590 | 1851 |
| Gene density (bp per gene) | 1816 | 2194 | 2059 | 2199 | 4374 | 2411 |
| Coding regions (%) | 73 | 68 | 68 | 73 | 53 | 76 |
| Coding regions including introns (%) | 81 | 73 | 80 | 82 | 59 | 77 |
| Number of genes with introns (%) | 70 | 60 | 75 | 71 | 54 | 4 |
| Exons | ||||||
| Number per gene | 3.3 | 2.8 | 2.7 | 3.9 | 2.6 | 1.1 |
| Mean length (bp) | 397 | 547 | 514 | 416 | 904 | 1748 |
| Total length (%) | 73 | 68 | 68 | 73 | 53 | 77 |
| Introns | ||||||
| Number per gene | 2.3 | 1.7 | 2.6 | 2.9 | 1.6 | 0.1 |
| Number per gene presenting intron | 3.4 | 2.9 | 3.5 | 4 | 2.9 | 1.3 |
| Mean length (bp) | 61 | 60 | 94 | 70 | 167 | 96 |
| Total length (%) | 8 | 5 | 12 | 9 | 6 | 0.02 |
| Intergenic regions | ||||||
| Mean length (bp) | 346 | 585 | 405 | 398 | 1784 | 561 |
| Total length (%) | 19 | 27 | 20 | 18 | 41 | 23 |
| RNAs | ||||||
| Number of tRNA genes c | 44 | 70 | 71 | 47 | 72 | 45 |
| Number of 5S rRNA genes | 2 | ND | 1 | 3 | 3 | 6 |
| Number of 5.8S/18S/28S rRNA units | 2 | 5 | 8 | 1 | 13 | 9 |
Quantitative data have been calculated using Artemis genome browser.
a Estimated size including gaps.
b Data from Plasmodb 8.1 data for Apicomplexa. B. microti : this work.
c Data from piroplasmoDB v1.1 and plasmoDB v8.1.
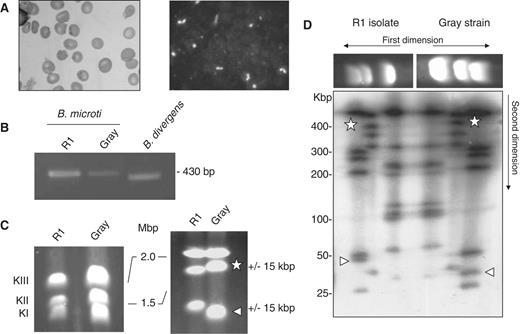
Babesia microti strain R1 characterization. (A) Light-microscopy analysis of B. microti infected blood. Left: blood smear. Right: immunofluorescence analysis using serum from a hamster infected with the B. microti Gray standard laboratory strain. Similar serum was used for serological assays. ( B) PCR amplification of the ssu gene using PIRO-A and PIRO-B primers ( 77 ). These primers amplify a 436 bp fragment in B. microti Gray and R1 strains and a 408 bp fragment in B. divergens Rouen strain. The integrity of the PCR fragment was confirmed by DNA sequencing. ( C)B. microti karyotype. PFGE conditions used are: left, 0.7% agarose, 400 s pulses for 55 h; right, 1% agarose, 200 s pulses for 65 h. Length polymorphism between the R1 isolate and the Gray strain is observed on chromosome 1 and 2. ( D) 2D-PFGE Not I Restriction Fragment Length Polymorphism (RFLP) analysis of B. microti . Each chromosome length polymorphism results from a single RFLP of 15 kbp (see triangle and star for chromosome I and II respectively). The genome structure of Gray and R1 strain may differ from each other only with a single recombination event. PFGE conditions used are: 1.2 % agarose gel, pulse conditions, 5 s for 8 h, 15 s for 7 h and 30 s for 6 h, tension, 6.5 V/cm.
More than 98% of the nuclear genome of B. microti has been assembled and annotated. Approximately 3500 CDS are present in the genome, making it the smallest set of genes found in Apicomplexa. Comparison of the B. microti genome with that of other piroplasms revealed an unexpected diversity. Notably, B. bovis and Theileria parva genomes contain ∼7% more genes and T. annulata 16% more genes than B. microti ( Table 1 ). No synteny blocks encompassing more than five genes could be found between B. microti and other Apicomplexa. Between 60 and 80% of the additional genes found in B. bovis and T. parva are members of large multigenic families (e.g. vesa and SmORF or svsp and Tpr , respectively). These families do not exist or have not expanded in the B. microti genome. B. microti contains only one large lineage-specific multigenic family encoding the sero-reactive antigen BMN ( 39 , 40 ). Twenty-four bmn genes are found in the B. microti genome. The gene structure of B. microti is substantially divergent from that of other Apicomplexa. The number of introns is high with nearly 70% of B. microti genes interrupted with short introns of 20–25 bp in length.
Functional annotation revealed that 60% of the predicted proteins of B. microti share homology with proteins of known or putative functions and about half of them have an assigned biological function in the KEGG database ( Supplementary Table S1 ). Approximately 12% of the proteins with unknown function are specific to B. microti . Two rDNA units are found in tandem on chromosome III and 2 rRNA 5S encoding genes and 44 tRNA genes are also present and these are distributed throughout the three chromosomes.
The DNA sequence of B. microti has an average of 36% G + C content. The G + C content is much lower at chromosome ends. An A + T rich sequence of 1 kbp is present in four copies in the genome: one on both chromosome I and II and two on chromosome III. This sequence may function as a centromeric region (CEN, Supplementary Figure S4 ). Analysis of the subtelomeric regions revealed the presence of a small set of multigene families associated with repeated blocks of sequences of 0.8–3 kbp ( Figure 2 ). Some of these genes are members of the B. microti sero-reactive antigen family ( bmn ) ( 39 , 40 ). The presence of genes encoding surface antigens is a common feature of many eukaryotic genomes and it represent a important source of antigenic variation for eukaryotic parasites ( 41 , 42 ). B. microti subtelomeric regions contain pseudo-genes but also four copies of a sequence homologous to members of the Theileria specific Tpr multigenic family and three copies of a sequence homologous to members of the B. bovis vesa multigene family ( Figure 2 ) . Tpr - and vesa -like genes of B. microti are divergent from those found in other piroplasms and the presence of both multigene families in the same genome is unique to B. microti . Interestingly, whereas in Theileria the Tpr genes are not associated with the telomeres and are dispersed throughout the chromosomes, in B. microti Tpr-like sequences are found near the telomeres. Moreover, members of the bmn gene family are present near the telomeres as well as in the coding core of the chromosomes. Four of the B. microti repeated telomeric sequences are found in the middle of chromosome III (IIIc cluster, Figure 2 ), suggesting that this chromosome likely derived from a fusion event between two ancestral chromosomes.
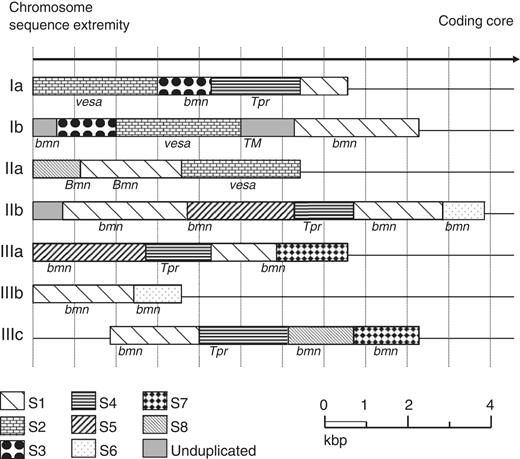
Mosaic organization of the Babesia microti chromosome extremities. Chromosome ends are labeled according to Supplementary Data . These regions are characterized by the presence of duplicated sequences scattered among the different chromosome extremities (S1–S8). Limits of sequence homologies have been calculated using miropeat and BLASTN analyses. Annotated genes are indicated on the figure. Most repeated genes are part of the bmn gene family and included truncated genes and pseudogenes. Several bmn genes are in transition regions between two duplicated sequences. The S2 sequence encoding a putative VESA antigen is repeated three times. The S4 sequence encodes Tpr orthologues and is repeated four times, two copies of which on chromosome ends IIb and IIIa are significantly shorter. The GC content in the chromosome ends is significantly lower than in the coding core. The sequence between S1 and S4 at extremity Ib encodes a putative sugar transporter ( TM ). The sequence is not duplicated but does not show any base composition bias compared to adjacent regions. It was not possible to precisely map the recombination sites associated with the rearrangements that took place at chromosome ends (average resolution of 100 bp).
Unlike other Apicomplexa that have a linear mitochondrial genome, B. microti has a circular 11 kbp mitochondrial genome with two inverted repeats of 2.5 kbp encompassing almost half of its size ( Figure 3 ). The genetic information carried in this genome is similar to that found in other Apicomplexa and encodes three proteins, cytb of the cytochrome bc complex (complex III) and coxI and coxIII of the cytochrome c oxidase (complex IV). The ribosomal lsu and ssu genes of B. microti are fragmented. Their presence and organization suggest a distinct evolution from that of other Apicomplexa. The six ribosomal genes encoding lsu fragments show an organization similar to that of previously annotated mitochondrial genomes from other piroplasms ( 43 ). Interestingly, unlike other species of the Babesia and Theileria families that lack detectable ssu genes, two ribosomal genes encoding ssu fragments were found in the B. microti mitochondrial genome.
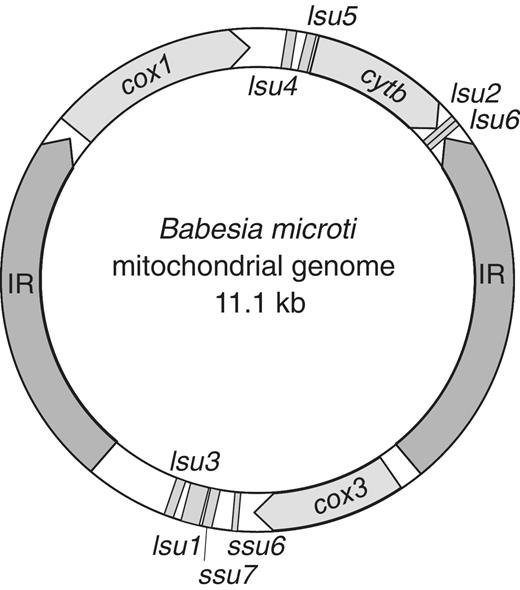
Circular organization of the Babesia microti mitochondrial genome. IR: inverted repeats; cox1 : cytochrome c oxidase subunit 1, cox3 : cytochrome c oxidase subunit 3, cytb : cytochrome bc complex subunit. The numbering of the ribosomal lsu and ssu genes fragments is performed according to the P. falciparum nomenclature.
B. microti defines a new Apicomplexan family
For many years, the classification of B. microti as a member of either the Babesia or Theileria families has been controversial. Although many taxonomists have classified B. microti as a member of the Babesia family, transmission electron microscopy analyses and recent molecular studies using two different genetic markers suggested that this organism may be distinct from the other species of this family ( 44–47 ). This limited set of molecular markers was, however, insufficient to ascertain the taxonomic position of this parasite among Apicomplexa. To address the evolution of B. microti among Apicomplexa, a set of 316 genes was selected from a total of 1002 CDS that share significant homology with CDS in the genomes of B. bovis , T. annulata and P. falciparum . These 316 genes belong to KEGG orthology groups and are present as single copy genes in the B. microti genome as well as that of eight Apicomplexan genomes selected for genome-scale phylogenetic analysis. These include B. bovis , T. annulata, T. parva , P. falciparum , Plasmodiumknowlesi , Plasmodiumvivax , Toxoplasma gondii and Cryptosporidium parvum . To locate the root of the phylogenetic tree, T.thermophila was used as an outgroup species. Two independent phylogenetic analyses produced in the same phylogenetic tree, placing B. microti at the root of piroplasms and separating it from the B. bovis and Theileria clades ( Figure 4 ). Approximately 88% of the selected proteins support the evolutionary separation of B. microti from B. bovis and Theileria species. The data indicate that Babesia is a paraphyletic group, the taxonomy of which must be revised. Furthermore, this analysis revealed that B. microti has evolved early in the evolution of piroplasms. These findings emphasize the need to create a new genus for the B. microti group of strains.
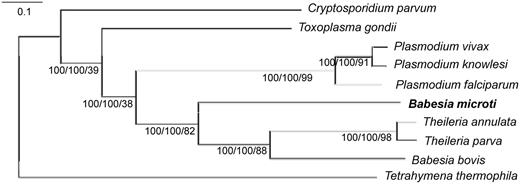
Babesia microti defines a new clade in the Apicomplexan phylum. The tree is inferred using a maximum likelihood approach on a concatenated alignment of 316 single-copy genes. Tetrahymena thermophila was included as outgroup. The same tree topology is inferred by a supertree approach compiling the 316 trees inferred from the 316 genes. Labels indicate the boostrap support from both the Supermatrix and Supertree analyses and the level of tree supporting the clade (%).
The minimal Apicomplexan metabolism of B. microti
Reconstruction of B. microti metabolism from its genomic information suggests high dependence of the parasite on glucose fermentation for energy production and redox regulation ( Figure 5 ). Genes encoding enzymes required for β-oxidation and hemoglobin degradation were not found in the genome of this organism. The analysis further indicates that the parasite may not synthesize heme or fatty acids de novo and lacks both mitochondrial and apicoplast pyruvate dehydrogenases. Furthermore, the function of the apicoplast seems to be limited to the production of the precursors of isoprenoids through the 2-C-methyl-D-erythritol 4-phosphate/1-deoxy-D-xylulose 5-phosphate pathway (MEP/DOXP) pathway, a metabolic function used by P. falciparum during its intraerythrocytic development ( 48 ). A ferredoxin/ferrodoxin:NAD oxidoreductase system is also present in the apicoplast and may provide electrons to the MEP/DOXP pathway resulting in the formation of isopentenyl diphosphate (IPP) and dimethylallyl diphosphate (DMAPP). Export of IPP and DMAPP to the cytosol has been shown to be an essential step in the synthesis of ubiquinones and dolichol and plays a critical role in Plasmodium development ( 49 ). B. microti lacks a mitochondrial superoxide dismutase, suggesting a minimally active respiratory chain. Consistent with this model, the mitochondrial antioxidant system is underdeveloped. A F 0 F 1 -ATPase made of a reduced number of subunits is found in the genome, suggesting that oxidative phosphorylation might take place in this organism. The gene encoding frataxin, an enzyme involved in the assembly of iron-sulfur clusters, was not identified in the genome, indicating that it is either lacking or highly divergent from known eukaryotic orthologs.
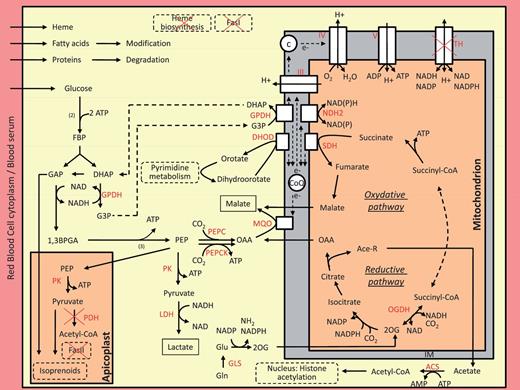
An integrated model for central metabolism of Babesia microti . Arrows show the direction of net fluxes. Lactate and malate are expected to be the major end products of the central metabolism. The gene encoding the lactate dehydrogenase is likely obtained by lateral transfer from a mammalian host. The apicoplast is devoted to the production of isoprenoids precursors. Numbers in brackets represent the biochemical steps in the reaction. Dashed arrows connect two reactions using the same metabolite. For simplicity, the membranes of the apicoplast and the outer membrane of the mitochondrion is not shown. Abbreviation used are: (1) Metabolites: 1,3BPGA, 1,3-bisphosphoglycerate; 2OG, 2-oxoglutarate; Ace-R: acetate/acetyl-CoA, c, cytochrome c; CoQ, Coenzyme-Q, ubiquinone; DHAP, dihydroxyacetone phosphate; e-, electrons; FBP, fructose 1,6-bisphosphate; G3P, glycerol-3-phosphate; GAP, glyceraldehyde-3-phosphate; Glu, glutamate; Gln, glutamine; OAA, oxaloacetate, PEP, phosphoenolpyruvate. (2) Enzymes (in red): ACS, AMP-forming acetyl-CoA synthetase; DHOD, dihydroorotate dehydrogenase; GLS, glutamine synthetase; GPDH, glycerol-3-phosphate dehydrogenase (FAD- or NAD-dependent enzymes); LDH, lactate dehydrogenase; MQO, malate:quinone oxidoreductase; NDH2, type II NADH:quinone oxidoreductase; OGDH, oxoglutarate dehydrogenase; PEPC, PEP carboxylase; PEPCK, PEP carboxykinase; PDH, pyruvate dehydrogenase; PK, pyruvate kinase; SDH, succinate dehydrogenase; TH, transhydrogenase; III, complex III of the respiratory chain; IV, complex IV of the respiratory chain; V, F 0 F 1 -ATP synthase.
Another unique feature of B. microti central metabolism is the acquisition of two genes by lateral gene transfer: Bm ldh encoding lactate dehydrogenase (LDH) and Bm tpk encoding thiamine pyrophosphokinase (TPK). Bm ldh and Bm tpk genes lack introns and are adjacent to each other on chromosome I but in opposite orientation. This chromosomal site is at the junction between the subtelomere and the coding region, a genomic location known in many organisms to be susceptible to double-stranded breaks and thus suitable for insertion of foreign DNA. Reverse transcriptase–PCR analyses show that Bm ldh is transcribed during the blood stage of the parasite, whereas the Bm tpk is not ( Supplementary Figure S5 ). In Apicomplexa, LDH activity is believed to have originated from a duplication of the LDH-like malate dehydrogenase ( mdh ) gene ( 50–52 ). However, sequence and phylogenetic analyses showed that the B. microti LDH is not related to Apicomplexan LDH. Both the protein and the gene share strong homology with their mammalian counterparts, supporting a lateral gene transfer mode of acquisition from the host ( Supplementary Figure S6A ). Equally unique among piroplasms, B. microti expresses a TPK protein (BmTPK) with 70% identity to TPKs from Bartonella species, which are transmitted by the same tick vector that transmits B. microti . BmTPK shares very low sequence similarity with TPKs from other eukaryotes ( Supplementary Figure S6B ). Together, these findings suggest that the Bm tpk gene may have been acquired from bacteria by B. microti through lateral transfer, an event that likely occurred during co-infection either in the tick vector or in a mammalian host following a tick bite. The presence of the Bm tpk gene in the B. microti genome may enable the parasite to produce thiamine pyrophosphate (ThiaPP), an essential cofactor for key enzymes of the tricarboxylic acid (TCA) cycle.
The organization of the glycolytic machinery and TCA cycle of piroplasm suggests a central role of the inner membrane of the mitochondria in parasite metabolism under anaerobic and microaerophilic conditions ( 53 ). B. microti has several novel metabolic pathways consistent with this model ( Figure 5 ). In B. microti , no mdh gene could be identified, suggesting that the malate:quinone oxydoreductase (MQO) alone is responsible for the production of malate. In the cytosol, the electron acceptor, oxaloacetate may be obtained either from glycolysis or the TCA cycle. A glycerol-3-phosphate (G3P)/dihydroxyacetone phosphate (DHAP) shuttle might provide electrons to feed MQO for malate production. However, there is no evidence that this electron transfer is fully responsible for the regulation of the redox balance in the cell. It is likely that B. microti produces pyruvate during its life cycle in addition to lactate and malate.
Genome analysis has also revealed that B. microti encodes a glutamine synthetase (GLS). The presence of the GLS enzyme in B. microti is unique among piroplasms and could provide the parasite with an alternative route for nitrogen assimilation and ATP production. GLS and glutamate dehydrogenase (GDH) enzymes are essential to convert glutamine and glutamate into 2-oxoglutarate (2OG) in order to feed the TCA cycle. Interestingly, the GLS and GDH enzymes of B. microti lack a signal peptide, suggesting that 2OG synthesis may take place in the cytosol followed by subsequent transport into the mitochondria where the TCA cycle might work in a reductive way from 2OG to malate. Because of the absence of TPK activity during intraerythrocytic development, the acquisition of ThiaPP from the host might be critical to equilibrate the redox balance of the mitochondria through the activity of oxoglutarate dehydrogenase. Based on the available genomic information, the oxidative TCA cycle of B. microti appears to be similar to that of P. falciparum ( 54 ). Complex III and IV of the respiratory chain are also present in B. microti . The proton gradient might be used by the F 0 F 1 -ATPase to produce ATP when the respiratory chain is highly active.
Glycosylphosphatidylinositol biosynthesis and glycosylphosphatidylinositol proteome
Glycosylphosphatidylinositol (GPI) anchors are glycolipids attached to many cell surface glycoproteins of lower and higher eukaryotes including important cell surface parasite antigens ( 55–59 ). They have been shown to play a critical function in the pathogenesis of Apicomplexan parasites ( 60–62 ). GPI anchors are synthesized in a stepwise manner in the membrane of the endoplasmic reticulum and attached to the C-terminal end of proteins following translation. The GPI biosynthetic pathways in eukaryotes are highly conserved and produce a core structure of ethanolaminephosphate-6Manα1–2Manα1–6Manα1–4GlcNα1–6-D-myoinositol-1-HPO4-lipid, where the lipid is diacylglycerol, alkylacylglycerol or ceramide. This minimal GPI structure may be embellished with various side chain modifications such as additional sugars or ethanolamine phosphate in a species-specific manner. In addition to GPI, various intermediate molecules of the biosynthesis pathway are also accumulated in the cytosol of parasites and some of them are secreted and can induce inflammatory responses ( 63–65 ). Genes encoding enzymes of the GPI biosynthetic pathway were found in the B. microti genome indicating that the synthesis of GPIs occurs in this organism ( Supplementary Table S3 ). The pagp1 deacylase was not found in the B. microti genome, suggesting that GPI anchors made by this parasite are likely to contain fully acylated inositol. Unlike Plasmodium or Toxoplasma species , which express three mannosyltransferases and harbor GPIs with three mannose residues in their core structure ( 66 ), the B. microti genome encodes only two mannosyltransferases: PIGM and PIGV, and no homologues of PIGB mannosyltransferase could be found. These findings indicate that the core structure of the B. microti GPI anchors may be different from that found in other eukaryotes. The implications of such a unique GPI structure on host immune response and parasite evasion remain to be determined.
Twelve putative GPI-anchored merozoite surface protein-encoding genes have been identified in the B. microti genome. All genes lack introns and their encoded proteins have no homologs in other organisms, including other Apicomplexa.
New insights into babesiosis therapy
Babesiosis therapy has generally been based on empirical trials and medical consensus that combines antimalarial drugs such as quinine or atovaquone with antibiotics such as clindamycin or azithromycin ( 67–69 ). Treatment failure due to suspected parasite drug resistance has been reported ( 70 ). Genome analysis indicates that B. microti lacks the proteases necessary to digest host hemoglobin. This result along with the lack of hemozoin formation by this parasite may explain the ineffectiveness of chloroquine in Babesiosis therapy and suggests that other compounds of the aminoquinoline family are unlikely to be effective. Conversely, because the B. microti genome encodes a 1-deoxy-D-xylulose 5-phosphate reductoisomerase (Dxr) enzyme, the parasite may be sensitive to the antibiotic fosmidomycin. Of the three major metabolic pathways in the apicoplast, the DOXP pathway seems to be the most conserved among Apicomplexa ( 71 ). Sequence analyses revealed the presence of DOXP genes in several Apicomplexa parasites including Babesia sp, Plasmodium, T. gondii, Theileria and Neospora caninum . The encoded Dxr proteins and their fosmidomycin binding sites are well conserved ( 72–75 ). Studies in P. falciparum and Babesiadivergens have shown that both fosmidomycin and FR900098 inhibit DOXR reductoisomerase and block parasite proliferation ( 75 ). These compounds have no effect on the growth of T. gondii or Theileria, even though both parasites express Dxr enzymes, which in vitro have been shown to be inhibited by Fosmidomycin and FR900098. Cell biological analyses demonstrated that the effectiveness of these drugs against Plasmodium and Babesia species is largely due to the new permeation pathways (NPP) induced by the parasites on the erythrocyte membrane ( 71 , 73 ). Although we do not yet know whether B. microti induces NPP-like pathways on its host plasma membrane and whether it expresses transporters on the plasma and apicoplast membranes capable of transporting these drugs, our metabolic reconstruction analysis suggests that such transport mechanisms may exist in B. microti . Therefore, we predict that both fosmidomycin and FR900098 are likely to inhibit B. microti development and thus could be effective drug candidates to treat human babesiosis. Fosmidomycin is currently in phase two clinical trials for treatment of uncomplicated malaria and has been shown to have an excellent safety profile in humans ( 75 ). Studies aimed to assay the sensitivity of B. microti to fosmidomycin and FR900098 in mice and hamsters are warranted and will set the stage for the advancement of this class of compounds for the treatment of human babesiosis. The B. microti genome also encodes dihydropteroate synthase and dihydrofolate reductase enzymes that are the target of sulfadoxine and pyrimethamine, suggesting that antifolates may be useful in the treatment of human babesiosis ( 76 ).
In conclusion, sequencing of the B. microti genome reveals new insights into the evolution of this parasite and other Apicomplexan pathogens and opens new avenues for future design of improved diagnostic assays and antimicrobial drugs. Erythrocytes are the only cells invaded by this parasite ( 37 ). Thus, the unique and rudimentary metabolism of this human pathogen indicates that the genome of B. microti contains the minimal genomic requirement for successful intraerythrocytic parasitism.
ACCESSION NUMBERS
The genome sequence data have been submitted to EMBL and are available under accession numbers: FO082868, FO082871, FO082872 and FO082874.
FUNDING
Intervet Schering Plough Animal Health; French ministry of research; French Agence Nationale de la Recherche ‘Investissements d’avenir/Bioinformatique’ (ANR-10-BINF-01-02, ‘Ancestrome’ to V.R.); National Institutes of Health [AI007603 to C.B.M.]; the Burroughs Wellcome Fund award [1006267 to C.B.M.]. Funding for open access charge: Intervet Schering Plough Animal Health; French ministry of research.
Conflict of interest statement . None declared.
ACKNOWLEDGMENTS
We thank Ronan Peyroutou and Zoé Gallice for analyzing the raw sequencing data. B.N., B.V., A.D., P.W. and V.B. sequenced, assembled and finished the sequence. K.H.-K., A.D. and B.N. annotated the genome sequence. E.C., K.H.-K., Y.A., V.R. and C.B.M. performed general functional annotation. S.R., B.C., S.D., K.M.-M., C.P.V. and A.G. isolated and characterized the R1 strain. V.R. and V.B. performed phylogenetic analysis. E.C. and F.B. reconstructed the central metabolism. F.D.-G., H.S.-E., R.T.S., E.C., V.B. and K.H.-K. analyzed the GPI biosynthesis pathway and GPI proteome. P.J.K., V.B., E.C. and C.B.M. analyze drug targets. T.P.S. strongly supported this work. E.C. and C.B.M. wrote the article. Most authors discussed the results and commented on the manuscript.
REFERENCES
Author notes
†Eric Précigout initiated the project but left us suddenly in 2006
The authors wish it to be known that, in their opinion, the first three authors should be regarded as joint First Authors.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments