Herpesviral Latency—Common Themes


Department of Reproductive Immunology and Pathology, Institute of Animal Reproduction and Food Research of Polish Academy of Sciences, Tuwima Str. 10, 10-748 Olsztyn, Poland
*
Author to whom correspondence should be addressed.
Pathogens 2020, 9(2), 125; https://doi.org/10.3390/pathogens9020125
Submission received: 22 January 2020 / Revised: 9 February 2020 / Accepted: 14 February 2020 / Published: 15 February 2020
(This article belongs to the Section Human Pathogens)
Abstract
:
Latency establishment is the hallmark feature of herpesviruses, a group of viruses, of which nine are known to infect humans. They have co-evolved alongside their hosts, and mastered manipulation of cellular pathways and tweaking various processes to their advantage. As a result, they are very well adapted to persistence. The members of the three subfamilies belonging to the family Herpesviridae differ with regard to cell tropism, target cells for the latent reservoir, and characteristics of the infection. The mechanisms governing the latent state also seem quite different. Our knowledge about latency is most complete for the gammaherpesviruses due to previously missing adequate latency models for the alpha and beta-herpesviruses. Nevertheless, with advances in cell biology and the availability of appropriate cell-culture and animal models, the common features of the latency in the different subfamilies began to emerge. Three criteria have been set forth to define latency and differentiate it from persistent or abortive infection: 1) persistence of the viral genome, 2) limited viral gene expression with no viral particle production, and 3) the ability to reactivate to a lytic cycle. This review discusses these criteria for each of the subfamilies and highlights the common strategies adopted by herpesviruses to establish latency.
1. Introduction
Herpesviruses are a family of enveloped viruses with large double-stranded DNA genomes [1]. Their life cycle is characterized by two phases: the lytic cycle—when the virus is actively replicating and the latency, which enables these viruses to persist for the lifetime of the host. Latency is defined as a state in which: 1—the viral genome persists; 2—viral gene expression is limited and there is no viral particle production, and 3—there is a possibility of reactivation back to the lytic replication [2]. These three criteria set latency apart from a persistent infection, during which new viral particles are produced; and abortive infection, in which the virus is unable to reactivate [1]. The name of this virus family derives from the Greek word herpes (to creep) and originally referred to the spreading lesions observed during herpes simplex virus (HSV) or varicella zoster virus (VZV) infection [3], but it could also refer to the quiescent nature of herpesviral infections, since latency is the hallmark of their life cycle.
Herpesviruses include three subfamilies: Alpha-, Beta-, and Gammaherpesvirinae, each one of them having different characteristics with regard to tropism, length of the replicative cycle and associated diseases. The Alphaherpesvirinae, including HSV-1 and 2 and VZV, show relatively broad host and cell tropism infecting both human and non-human cells. With regard to the cell types, in vitro virtually all types can become infected including fibroblasts, epithelial cells, and neurons [4]. The replicative cycle of alpha-herpesviruses is very short (hours), they spread fast in culture and latency is established in sensory neurons [5]. The Betaherpesvirinae include the human cytomegalovirus (HCMV), human herpesvirus 6A and B (HHV-6A and B), and human herpesvirus 7 (HHV-7) and have species-specific tropism and a longer (days) replication cycle [6]. They spread slower and the infection results in characteristic cell enlargement [3,7]. This subfamily infects epithelial cells, endothelial cells, smooth muscle cells, monocytes, T lymphocytes, and fibroblasts and establishes latency in CD34+ hematopoietic stem cells and CD14+ monocytes or T lymphocytes, but recent studies suggest that HCMV might also be able to persist in neuronal cells [3,7,8,9,10]. The Gammaherpesvirinae, which include Epstein–Barr virus (EBV) and Kaposi’s sarcoma-associated herpesvirus (KSHV), have very narrow tropism both with regard to the host and the cell type, they are species-specific and infect lymphocytes, epithelial and endothelial cells, and establish latency in B lymphocytes. Concerning comparison of the life cycles of the three herpesvirus subfamilies, it is important to state that for the alpha- and the beta-herpesviruses the lytic cycle is the default path, while for the gammaherpesviruses it is the latency that predominates. Consequently, the latency of the Gammaherpesvirinae has been studied the most and is therefore best understood. One more unique feature of the latent gammaherpesviruses is their association with oncogenesis. EBV is associated with large number of malignancies, among others Burkitt’s lymphoma, Hodgkin’s disease, nasopharyngeal carcinoma, and post-transplant lymphoproliferative disease (PTLD) [11]. KSHV is the causative agent of Kaposi sarcoma (KS), plasmablastic variant of Multicentric Castleman’s disease (MCD) and primary effusion lymphoma (PEL) [12]. Both of the viruses induce tumor formation through persistence of the viral genome and therefore continuous expression of viral latent proteins (see also below), which have the ability to inhibit apoptosis, evade anti-viral immune response, and induce proliferation [13].
2. Latent Genome Persistence
As mentioned earlier, herpesviruses have large dsDNA genomes that are linear when inside of the viral particle, however they become circularized upon entering the infected cell [1]. During the lytic phase herpesviruses use the ‘rolling circle mechanism’ to replicate their genome and most of the proteins needed for this process are encoded in the viral genomes. Lytically replicating virus practically converts the infected cell into ‘a virus-producing factory’, redirecting all the cellular processes towards viral protein and DNA synthesis. In contrast, during latency there is no particle production and the viral gene expression is limited to a minimum. Beta and gamma herpesviruses had to develop a strategy to maintain their latent genomes in dividing cells (hematopoietic progenitors or lymphoid cells, respectively). The genomes persist in the form of circular molecules—episomes, attached to cellular chromosomes with the help of viral and cellular proteins [1,14,15]. Alphaherpesviruses, on the other hand, establish latency in terminally differentiated, non-dividing neurons, therefore in this case, there is no risk of the viral genome being lost and consequently also no need to tether the episome to cellular chromatin. Among the human herpesviruses an exception with regard to the maintenance of the latent genome as an episome is HHV-6, which integrates in the telomere sequences of the host chromosomes [16,17,18,19,20,21]. The herpesviruses that establish latency in proliferating cells duplicate their latent episomes in the nucleus once per cell cycle, in synchronization with the cellular replication in the S phase [22]. In contrast to lytic replication that is executed mostly by the viral proteins, latent replication is performed with the help of cellular machinery. Additionally, gammaherpesviruses encode origin binding proteins (OBPs), latency-associated nuclear antigen 1 (LANA1) for KSHV and Epstein–Barr nuclear antigen 1 (EBNA1) for EBV, that are responsible for recruiting the cellular replication proteins to viral genomes [23,24,25,26]. Viral OBPs bind specific DNA sequences within the viral genome (latent origin of replication) and at the same time interact with cellular chromatin binding proteins, thus mediating the tethering of viral genomes to cellular chromosomes [26,27]. Even though both the beta- and the gammaherpesviruses persist in dividing cells, for a long time the OBPs have been known only for the gammaherpesviruses. However, recently, the existence of a latency protein responsible for both genome tethering and latent replication has been reported for HCMV [28]. Future research will show how similar in its functions is this protein to gammaherpesviral OBPs.
Herpesviral genomes inside the viral particle are compacted with the help of spermine, but do not contain nucleosomes [29,30,31]. In contrast, latent genomes of herpesviruses are fully chromatinized, in a manner similar to the cellular DNA [32,33,34,35,36]. As a consequence, transcriptional activity of the viral latent chromatin can be regulated by histone modifications and their changes, using mechanisms as in the case of cellular chromatin.
3. Viral Gene Expression Patterns in Latency
The original definition of latent and lytic states in case of herpesviruses describes the lytic cycle as the phase in which transcription from the viral genomes is very high, all or almost all the viral genes are expressed, and latency as a condition when only a limited number of genes is expressed. However, the more sensitive assays we use and the more we know about the latent expression the less limited it appears to be. The subfamily of herpesviruses that is most studied with respect to latency are the gammaherpesviruses; therefore, we will start the overview of the latent programs from them.
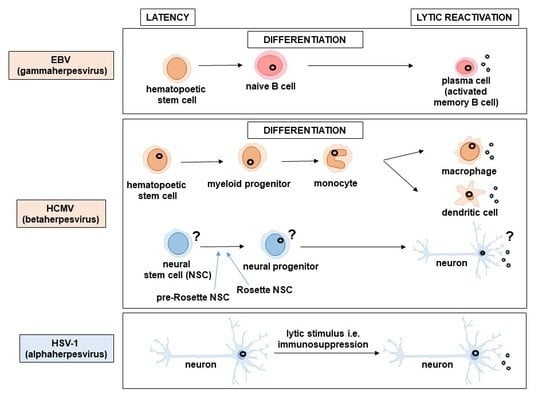
EBV infects naïve B cells, which are the most abundant cells circulating in the blood, and uses B cell differentiation to promote viral latency establishment in the long-lived memory B cells (Figure 1). Studies of EBV-induced tumors (Burkitt lymphoma, nasopharyngeal carcinoma, EBV positive Hodgkins disease) revealed that the virus engages different latency programs in different tumors [26]. Similarly, in the B cells of healthy EBV positive individuals, different viral transcription programs are found to be associated with specific differentiation stages of the B cell [37]. The EBV latency proteins include a set of EBNAs and three latency-associated membrane proteins (LMPs) (Table 1) [26]. The latency program with the highest expression is the type III latency found in the lymphoblastoid cell lines immortalized by EBV and the proteins expressed in these cells include: EBNA-1, -2, -3a, -3c, -3b, –LP, and LMP-1, -2a, and -2b [26]. This program is also found in infected naïve B cells [37]. In nasopharyngeal carcinoma and EBV positive Hodgkins disease the type II latency was detected, which is characterized by expression of EBNA-1 and LMP-1 and/or -2 [26]. Type II latency program was also found in germinal center B cells [37]. Cells of Burkitt lymphoma represent EBV type I latency expressing only EBNA1 [26]. This program is found also in proliferating memory B cells [37]. The quiescent memory B cells show the so-called type 0 latency, where only the viral genome is detected, but not the viral proteins. Finally, when the memory B cells become activated and turn into plasma cells, EBV enters the lytic cycle [37].
The functions of the EBV latent proteins allow the virus to successfully persist in the infected cells. EBNA-LP and EBNA-2 are responsible for the transcriptional regulation of expression of other latency proteins [38]. EBNA1 plays an important role in virus persistence, by tethering the viral genome to cellular chromosomes and participating in viral genome segregation and latent replication [39,40]. Additionally, EBNA1 acts also as a transcriptional activator [41,42]. LMP-1 belongs to the tumor necrosis factor (TNF) superfamily of proteins and mimics the CD4+ T cell signals. It promotes B cell survival by constitutively upregulating NF-κB signaling [43,44]. The membrane protein LMP-2a mimics B cell receptor survival signals [45].
In addition to protein expression, also non-coding RNAs are expressed in all types of EBV latency, these include EBV-encoded small RNAs (EBER 1 and EBER2), miRNAs encoded in the BamHI A rightward transcript (BART) and the BHRF1 locus as well as recently identified circular RNAs [46,47,48,49]. EBERs confer oncogenic properties on the virus infected cells and antagonize the function of PKR (protein kinase RNA-dependent), thereby preventing apoptosis of the infected cells and influencing the innate immune responses [50,51,52,53]. BHRF1 miRNAs are expressed in the type III latency cells, they inhibit apoptosis and promote passage through the cell cycle [54]. On the other hand, the BART miRNAs are expressed in all types of latency and have been shown to be important for latency maintenance [55].
The second human gammaherpesvirus, KSHV, so far is not known to have multiple latency programs, the way it has been described for EBV. Instead, there are four major open reading frames (ORFs) expressed during KSHV latency: ORF73 (LANA), ORF72 (viral cyclin—vCyc), ORF71 (viral FLICE inhibitory protein—vFLIP), and ORF K12 (Kaposins) (Table 1) [27]. LANA is the origin binding protein of KSHV and is therefore responsible for latent replication of the viral genome as well as tethering the genome to cellular chromosomes [24,56,57]. Additionally, LANA also inhibits the action of p53 [58] promoting the survival of infected cells and inhibits the function of Rb [59]. LANA was shown to promote cell cycle progression by binding and sequestering glycogen synthase kinase-3β (GSK3β) [60]. KSHV vCyc is a homologue of the cellular cyclin D2 [61] and was shown to contribute to the tumorigenic properties of KSHV [62]. vFLIP similarly to cFLIP promotes cell survival, by inhibiting caspase 8. It also inhibits autophagy promoting cell survival [63]. It induces NF-κB signaling and therefore promotes latent state and prevents lytic reactivation [64]. The fourth latent ORF encodes a family of Kaposin proteins (Kaposin A, Kaposin B, and Kaposin C) [65]. Kaposin A is able to induce cell transformation [66] and was reported to inhibit apoptosis through interaction with septin 4 [67]. Kaposin B was shown to stimulate cytokine release and therefore contribute to the proinflammatory environment of KSHV-associated tumors [68]. The function of Kaposin C is not known yet.
In addition to the four major latent KSHV ORFs expressed in the KSHV-associated tumors: PEL (primary effusion lymphoma) and KS (Kaposi’s sarcoma) cells, there is also vIRF3 (also called LANA2), which is expressed only in PEL and multicentric Castleman’s disease (MCD) [69]. This protein is a homologue of the cellular interferon regulatory factor (IRF) and inhibits interferon (IFN) induction and apoptosis [69,70,71]. Another protein expressed at low levels in latent PEL cells, but not in KS is vIL-6, which is a constitutively active homologue of cellular IL-6 [72,73,74,75,76]. vIL-6 promotes proliferation and prevents apoptosis of PEL cells [76,77,78]. The cell type specific expression of vIRF3 and vIL-6 suggests that KSHV might have different latency programs depending on the type of infected cell. In fact this has been observed in a recent study [79] comparing latency in TREx-BCBL1RTA and iSLK.219. Another type of exception to the canonical latency program of the four major latency proteins is the K1 protein, which is expressed in latency at low levels and is induced in the lytic cycle [72,80]. K1 is a membrane protein that mimics B cell receptor signaling. It is able to promote cell growth and transformation [81,82] and prevent apoptosis of infected cells [83,84]. K1 is a functional homologue of EBV LMP-2A.
Similar to EBV, KSHV also encodes non-coding RNAs, including miRNAs [85,86], long non-coding RNA-polyadenylated nuclear RNA (PAN) expressed at only low levels in latency [87], and circular RNAs encoded in vIRF4 and PAN loci [49]. KSHV miRNAs have been shown to promote angiogenesis [88], affect differentiation of infected cells [89,90], and prevent lytic reactivation through different mechanisms [91,92].
Initially, the latency program for HCMV was thought to include, as in the case of other herpesviruses, a limited number of genes. Therefore, also in the case of this virus several genes associated with latency were identified, including: UL138, UL111A, latency unique nuclear antigen (LUNA), US28, a splice variant of IE1—IE1x4, lncRNA2.7, 4.9, and UL144. UL138 is required for HCMV latency establishment (Table 1) [28,93], it induces TNF receptor type I (TNFRI) [94] and downregulates multidrug resistance-associated protein-1 (MRP1) [95]. Additionally, it was shown to increase surface expression and activate epidermal growth factor receptor (EGFR), a receptor that is critical for cell survival, proliferation, and differentiation and therefore promote virus persistence [96]. LUNA, latency unique nuclear antigen (also called UL81-82ast) is an anti-sense transcript encoded in the UL81–82 locus [93,97,98]. LUNA is required for reactivation [98] and more recently it was published to have de-SUMOylating activity, which allows it to disrupt promyelocytic leukemia (PML) nuclear bodies [99]—cellular structures with an established anti-viral function. US28 is a chemokine receptor (vGPCR)—that has been detected in HCMV latency [93,100,101,102]. It was shown to affect differentiation of infected cells and to be required for latency establishment in myeloid progenitors [102,103,104,105]. US28 affects several signaling pathways, which results in silencing of the HCMV major immediate early promoter (MIEP) [103].
Some of the HCMV proteins show expression of specific splice variants depending on the phase of the viral life cycle. One such example is UL111A, which encodes a viral homolog of IL-10, which is expressed in the lytic cycle, while in latency a shorter splice variant LAcmvIL-10 is detected [106,107]. Cellular IL-10 has broad immunosuppressive properties, but the LAcmvIL-10, so far has only been shown to downregulate MHC II in myeloid cells [108], which results in impaired recognition of latently infected cells by CD4+ T cells [109]. Another protein that shows splice-variant-specific expression in latency is IE1x4. In the lytic cycle, IE1 full length protein is the major immediate early transcript, in latency a shorter variant IE1x4 is expressed and has been shown to be important for latent genome replication and maintenance [28]. Additionally, two long non-coding RNAs are expressed during HCMV latency: RNA2.7 and RNA4.9. The lnc4.9 binds polycomb repressor complex (PRC2) and is responsible for the repression of lytic gene expression. In the lytic cycle, lnc2.7 has anti-apoptotic properties; however, its function in latency has not been studied so far [110]. One more level of complexity in the regulation of latent transcription is demonstrated by UL144, whose expression in latency is virus-isolate-specific [111]. UL144, also called herpesvirus entry mediator (HVEM/TNFRSF14) belongs to the TNF receptor superfamily of proteins and therefore could participate in regulation of immune cell function, but its function in latency still needs to be investigated [112].
All of the HCMV transcripts described above play undoubtedly crucial roles in latency, however, an increasing number of studies show that the expression pattern in latency includes many more genes [93,101,113,114,115]. Rosetto et al. have shown the expression of mRNA of lytic replication genes (UL84 and UL44) in both naturally infected cells from HCMV positive donors and in latent models [113]. Another study which also used both natural and model latency systems has identified an even larger group of genes expressed in latency [114]. The authors classified HCMV genes into two groups. The first one contained highly expressed genes, which were not differentially regulated in latency vs. lytic replication. 30 genes classified to the second group were expressed at overall lower level and were differentially expressed between these two states. This group of genes might be important in defining the distinct infection patterns. Most recent results suggest that in the case of HCMV the difference between the lytic and latent transcriptome might be rather quantitative than qualitative and that the set of viral genes expressed in latency resembles the genes expressed in the late stage of the lytic cycle [115]. Certainly, it is well documented that after initial infection associated with high transcription levels, the HCMV genome becomes chromatinized and progressively repressed, which promotes the establishment of latency characterized by overall lower gene expression [6]. Latent transcription program of HCMV remains a controversial topic and further research will be needed to fully define it.
Most of the human herpesviruses maintain their genomes during latency in an extrachromosomal form of episomes. However, as mentioned earlier, HHV-6A/B represents a unique example of genome integration into telomeric regions of chromosomes [116]. Since HHV-6 has a wide cell tropism, the integration might occur also in gametes (germinal cells). According to Mendelian law, the integrated HHV-6 genome can be transferred to 50% of the offspring. This situation can result in an individual carrying an integrated copy of the HHV-6 genome in all the cells of the body. This condition is called inherited chromosomally integrated HHV-6 (iciHHV-6) and occurs with an approximate frequency of 1% among the world human population [117,118].
The transcriptional activity of integrated HHV-6A/B genome has not been studied extensively. In fact, comprehensive annotation of HHV-6A and HHV-6B genomes has been carried out only very recently [119]. Long before this extensive transcriptome and translatome analysis were performed, four latency associated-transcripts of HHV-6 (H6LTs) were described in HHV-6B-latently infected macrophages and these transcripts were suggested to give rise to three proteins: ORF99, ORF142, and ORF145 [120]. However, more sensitive assay revealed that these H6LTs were only detectable during latency establishment and upon reactivation, but not during latency as such [121]. In HHV-6A-infected peripheral blood mononuclear cells expression of U94 transcript was detected during latency. U94 was shown to inhibit viral lytic replication and was therefore proposed to promote latency establishment and maintenance [122,123]. More recent RNA-seq analysis did not detect any transcripts in patient derived iciHHV-6A cells and in in vitro generated 293-HHV-6A cells. These results might suggest that the latent HHV-6 genome is maintained in a transcriptionally quiescent and highly condensed chromatin state confirmed by detection of repressive histone modifications H3K9me3 and H3K27me3 [124]. Future studies will hopefully reveal the reason for differences in detection of latent transcripts in HHV-6A infected cells. Recent publications highlighted the role of small non-coding RNAs (sncRNAs) including miRNAs in herpesvirus latency maintenance and in regulation of the lytic switch [125,126]. In the case of HHV-6, transcription of miR-U86 was detected in latency and observed to be upregulated in the so-called ‘transactivation’, which is the very early stage of lytic reactivation characterized by the expression of some lytic transcripts, but lack of expression of viral proteins [127]. Upregulation of miR-U86 upon transactivation remains in agreement with the previous suggestion that miR-U86 inhibits the expression of U86 and therefore prevents lytic replication and supports latency [128]. Taken together, detection of the H6LTs only during latency establishment and upon reactivation, but not in latency as such [121] and detection of several transcripts including miR-U86 upon transactivation [127], led to the hypothesis that H6LTs and miRNAs could be detected only at specific time points, i.e., at the point of latency establishment and during transactivation [124,127].
Unfortunately, mechanisms of HHV-7 latency establishment and maintenance are still under investigation and latency genes of HHV-7 have not yet been identified.
Initially, HSV latent transcription was considered to be limited to the latency associated transcripts (LATs), which are found on the opposite strand of the ICP0 locus, encoding the major immediate early protein (Table 1) [129,130]. Of note, latency-associated transcripts (LATs) of all alphaherpesviruses originate from genome regions encoding ICP0 homologs [131]. The LATs are not required for latency establishment [132,133,134], but they participate in repression of lytic genes and therefore contribute to latency establishment [135,136,137,138]. LATs also inhibit apoptosis of latently infected cells [139,140,141]. The LAT transcripts include short non-coding RNAs—sRNA1 and sRNA2 [129] and the long non-coding RNAs (lncRNAs) [132]. Out of 17 miRNAs encoded by HSV-1, 11 are found within or close to LAT locus and 7 are expressed in latency [142,143,144,145]. The latently expressed miRNAs repress transcription of lytic genes and affect viral replication in neuronal cells [145,146]. Over the years, evidence has accumulated showing that the latency of HSV-1 might be much less “silent” than previously thought and that expression of lytic genes can be detected in latent cells [147,148,149,150,151,152,153,154,155,156,157,158,159,160]. It was shown that reactivation of HSV in latency is quite frequent and spontaneous [161,162]. It has also been observed that only ~30% of latently infected neurons expresses LATs, but the population of cells that express LATs changes over time, such that each cell expresses LATs at some point [163,164,165,166]. Additionally, ICP0—the lytic protein, was shown to regulate transcription of LATs and chromatin structure of the latent genome [157]. These observations challenge the binary view on HSV transcription, which assumes either highly regulated lytic program or continuous expression of latent genes (LATs) [160,167]. One of the hypotheses explaining the detection of lytic transcripts in latency says that they result from frequent abortive reactivation episodes in only few cells [168]. Alternatively, detection of lytic transcripts in latent models could imply changes (possibly also cyclic changes) in viral gene expression signifying regulation of viral transcription in latency, i.e., a possible “latency program” as others put forward [160,169]. It remains to be determined whether detection of lytic gene expression in latency corresponds to frequent abortive infection or is a result of dynamic expression changes.
In VZV-latently infected human ganglia, several transcripts have been detected including ORF21, 29, 62, 63, and 66 [170,171]. Further quantitative assays revealed that only ORF63 was detected consistently in latently infected trigeminal ganglia, it was also the most abundant transcript [172,173]. ORF63 was shown to be important for latency establishment and to suppress apoptosis in human ganglia under experimental conditions [174,175]. Subsequently, RNA as well as the protein of an additional gene, ORF4 were identified in latently infected human ganglia and shown to be important for latency establishment [176]. Recently identified VZV latency-associated transcript (VLT) provides a novel aspect and might bring breakthroughs in our understanding of VZV latency and reactivation [177,178]. VLT is expressed in a few copies per neuron during the latent phase, but alternatively spliced VLT variants can be detected during lytic cycle and are termed VLTly. VLT in contrast to VLTly, is possibly expressed from a neuron specific promoter, which would explain a single unique VLT isoform predominating in latently infected ganglia [177]. The VLT resembles the HSV LAT and is encoded antisense to VZV ORF61 (a homolog of HSV ICP0). Both proteins VZV ORF61 and HSV ICP0 (infected cell polypeptide 0) belong to the immediate early category (IE) and are required for the viral lytic cycle. Therefore, it is hypothesized that VLT plays a role in latency maintenance by inhibiting ORF61 expression and function [131,177]. In conclusion, VZV latency in human ganglia is in most cases restricted to the expression of both latency associated transcripts: VLT and ORF63; however, not all of the TG (trigeminal) neurons are transcriptionally active. Despite the potential of both of these transcripts to be translated during the latent phase, the viral proteins remain undetected in latently infected human TG neurons by immunohistochemistry [177].
4. Virus Reactivation
Herpesviruses from all the three subfamilies can periodically reactivate to cause symptomatic recurrent infection or be asymptomatically shed to new hosts. Reactivation and re-entering a lytic cycle can be triggered by a broad range of physiological and environmental factors (Table 1).
Physiological triggers that induce gammaherpesvirus reactivation are not as clear as in the case of other herpesviruses (see below). Infections by other viruses, such as HIV, HSV-1, HSV-2, HHV-6, HHV-7, HCMV, and papillomavirus can contribute to lytic reactivation of KSHV [179,180,181,182]. Further research has shown that activation of Toll-like receptors 6 and 7 (TLR6 and TLR7) by other viruses can trigger KSHV reactivation [180]. Similarly, previous studies suggest that HIV-1, HPV, HHV-6, Plasmodium falciparum, Leptospirosis, and Group A Streptococci stimulate EBV reactivation [183,184,185,186,187,188]. In the case of EBV, it was also highlighted that the lytic cycle can be induced by Treponema pallidum through TLR2 and B-cell receptor (BCR) activation [189]. The increased number of antigens or decreased level of circulating antibodies are also likely to trigger EBV reactivation in memory B-cells through BCR [190]. Moreover, proinflammatory cytokines (Oncostatin M-OSM, hepatocyte growth factor/scattered factor—HGF/SF and IFNγ) produced in response to infection can facilitate lytic replication of KSHV [180,191,192,193]. In the case of EBV prostaglandin E2 (PGE2) was shown to induce lytic reactivation [194]. Taken together, infection with other viruses or bacteria can induce lytic reactivation of gammaherpesviruses through the activation of Toll-like receptors, BCR as well as cytokines produced during infection. Hypoxia is another possible cofactor of KSHV reactivation since it was found that KS tumors are more likely to appear in body parts that are relatively weakly supplied with blood and oxygen [180,195]. Low oxygen conditions can also facilitate EBV reactivation [196]. Hypoxia inducible factor 1α (HIF1α) can directly induce EBV reactivation by activating the expression of BZLF1, a lytic switch protein [197]. In fact, reactivation of KSHV by hypoxia and proinflammatory cytokines involves oxidative stress and reactive oxygen species (ROS). Hydrogen peroxide can induce the lytic cycle of KSHV through activation of mitogen-activated protein kinase (MAPK) pathways [198]. Upregulated ROS production also induces KSHV reactivation through inhibition of NF-κB pathway [199]. Stress and changes in catecholamines levels can induce EBV reactivation [200]. Elevated levels of stress hormones together with dysregulation in cell-mediated immunity have been implicated in EBV reactivation in astronauts during spaceflight [201,202,203,204]. Exposure of astronauts to unique non-terrestrial stressors—such as variable gravitational forces, cosmic radiation, and microgravity—dysregulates both immune and endocrine systems facilitating herpesvirus reactivation, although it remains asymptomatic in most cases [204]. Other environmental stressors such as chemotherapy and radiotherapy can induce lytic reactivation of EBV, probably due to suppressed immune system [205,206].
A variety of chemical and biological factors can induce gammaherpesvirus reactivation in cell culture latency systems. Strong inducers of KSHV and EBV lytic reactivation are the phorbol ester TPA (12-O-tetradecanoylphorbol-13-acetate) that broadly activates signal transduction cascades, as well as two histone deacetylase inhibitors: sodium butyrate (NaBu) and trichostatin A (TSA), [207,208,209,210]. TPA has been used for induction of EBV reactivation with higher efficacy than hypoxia [196]. TPA induces both EBV and KSHV lytic cycle in latently infected cells via MAPK/ERK and protein kinase C (PKC) pathways [208,211]. HDAC inhibitors reduce the overall histone deacetylation and therefore lead to global transcriptional activation. Valproic acid (VPA, 2-propyl-pentanoic acid), another histone deacetylase (HDAC) inhibitor, exerts opposite effects on KSHV and EBV reactivation. It strongly induces KSHV lytic cycle, whereas it inhibits lytic reactivation of EBV. The explanation for these varying outcomes might be different pathways leading to EBV vs KSHV reactivation [209].
The reactivation of viruses belonging to Betaherpesvirinae is mainly mediated by immune response and cytokines that are released and stimulate the terminal differentiation of infected cells. The important reservoirs of latent HCMV are the CD34+ hematopoietic stem cells and CD33+ myeloid progenitors, which develop into latently-infected CD14+ blood monocytes. Latent HCMV can reactivate in these cells as a consequence of differentiation towards macrophages and myeloid dendritic cells (DCs) driven by proinflammatory cytokines (IFNγ, TNFα, IL-4, GM-CSF) (Figure 1) [8,212]. In comparison to neurons that represent a life-long reservoir of latent alphaherpesviruses, the hematopoietic reservoir of latent HCMV is not long-lasting and it rather comprises a temporary stage due to much shorter life span of infected cells [212]. Interestingly, recent study demonstrated that HCMV induces differentiation of hematopoietic progenitor cells (HPCs) into a long-lived and immunosuppressive subpopulation of monocytes to achieve latency [213]. Betaherpesviruses are frequently reactivated in allograft recipients [214,215,216,217]. Immunosuppressive therapy that is applied in these patients to prevent and treat graft rejection and graft versus host disease potently suppresses cellular immunity, making these individuals more prone to viral reactivation [218]. A reduced number of CD8+ T cells that play a crucial role in controlling HCMV latent infection contributes to HCMV reactivation in immunosuppressed patients [219]. In cell culture systems, which model the situation in transplant patients, the presence of allogeneic peripheral blood mononuclear cells (PBMCs) can reactivate HCMV from latently infected monocytes. These data suggest that cytokines released as a result of allogeneic activation of T cells induce the differentiation of monocytes towards macrophages and consequently lead to HCMV lytic reactivation in those cells [214]. Although generally, HCMV reactivation does not comprise a common clinical problem, it might have serious clinical consequences in immunosuppressed patients such as those infected with HIV or undergoing immunosuppressive therapy after transplantation [220]. Alloreactivity is responsible for induction of virus reactivation not only as a result of recognizing foreign (transplanted) cells, but functions also to recognize presence of pathogens. In many cases, infection of cells positive for a herpesvirus with other pathogen induces reactivation of that herpesvirus. Additionally, reactivation of HHV-7 was shown to induce lytic replication of HCMV in allograft recipients [221]. Moreover, HCMV tends to reactivate in astronauts during and after spaceflight due to immune system dysregulation as well as activation of hypothalamic-pituitary-adrenal (HPA) and sympathetic-adrenal-medullary (SAM) axes of endocrine response. Thus, astronauts shedding HCMV might pose a risk to newborns or immunocompromised individuals after landing on Earth [201,202,204,222].
Although the most significant site of HCMV persistence is the myeloid lineage, HCMV can also establish latency in other cell types. There are studies confirming HCMV latency establishment in neural lineage, particularly in primitive neural stem cells, therefore suggesting a link to pathology of central nervous system observed in children with congenital CMV disease [10]. Similar to HCMV latency in myeloid lineage, reactivation of HCMV from latently infected neural stem cells seems to be differentiation dependent. However, contradictory results have been reported with regard to the permissivity of cells derived from different stages of neural differentiation (neural progenitor cells, neural precursor cells, and neurons) for the HCMV lytic cycle [10,223,224,225,226].
In cultured latently infected peripheral blood monocytes (PBMs), the differentiation-dependent shift of HCMV to the lytic cycle can be induced by granulocyte colony stimulating factor (G-CSF) and hydrocortisone. Although the above mentioned substances were observed to induce the expression of viral immediate early (IE) and early (E) genes, the production of infectious virus was not detected [227]. However, using a protocol for differentiation of CD34+ progenitors to mature dendritic cells (DCs) (involving transforming growth factor-β (TGFβ), TNFα, stem cell factor, granulocyte-macrophage colony-stimulating factor (GM-CSF), and FMS-like tyrosine kinase 3 ligand (Flt-3L) and lipopolysaccharide, it is possible to induce viral particle production [228]. As it was mentioned before, cytokines produced by allogeneic T-cell stimulation can also induce HCMV reactivation in monocytes [214]. Thus, myeloid cell differentiation is essential for HCMV reactivation in both latently infected organisms and cell culture systems. However, the regulation of viral lytic gene expression by the process of differentiation is not fully understood. The chemical compound that is effectively used to reactivate HCMV from latency is the phorbol ester TPA [229].
HHV-6A, HHV-6B, and HHV-7 that belong to the Roseolovirus genus of betaherpesviruses share common mechanisms of establishing latency and reactivation. Similar to HCMV, HHV-6 also establishes latency in CD34+ hematopoietic stem cells [9] and in myeloid cell lines [230]. Additionally, under experimental conditions latency can also be established in oligodendrocytes (HHV-6A) and in astrocytes (HHV-6B) [231,232]. Latent HHV-7 is carried in peripheral blood mononuclear cells (PBMCs) and can be reactivated as a result of T-cell activation by anti-CD3 monoclonal antibody and IL-2, as well as anti-CD3 and anti-CD28 monoclonal antibodies. Latent HHV-6B present in PBMCs is resistant to this type of stimulus. However, it can be reactivated by superinfection with HHV-7 [233]. The knowledge about cell types in which HHV-7 can establish latency is limited.
Among the Roseolovirus genus, latent HHV-6B and HHV-7 present in cultured cells can be reactivated by TPA [234,235], while lytic cycle of HHV-6A in PBMCs can be induced by TPA, hydrocortisone and trichostatin A (TSA) [21].
The frequency of clinical reactivation varies among alphaherpesviruses. Symptomatic reactivation of HSV may occur repeatedly and mostly in young people, whereas clinical reactivation of VZV typically occurs once in a lifetime and mostly affects elderly people [236]. The reactivation of HSV and the emergence of cold sores are related to various stimuli. It has been known for over a century that local injury (e.g., surgery) introduced to the nerve results in an incidence of cold sores (re-emergence of herpetic lesions) in the dermatome innervated by the injured nerve [237]. Other signals for reactivation of HSV can be sunlight and UV radiation [238]. It was suggested that immunosuppression and reduced cellular immunity due to UV radiation also contributes to reactivation of VZV, resulting in a higher frequency of zoster outbreaks in the summer [239,240]. Moreover, spacecraft crewmembers can experience VZV reactivation, which is usually asymptomatic and caused by elevated cortisol level together with dysregulation of immune system [201,202,204]. Long duration space mission can also lead to HSV-1 lytic reactivation although less frequently [204]. Changes in core body temperature, such as hyperthermia caused by fever, as well as hypothermia also facilitate the reactivation of HSV [241]. Prostaglandin PGE2 and cytokines such as IL-6 that are produced during high fever can directly stimulate neurons with latent HSV reservoirs [242,243,244]. Persistent psychological stress and fatigue are often responsible for re-emerging herpes symptoms, which partially correlates with reduced CD8+ T cell surveillance of latently infected neurons (Figure 1) [245]. Stressful life events and depression have also been shown to be associated with VZV reactivation (herpes zoster) [246,247,248]. Hormonal changes and hypoxic conditions can also cause HSV reactivation [249,250]. Moreover, it has been observed that in the context of HSV recurrence different triggers can lead to reactivation from different subtypes of neurons [251].
Under experimental conditions, several molecular pathways have been targeted to reactivate alphaherpesviruses. Continuous nerve growth factor (NGF) signaling has been known to be essential for maintaining HSV latency in neuron model systems of mice and rats. Thus, withdrawal of NGF from latently infected cultures leads to reactivation of the lytic cycle [252,253]. Inhibition of NGF signaling also induces VZV DNA production in axotomized cadaver ganglia [254]. Recently, it was shown, that NaBu, is able to reactivate alphaherpesviruses in cell culture [255,256]. Taking advantage of naturally occurring mechanism of HSV reactivation mediated by catecholamines and glucocorticosteroids released under influence of emotional stress, dexamethasone, a synthetic corticosteroid is efficiently used to reactivate HSV-1 both ex vivo and in primary neuronal latent systems [257,258]. The treatment of cells latently infected with VZV with phosphoinositide-3-kinase (PI3K) inhibitor (LY7294002) induces viral replicative cycle [256].
Host genetic variation, which is a component of infectious disease pathogenesis, has also been found to be associated with frequency of reactivation of herpesviruses. It was suggested that impaired surveillance of latent KSHV infection in individuals from Africa might be associated with 2 HLA-A alleles, A*6801 and A*4301, and 1 HLA-DRB1 allele group, DRB1*04, resulting in enhanced KSHV shedding in saliva [259]. Moreover, single nucleotide polymorphisms (SNPs) in vascular endothelial growth factor A (VEGFA) gene regions have been associated with KSHV viremia in kidney transplant recipients [260]. A long list of host genetic variants underlying control of humoral immune response to EBV has been shown to be associated with an increased lytic reactivation (reviewed in [261]). Similarly, also in the case of HCMV the role of host genetics in immune response to the virus was revealed and a large collection of associated genes was identified eg. TLRs, NKG2D, PD-1, IFNG, IL-6 just to name a few (reviewed in [262]). The clinical course of HSV-1 infection can be modified by MHC class I allotypes (B*18, C*15, and the group of alleles encoding A19), the high-affinity receptor/ligand pair KIR2DL2/HLA-C1, and the CD16A-158V/F dimorphism. Since these polymorphic genes are involved in the adaptive immunity, these findings confirm an important role of CD8+ T cells and NK cells in host defense against HSV-1 infection [263]. Frequency and severity of cold sore episodes due to HSV-1 reactivation can be associated with the haplotype of the Cold Sore Susceptibility Gene-1 (CSSG-1). Individuals with H1, H2, and H4 haplotypes of CSSG-1 experience more frequent and severe herpetic lesions [264]. Genetic variation in the HLA region (HLA Complex P5—HCP5) was linked to susceptibility to herpes zoster resulting from VZV reactivation [265].
Greater focus on host genetics of herpesviral infection is needed to predict which individuals are at higher risk of viral reactivation and complications caused by recurrent infection. In the future, larger genomic studies can bring breakthroughs in understanding the host response to herpesviruses.
On the organism level, it is thought that the reactivation of herpesviruses is a combination of both immunosuppression and factors that act directly on latently infected cells. On the cellular level, virus reactivation is believed to be the result of a disturbed ‘balance’ between signaling pathways that activate and inhibit viral lytic replication. Generally, harsh conditions can lead to virus reactivation and progeny production in order to be spread to new hosts. Although certain environmental stimuli are well defined, our knowledge of the exact mechanisms and conditions leading to lytic switch is incomplete. However, the regulation of the latency to lytic switch is essential to develop novel therapeutic strategies that could target the latent human herpesviruses.
5. Conclusions
Different herpesviruses establish latency in different cells, which affects the characteristics of the process. The common feature for all herpesviruses is the persistence of the genome in form of an episome, the difference with regards to persistence lies in that the beta- and gammaherpesviruses, encode a protein participating in latent replication and genome tethering, and alphaherpesviruses, do not have such a protein, as they persist in non-dividing neurons. Genomes of all herpesviruses become chromatinized upon entering the cell, therefore transcriptional activity of all of them is subject to regulation by chromatin modifications.
With regard to gene expression in latency, at first glance each subfamily seems to have its own distinctive pattern (Table 1). Alphaherpesviruses express only LATs or VLTs (for HSV and VZV respectively), the betaherpesviruses have been shown to express a set of latent genes and the EBV, gammaherpesviruses has a whole series of latent expression programs differing in the number of expressed proteins. KSHV so far is not known to have different latency programs, but it was shown that the expression of some latent proteins is cell type specific, so the existence of different transcriptional programs cannot be excluded. Expression of HSV-1 LAT in all of the cells of the population, but only in 30% of the cells at one timepoint, suggests that also alphaherpesviruses might have a latency transcriptional program. Traditionally latency is viewed as a state with limited expression of genes. However, more recent studies show the expression of proteins classified as lytic during latency of viruses belonging to all three subfamilies. Further research is needed to fully understand the phenomenon of lytic gene expression during latency.
Herpesviruses have the ability to reactivate from latency to lytic cycle. Immunosuppression—due to i.e., exposure to UV radiation, stress, or chemotherapy—is a common inducer of the lytic cycle of all herpesviruses. The presence of proinflammatory cytokines is another signal that tends to reactivate herpesviruses. In the case of HCMV the reactivation is observed during the process of myeloid differentiation, which is associated with cytokine secretion. The final differentiation step of memory B cells to plasma cells is also a signal for EBV to enter the lytic cycle. Hypoxia is an important stimulus inducing the lytic cycle in Alphaherpesvirinae (HSV-1) and Gammaherpesvirinae (KSHV, EBV). Finally, infection with another pathogen can also induce reactivation of the latent beta- (HHV-6B) or gammaherpesviruses (KSHV, EBV).
Even though latency looks a bit different in case of each of the subfamilies of the Herpesviridae with increasing number and sophistication of the model systems as well as improved sensitivity of assays the common features of the hallmark of herpesviruses—their latency state—start to emerge (Figure 1 and Table 1).
Author Contributions
Conceptualization, M.W.-G.; Writing—original draft preparation, M.W.-G. and E.K.-K.; Writing—review and editing, M.W.-G., E.K.-K., and M.S.; Visualization, M.W.-G., E.K.-K., and M.S.; Supervision, M.W.-G.; Project administration, M.W.-G. and E.K.-K.; Funding acquisition, M.W.-G. All authors have read and agreed to the published version of the manuscript.
Funding
The work was supported by the National Science Centre in Poland grant no. 2017/26/E/NZ6/01124.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Pellett, P.E.; Roizman, B. Herpesviridae. In Fields Virology, 6th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; Volume 2. [Google Scholar]
- Speck, S.H.; Ganem, D. Viral latency and its regulation: Lessons from the gamma-herpesviruses. Cell Host Microbe 2010, 8, 100–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roizman, B.; Knipe, D.M.; Whitley, R.J. Herpes Simplex Viruses. In Fields Virology, 6th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; Volume 2. [Google Scholar]
- Spear, P.G.; Longnecker, R. Herpesvirus entry: An update. J. Virol. 2003, 77, 10179–10185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitley, R.J. Herpesviruses. In Medical Microbiology, 4th ed.; Baron, S., Ed.; University of Texas Medical Branch at Galveston: Galveston, TX, USA, 1996. [Google Scholar]
- Mocarski, E.S.; Shenk, T.; Pass, R.F. Cytomegalovirus. In Fields Virology, 6th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; Volume 2. [Google Scholar]
- Davison, A.J. Herpesviruses: General Features. Ref. Modul. Biomed. Sci. 2014. [Google Scholar] [CrossRef]
- Hahn, G.; Jores, R.; Mocarski, E.S. Cytomegalovirus remains latent in a common precursor of dendritic and myeloid cells. Proc. Natl. Acad. Sci. USA 1998, 95, 3937–3942. [Google Scholar] [CrossRef] [Green Version]
- Luppi, M.; Barozzi, P.; Morris, C.; Maiorana, A.; Garber, R.; Bonacorsi, G.; Donelli, A.; Marasca, R.; Tabilio, A.; Torelli, G. Human herpesvirus 6 latently infects early bone marrow progenitors in vivo. J. Virol. 1999, 73, 754–759. [Google Scholar] [CrossRef] [Green Version]
- Belzile, J.P.; Stark, T.J.; Yeo, G.W.; Spector, D.H. Human cytomegalovirus infection of human embryonic stem cell-derived primitive neural stem cells is restricted at several steps but leads to the persistence of viral DNA. J. Virol. 2014, 88, 4021–4039. [Google Scholar] [CrossRef] [Green Version]
- Means, R.E.; Lang, S.M.; Jung, J.U. Human gammaherpesvirus immune evasion strategies. In Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis, 1st ed.; Arvin, A., Campadelli-Fiume, G., Mocarski, E., Moore, P.S., Roizman, B., Whitley, R., Yamanishi, K., Eds.; Cambridge University Press: Cambridge, UK, 2007. [Google Scholar]
- Parravicini, C.; Chandran, B.; Corbellino, M.; Berti, E.; Paulli, M.; Moore, P.S.; Chang, Y. Differential viral protein expression in Kaposi’s sarcoma-associated herpesvirus-infected diseases: Kaposi’s sarcoma, primary effusion lymphoma, and multicentric Castleman’s disease. Am. J. Pathol. 2000, 156, 743–749. [Google Scholar] [CrossRef]
- Jha, H.C.; Banerjee, S.; Robertson, E.S. The Role of Gammaherpesviruses in Cancer Pathogenesis. Pathogens 2016, 5. [Google Scholar] [CrossRef] [Green Version]
- Jacob, R.J.; Morse, L.S.; Roizman, B. Anatomy of herpes simplex virus DNA. XII. Accumulation of head-to-tail concatemers in nuclei of infected cells and their role in the generation of the four isomeric arrangements of viral DNA. J. Virol. 1979, 29, 448–457. [Google Scholar] [CrossRef] [Green Version]
- Poffenberger, K.L.; Roizman, B. A noninverting genome of a viable herpes simplex virus 1: Presence of head-to-tail linkages in packaged genomes and requirements for circularization after infection. J. Virol. 1985, 53, 587–595. [Google Scholar] [CrossRef] [Green Version]
- Luppi, M.; Marasca, R.; Barozzi, P.; Ferrari, S.; Ceccherini-Nelli, L.; Batoni, G.; Merelli, E.; Torelli, G. Three cases of human herpesvirus-6 latent infection: Integration of viral genome in peripheral blood mononuclear cell DNA. J. Med. Virol. 1993, 40, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Daibata, M.; Taguchi, T.; Taguchi, H.; Miyoshi, I. Integration of human herpesvirus 6 in a Burkitt’s lymphoma cell line. Br. J. Haematol. 1998, 102, 1307–1313. [Google Scholar] [CrossRef] [PubMed]
- Nacheva, E.P.; Ward, K.N.; Brazma, D.; Virgili, A.; Howard, J.; Leong, H.N.; Clark, D.A. Human herpesvirus 6 integrates within telomeric regions as evidenced by five different chromosomal sites. J. Med. Virol. 2008, 80, 1952–1958. [Google Scholar] [CrossRef] [PubMed]
- Ward, K.N.; Leong, H.N.; Nacheva, E.P.; Howard, J.; Atkinson, C.E.; Davies, N.W.; Griffiths, P.D.; Clark, D.A. Human herpesvirus 6 chromosomal integration in immunocompetent patients results in high levels of viral DNA in blood, sera, and hair follicles. J. Clin. Microbiol. 2006, 44, 1571–1574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka-Taya, K.; Sashihara, J.; Kurahashi, H.; Amo, K.; Miyagawa, H.; Kondo, K.; Okada, S.; Yamanishi, K. Human herpesvirus 6 (HHV-6) is transmitted from parent to child in an integrated form and characterization of cases with chromosomally integrated HHV-6 DNA. J. Med. Virol. 2004, 73, 465–473. [Google Scholar] [CrossRef] [PubMed]
- Arbuckle, J.H.; Medveczky, M.M.; Luka, J.; Hadley, S.H.; Luegmayr, A.; Ablashi, D.; Lund, T.C.; Tolar, J.; De Meirleir, K.; Montoya, J.G.; et al. The latent human herpesvirus-6A genome specifically integrates in telomeres of human chromosomes in vivo and in vitro. Proc. Natl. Acad. Sci. USA 2010, 107, 5563–5568. [Google Scholar] [CrossRef] [Green Version]
- Verma, S.C.; Choudhuri, T.; Robertson, E.S. The minimal replicator element of the Kaposi’s sarcoma-associated herpesvirus terminal repeat supports replication in a semiconservative and cell-cycle-dependent manner. J. Virol. 2007, 81, 3402–3413. [Google Scholar] [CrossRef] [Green Version]
- Grundhoff, A.; Ganem, D. The latency-associated nuclear antigen of Kaposi’s sarcoma-associated herpesvirus permits replication of terminal repeat-containing plasmids. J. Virol. 2003, 77, 2779–2783. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Garber, A.C.; Renne, R. The latency-associated nuclear antigen of Kaposi’s sarcoma-associated herpesvirus supports latent DNA replication in dividing cells. J. Virol. 2002, 76, 11677–11687. [Google Scholar] [CrossRef] [Green Version]
- Lim, C.; Sohn, H.; Lee, D.; Gwack, Y.; Choe, J. Functional dissection of latency-associated nuclear antigen 1 of Kaposi’s sarcoma-associated herpesvirus involved in latent DNA replication and transcription of terminal repeats of the viral genome. J. Virol. 2002, 76, 10320–10331. [Google Scholar] [CrossRef] [Green Version]
- Longnecker, R.; Kieff, E.; Cohen, J.I. Epstein–Barr Virus. In Fields Virology, 6th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; Volume 2. [Google Scholar]
- Damania, B.; Cesarman, E. Kaposi’s Sarcoma–Associated Herpesvirus. In Fields Virology, 6th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; Volume 2. [Google Scholar]
- Tarrant-Elorza, M.; Rossetto, C.C.; Pari, G.S. Maintenance and replication of the human cytomegalovirus genome during latency. Cell Host Microbe 2014, 16, 43–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibson, W.; Roizman, B. Compartmentalization of spermine and spermidine in the herpes simplex virion. Proc. Natl. Acad. Sci. USA 1971, 68, 2818–2821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, J.; Fraser, N.W. Temporal association of the herpes simplex virus genome with histone proteins during a lytic infection. J. Virol. 2008, 82, 3530–3537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groves, I.J.; Reeves, M.B.; Sinclair, J.H. Lytic infection of permissive cells with human cytomegalovirus is regulated by an intrinsic ‘pre-immediate-early’ repression of viral gene expression mediated by histone post-translational modification. J. Gen. Virol. 2009, 90, 2364–2374. [Google Scholar] [CrossRef] [PubMed]
- Shaw, J.E.; Levinger, L.F.; Carter, C.W. Nucleosomal structure of Epstein–Barr virus DNA in transformed cell lines. J. Virol. 1979, 29, 657–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wensing, B.; Stühler, A.; Jenkins, P.; Hollyoake, M.; Karstegl, C.E.; Farrell, P.J. Variant chromatin structure of the oriP region of Epstein–Barr virus and regulation of EBER1 expression by upstream sequences and oriP. J. Virol. 2001, 75, 6235–6241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dyson, P.J.; Farrell, P.J. Chromatin structure of Epstein–Barr virus. J. Gen. Virol. 1985, 66 Pt 9, 1931–1940. [Google Scholar] [CrossRef]
- Deshmane, S.L.; Fraser, N.W. During latency, herpes simplex virus type 1 DNA is associated with nucleosomes in a chromatin structure. J. Virol. 1989, 63, 943–947. [Google Scholar] [CrossRef] [Green Version]
- Stedman, W.; Deng, Z.; Lu, F.; Lieberman, P.M. ORC, MCM, and histone hyperacetylation at the Kaposi’s sarcoma-associated herpesvirus latent replication origin. J. Virol. 2004, 78, 12566–12575. [Google Scholar] [CrossRef] [Green Version]
- Thorley-Lawson, D.A.; Duca, K.A.; Shapiro, M. Epstein–Barr virus: A paradigm for persistent infection—For real and in virtual reality. Trends Immunol. 2008, 29, 195–201. [Google Scholar] [CrossRef]
- Speck, S.H. Regulation of EBV Latency-Associated Gene Expression. In Epstein–Barr Virus; Robertson, E.S., Ed.; Caister Academic Press: Wymondham, UK, 2005. [Google Scholar]
- Yates, J.L.; Warren, N.; Sugden, B. Stable replication of plasmids derived from Epstein–Barr virus in various mammalian cells. Nature 1985, 313, 812–815. [Google Scholar] [CrossRef] [PubMed]
- Reisman, D.; Yates, J.; Sugden, B. A putative origin of replication of plasmids derived from Epstein–Barr virus is composed of two cis-acting components. Mol. Cell. Biol. 1985, 5, 1822–1832. [Google Scholar] [CrossRef] [PubMed]
- Sugden, B.; Warren, N. A promoter of Epstein–Barr virus that can function during latent infection can be transactivated by EBNA-1, a viral protein required for viral DNA replication during latent infection. J. Virol. 1989, 63, 2644–2649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.A.; Diamond, M.E.; Yates, J.L. Genetic evidence that EBNA-1 is needed for efficient, stable latent infection by Epstein–Barr virus. J. Virol. 1999, 73, 2974–2982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izumi, K.M.; Kieff, E.D. The Epstein–Barr virus oncogene product latent membrane protein 1 engages the tumor necrosis factor receptor-associated death domain protein to mediate B lymphocyte growth transformation and activate NF-kappaB. Proc. Natl. Acad. Sci. USA 1997, 94, 12592–12597. [Google Scholar] [CrossRef] [Green Version]
- Bornkamm, G.W.; Hammerschmidt, W. Molecular virology of Epstein–Barr virus. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2001, 356, 437–459. [Google Scholar] [CrossRef] [Green Version]
- Miller, C.L.; Burkhardt, A.L.; Lee, J.H.; Stealey, B.; Longnecker, R.; Bolen, J.B.; Kieff, E. Integral membrane protein 2 of Epstein–Barr virus regulates reactivation from latency through dominant negative effects on protein-tyrosine kinases. Immunity 1995, 2, 155–166. [Google Scholar] [CrossRef] [Green Version]
- Lerner, M.R.; Andrews, N.C.; Miller, G.; Steitz, J.A. Two small RNAs encoded by Epstein–Barr virus and complexed with protein are precipitated by antibodies from patients with systemic lupus erythematosus. Proc. Natl. Acad. Sci. USA 1981, 78, 805–809. [Google Scholar] [CrossRef] [Green Version]
- Pfeffer, S.; Zavolan, M.; Grässer, F.A.; Chien, M.; Russo, J.J.; Ju, J.; John, B.; Enright, A.J.; Marks, D.; Sander, C.; et al. Identification of virus-encoded microRNAs. Science 2004, 304, 734–736. [Google Scholar] [CrossRef]
- Edwards, R.H.; Marquitz, A.R.; Raab-Traub, N. Epstein–Barr virus BART microRNAs are produced from a large intron prior to splicing. J. Virol. 2008, 82, 9094–9106. [Google Scholar] [CrossRef] [Green Version]
- Toptan, T.; Abere, B.; Nalesnik, M.A.; Swerdlow, S.H.; Ranganathan, S.; Lee, N.; Shair, K.H.; Moore, P.S.; Chang, Y. Circular DNA tumor viruses make circular RNAs. Proc. Natl. Acad. Sci. USA 2018, 115, E8737–E8745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komano, J.; Maruo, S.; Kurozumi, K.; Oda, T.; Takada, K. Oncogenic role of Epstein–Barr virus-encoded RNAs in Burkitt’s lymphoma cell line Akata. J. Virol. 1999, 73, 9827–9831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nanbo, A.; Inoue, K.; Adachi-Takasawa, K.; Takada, K. Epstein–Barr virus RNA confers resistance to interferon-alpha-induced apoptosis in Burkitt’s lymphoma. EMBO J. 2002, 21, 954–965. [Google Scholar] [CrossRef] [PubMed]
- Iwakiri, D.; Zhou, L.; Samanta, M.; Matsumoto, M.; Ebihara, T.; Seya, T.; Imai, S.; Fujieda, M.; Kawa, K.; Takada, K. Epstein–Barr virus (EBV)-encoded small RNA is released from EBV-infected cells and activates signaling from Toll-like receptor 3. J. Exp. Med. 2009, 206, 2091–2099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samanta, M.; Iwakiri, D.; Takada, K. Epstein–Barr virus-encoded small RNA induces IL-10 through RIG-I-mediated IRF-3 signaling. Oncogene 2008, 27, 4150–4160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seto, E.; Moosmann, A.; Grömminger, S.; Walz, N.; Grundhoff, A.; Hammerschmidt, W. Micro RNAs of Epstein–Barr virus promote cell cycle progression and prevent apoptosis of primary human B cells. PLoS Pathog. 2010, 6, e1001063. [Google Scholar] [CrossRef] [Green Version]
- Lin, X.; Tsai, M.-H.; Shumilov, A.; Poirey, R.; Bannert, H.; Middeldorp, J.M.; Feederle, R.; Delecluse, H.-J. The Epstein–Barr Virus BART miRNA Cluster of the M81 Strain Modulates Multiple Functions in Primary B Cells. PLoS Pathog. 2015, 11, e1005344. [Google Scholar] [CrossRef]
- Barbera, A.J.; Chodaparambil, J.V.; Kelley-Clarke, B.; Luger, K.; Kaye, K.M. Kaposi’s sarcoma-associated herpesvirus LANA hitches a ride on the chromosome. Cell Cycle 2006, 5, 1048–1052. [Google Scholar] [CrossRef]
- Cotter, M.A.; Robertson, E.S. The latency-associated nuclear antigen tethers the Kaposi’s sarcoma-associated herpesvirus genome to host chromosomes in body cavity-based lymphoma cells. Virology 1999, 264, 254–264. [Google Scholar] [CrossRef] [Green Version]
- Friborg, J.; Kong, W.; Hottiger, M.O.; Nabel, G.J. p53 inhibition by the LANA protein of KSHV protects against cell death. Nature 1999, 402, 889–894. [Google Scholar] [CrossRef]
- Radkov, S.A.; Kellam, P.; Boshoff, C. The latent nuclear antigen of Kaposi sarcoma-associated herpesvirus targets the retinoblastoma-E2F pathway and with the oncogene Hras transforms primary rat cells. Nat. Med. 2000, 6, 1121–1127. [Google Scholar] [CrossRef] [PubMed]
- Fujimuro, M.; Wu, F.Y.; ApRhys, C.; Kajumbula, H.; Young, D.B.; Hayward, G.S.; Hayward, S.D. A novel viral mechanism for dysregulation of beta-catenin in Kaposi’s sarcoma-associated herpesvirus latency. Nat. Med. 2003, 9, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Moore, P.S.; Talbot, S.J.; Boshoff, C.H.; Zarkowska, T.; Godden-Kent; Paterson, H.; Weiss, R.A.; Mittnacht, S. Cyclin encoded by KS herpesvirus. Nature 1996, 382, 410. [Google Scholar] [CrossRef] [PubMed]
- Jones, T.; Ramos da Silva, S.; Bedolla, R.; Ye, F.; Zhou, F.; Gao, S.J. Viral cyclin promotes KSHV-induced cellular transformation and tumorigenesis by overriding contact inhibition. Cell Cycle 2014, 13, 845–858. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.S.; Li, Q.; Lee, J.Y.; Lee, S.H.; Jeong, J.H.; Lee, H.R.; Chang, H.; Zhou, F.C.; Gao, S.J.; Liang, C.; et al. FLIP-mediated autophagy regulation in cell death control. Nat. Cell Biol. 2009, 11, 1355–1362. [Google Scholar] [CrossRef] [Green Version]
- Chaudhary, P.M.; Jasmin, A.; Eby, M.T.; Hood, L. Modulation of the NF-kappa B pathway by virally encoded death effector domains-containing proteins. Oncogene 1999, 18, 5738–5746. [Google Scholar] [CrossRef] [Green Version]
- Sadler, R.; Wu, L.; Forghani, B.; Renne, R.; Zhong, W.; Herndier, B.; Ganem, D. A complex translational program generates multiple novel proteins from the latently expressed kaposin (K12) locus of Kaposi’s sarcoma-associated herpesvirus. J. Virol. 1999, 73, 5722–5730. [Google Scholar] [CrossRef] [Green Version]
- Muralidhar, S.; Pumfery, A.M.; Hassani, M.; Sadaie, M.R.; Kishishita, M.; Brady, J.N.; Doniger, J.; Medveczky, P.; Rosenthal, L.J. Identification of kaposin (open reading frame K12) as a human herpesvirus 8 (Kaposi’s sarcoma-associated herpesvirus) transforming gene. J. Virol. 1998, 72, 4980–4988. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.W.; Tu, P.F.; Hsiao, N.W.; Chang, C.Y.; Wan, L.; Lin, Y.T.; Chang, H.W. Identification of a novel septin 4 protein binding to human herpesvirus 8 kaposin A protein using a phage display cDNA library. J. Virol. Methods 2007, 143, 65–72. [Google Scholar] [CrossRef]
- McCormick, C.; Ganem, D. The kaposin B protein of KSHV activates the p38/MK2 pathway and stabilizes cytokine mRNAs. Science 2005, 307, 739–741. [Google Scholar] [CrossRef]
- Rivas, C.; Thlick, A.E.; Parravicini, C.; Moore, P.S.; Chang, Y. Kaposi’s sarcoma-associated herpesvirus LANA2 is a B-cell-specific latent viral protein that inhibits p53. J. Virol. 2001, 75, 429–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esteban, M.; García, M.A.; Domingo-Gil, E.; Arroyo, J.; Nombela, C.; Rivas, C. The latency protein LANA2 from Kaposi’s sarcoma-associated herpesvirus inhibits apoptosis induced by dsRNA-activated protein kinase but not RNase L activation. J. Gen. Virol. 2003, 84, 1463–1470. [Google Scholar] [CrossRef] [PubMed]
- Joo, C.H.; Shin, Y.C.; Gack, M.; Wu, L.; Levy, D.; Jung, J.U. Inhibition of interferon regulatory factor 7 (IRF7)-mediated interferon signal transduction by the Kaposi’s sarcoma-associated herpesvirus viral IRF homolog vIRF3. J. Virol. 2007, 81, 8282–8292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandriani, S.; Ganem, D. Array-based transcript profiling and limiting-dilution reverse transcription-PCR analysis identify additional latent genes in Kaposi’s sarcoma-associated herpesvirus. J. Virol. 2010, 84, 5565–5573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, P.S.; Boshoff, C.; Weiss, R.A.; Chang, Y. Molecular mimicry of human cytokine and cytokine response pathway genes by KSHV. Science 1996, 274, 1739–1744. [Google Scholar] [CrossRef]
- Aoki, Y.; Yarchoan, R.; Wyvill, K.; Okamoto, S.; Little, R.F.; Tosato, G. Detection of viral interleukin-6 in Kaposi sarcoma-associated herpesvirus-linked disorders. Blood 2001, 97, 2173–2176. [Google Scholar] [CrossRef]
- Neipel, F.; Albrecht, J.C.; Fleckenstein, B. Cell-homologous genes in the Kaposi’s sarcoma-associated rhadinovirus human herpesvirus 8: Determinants of its pathogenicity? J. Virol. 1997, 71, 4187–4192. [Google Scholar] [CrossRef] [Green Version]
- Nicholas, J.; Ruvolo, V.R.; Burns, W.H.; Sandford, G.; Wan, X.; Ciufo, D.; Hendrickson, S.B.; Guo, H.G.; Hayward, G.S.; Reitz, M.S. Kaposi’s sarcoma-associated human herpesvirus-8 encodes homologues of macrophage inflammatory protein-1 and interleukin-6. Nat. Med. 1997, 3, 287–292. [Google Scholar] [CrossRef]
- Burger, R.; Neipel, F.; Fleckenstein, B.; Savino, R.; Ciliberto, G.; Kalden, J.R.; Gramatzki, M. Human herpesvirus type 8 interleukin-6 homologue is functionally active on human myeloma cells. Blood 1998, 91, 1858–1863. [Google Scholar] [CrossRef]
- Chen, D.; Sandford, G.; Nicholas, J. Intracellular signaling mechanisms and activities of human herpesvirus 8 interleukin-6. J. Virol. 2009, 83, 722–733. [Google Scholar] [CrossRef] [Green Version]
- Ye, X.; Zhao, Y.; Karijolich, J. The landscape of transcription initiation across latent and lytic KSHV genomes. PLoS Pathog. 2019, 15, e1007852. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.S.; Lee, S.H.; Feng, P.; Chang, H.; Cho, N.H.; Jung, J.U. Characterization of the Kaposi’s sarcoma-associated herpesvirus K1 signalosome. J. Virol. 2005, 79, 12173–12184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.; Veazey, R.; Williams, K.; Li, M.; Guo, J.; Neipel, F.; Fleckenstein, B.; Lackner, A.; Desrosiers, R.C.; Jung, J.U. Deregulation of cell growth by the K1 gene of Kaposi’s sarcoma-associated herpesvirus. Nat. Med. 1998, 4, 435–440. [Google Scholar] [CrossRef] [PubMed]
- Prakash, O.; Tang, Z.Y.; Peng, X.; Coleman, R.; Gill, J.; Farr, G.; Samaniego, F. Tumorigenesis and aberrant signaling in transgenic mice expressing the human herpesvirus-8 K1 gene. J. Natl. Cancer Inst. 2002, 94, 926–935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomlinson, C.C.; Damania, B. The K1 protein of Kaposi’s sarcoma-associated herpesvirus activates the Akt signaling pathway. J. Virol. 2004, 78, 1918–1927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Dittmer, D.P.; Tomlinson, C.C.; Fakhari, F.D.; Damania, B. Immortalization of primary endothelial cells by the K1 protein of Kaposi’s sarcoma-associated herpesvirus. Cancer Res 2006, 66, 3658–3666. [Google Scholar] [CrossRef] [Green Version]
- Cai, X.; Lu, S.; Zhang, Z.; Gonzalez, C.M.; Damania, B.; Cullen, B.R. Kaposi’s sarcoma-associated herpesvirus expresses an array of viral microRNAs in latently infected cells. Proc. Natl. Acad. Sci. USA 2005, 102, 5570–5575. [Google Scholar] [CrossRef] [Green Version]
- Samols, M.A.; Hu, J.; Skalsky, R.L.; Renne, R. Cloning and identification of a microRNA cluster within the latency-associated region of Kaposi’s sarcoma-associated herpesvirus. J. Virol. 2005, 79, 9301–9305. [Google Scholar] [CrossRef] [Green Version]
- Sun, R.; Lin, S.F.; Gradoville, L.; Miller, G. Polyadenylylated nuclear RNA encoded by Kaposi sarcoma-associated herpesvirus. Proc. Natl. Acad. Sci. USA 1996, 93, 11883–11888. [Google Scholar] [CrossRef] [Green Version]
- Samols, M.A.; Skalsky, R.L.; Maldonado, A.M.; Riva, A.; Lopez, M.C.; Baker, H.V.; Renne, R. Identification of cellular genes targeted by KSHV-encoded microRNAs. PLoS Pathog. 2007, 3, e65. [Google Scholar] [CrossRef]
- Gottwein, E.; Mukherjee, N.; Sachse, C.; Frenzel, C.; Majoros, W.H.; Chi, J.T.; Braich, R.; Manoharan, M.; Soutschek, J.; Ohler, U.; et al. A viral microRNA functions as an orthologue of cellular miR-155. Nature 2007, 450, 1096–1099. [Google Scholar] [CrossRef] [PubMed]
- Skalsky, R.L.; Hu, J.; Renne, R. Analysis of viral cis elements conferring Kaposi’s sarcoma-associated herpesvirus episome partitioning and maintenance. J. Virol. 2007, 81, 9825–9837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellare, P.; Ganem, D. Regulation of KSHV lytic switch protein expression by a virus-encoded microRNA: An evolutionary adaptation that fine-tunes lytic reactivation. Cell Host Microbe 2009, 6, 570–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, X.; Bai, Z.; Ye, F.; Xie, J.; Kim, C.G.; Huang, Y.; Gao, S.J. Regulation of NF-kappaB inhibitor IkappaBalpha and viral replication by a KSHV microRNA. Nat. Cell Biol. 2010, 12, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Goodrum, F.D.; Jordan, C.T.; High, K.; Shenk, T. Human cytomegalovirus gene expression during infection of primary hematopoietic progenitor cells: A model for latency. Proc. Natl. Acad. Sci. USA 2002, 99, 16255–16260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le, V.T.; Trilling, M.; Hengel, H. The cytomegaloviral protein pUL138 acts as potentiator of tumor necrosis factor (TNF) receptor 1 surface density to enhance ULb’-encoded modulation of TNF-α signaling. J. Virol. 2011, 85, 13260–13270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weekes, M.P.; Tan, S.Y.; Poole, E.; Talbot, S.; Antrobus, R.; Smith, D.L.; Montag, C.; Gygi, S.P.; Sinclair, J.H.; Lehner, P.J. Latency-associated degradation of the MRP1 drug transporter during latent human cytomegalovirus infection. Science 2013, 340, 199–202. [Google Scholar] [CrossRef] [Green Version]
- Buehler, J.; Zeltzer, S.; Reitsma, J.; Petrucelli, A.; Umashankar, M.; Rak, M.; Zagallo, P.; Schroeder, J.; Terhune, S.; Goodrum, F. Opposing Regulation of the EGF Receptor: A Molecular Switch Controlling Cytomegalovirus Latency and Replication. PLoS Pathog. 2016, 12, e1005655. [Google Scholar] [CrossRef] [Green Version]
- Bego, M.; Maciejewski, J.; Khaiboullina, S.; Pari, G.; St Jeor, S. Characterization of an antisense transcript spanning the UL81-82 locus of human cytomegalovirus. J. Virol. 2005, 79, 11022–11034. [Google Scholar] [CrossRef] [Green Version]
- Keyes, L.R.; Hargett, D.; Soland, M.; Bego, M.G.; Rossetto, C.C.; Almeida-Porada, G.; St Jeor, S. HCMV protein LUNA is required for viral reactivation from latently infected primary CD14⁺ cells. PLoS ONE 2012, 7, e52827. [Google Scholar] [CrossRef]
- Poole, E.L.; Kew, V.G.; Lau, J.C.H.; Murray, M.J.; Stamminger, T.; Sinclair, J.H.; Reeves, M.B. A Virally Encoded DeSUMOylase Activity Is Required for Cytomegalovirus Reactivation from Latency. Cell Rep. 2018, 24, 594–606. [Google Scholar] [CrossRef] [Green Version]
- Beisser, P.S.; Laurent, L.; Virelizier, J.L.; Michelson, S. Human cytomegalovirus chemokine receptor gene US28 is transcribed in latently infected THP-1 monocytes. J. Virol. 2001, 75, 5949–5957. [Google Scholar] [CrossRef] [Green Version]
- Cheung, A.K.; Abendroth, A.; Cunningham, A.L.; Slobedman, B. Viral gene expression during the establishment of human cytomegalovirus latent infection in myeloid progenitor cells. Blood 2006, 108, 3691–3699. [Google Scholar] [CrossRef]
- Humby, M.S.; O’Connor, C.M. Human Cytomegalovirus US28 Is Important for Latent Infection of Hematopoietic Progenitor Cells. J. Virol. 2015, 90, 2959–2970. [Google Scholar] [CrossRef] [Green Version]
- Krishna, B.A.; Poole, E.L.; Jackson, S.E.; Smit, M.J.; Wills, M.R.; Sinclair, J.H. Latency-Associated Expression of Human Cytomegalovirus US28 Attenuates Cell Signaling Pathways To Maintain Latent Infection. MBio 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Krishna, B.A.; Humby, M.S.; Miller, W.E.; O’Connor, C.M. Human cytomegalovirus G protein-coupled receptor US28 promotes latency by attenuating c-fos. Proc. Natl. Acad. Sci. USA 2019, 116, 1755–1764. [Google Scholar] [CrossRef] [Green Version]
- Krishna, B.A.; Wass, A.B.; Sridharan, R.; O’Connor, C.M. The Requirement for US28 during Cytomegalovirus Latency Is Independent of US27 and US29 Gene Expression. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Jenkins, C.; Abendroth, A.; Slobedman, B. A novel viral transcript with homology to human interleukin-10 is expressed during latent human cytomegalovirus infection. J. Virol. 2004, 78, 1440–1447. [Google Scholar] [CrossRef] [Green Version]
- Kotenko, S.V.; Saccani, S.; Izotova, L.S.; Mirochnitchenko, O.V.; Pestka, S. Human cytomegalovirus harbors its own unique IL-10 homolog (cmvIL-10). Proc. Natl. Acad. Sci. USA 2000, 97, 1695–1700. [Google Scholar] [CrossRef] [Green Version]
- Jenkins, C.; Garcia, W.; Godwin, M.J.; Spencer, J.V.; Stern, J.L.; Abendroth, A.; Slobedman, B. Immunomodulatory properties of a viral homolog of human interleukin-10 expressed by human cytomegalovirus during the latent phase of infection. J. Virol. 2008, 82, 3736–3750. [Google Scholar] [CrossRef] [Green Version]
- Cheung, A.K.; Gottlieb, D.J.; Plachter, B.; Pepperl-Klindworth, S.; Avdic, S.; Cunningham, A.L.; Abendroth, A.; Slobedman, B. The role of the human cytomegalovirus UL111A gene in down-regulating CD4+ T-cell recognition of latently infected cells: Implications for virus elimination during latency. Blood 2009, 114, 4128–4137. [Google Scholar] [CrossRef] [Green Version]
- Reeves, M.B.; Davies, A.A.; McSharry, B.P.; Wilkinson, G.W.; Sinclair, J.H. Complex I binding by a virally encoded RNA regulates mitochondria-induced cell death. Science 2007, 316, 1345–1348. [Google Scholar] [CrossRef] [Green Version]
- Poole, E.; Walther, A.; Raven, K.; Benedict, C.A.; Mason, G.M.; Sinclair, J. The myeloid transcription factor GATA-2 regulates the viral UL144 gene during human cytomegalovirus latency in an isolate-specific manner. J. Virol. 2013, 87, 4261–4271. [Google Scholar] [CrossRef] [Green Version]
- Benedict, C.A.; Butrovich, K.D.; Lurain, N.S.; Corbeil, J.; Rooney, I.; Schneider, P.; Tschopp, J.; Ware, C.F. Cutting edge: A novel viral TNF receptor superfamily member in virulent strains of human cytomegalovirus. J. Immunol. 1999, 162, 6967–6970. [Google Scholar]
- Rossetto, C.C.; Tarrant-Elorza, M.; Pari, G.S. Cis and trans acting factors involved in human cytomegalovirus experimental and natural latent infection of CD14 (+) monocytes and CD34 (+) cells. PLoS Pathog. 2013, 9, e1003366. [Google Scholar] [CrossRef] [Green Version]
- Cheng, S.; Caviness, K.; Buehler, J.; Smithey, M.; Nikolich-Žugich, J.; Goodrum, F. Transcriptome-wide characterization of human cytomegalovirus in natural infection and experimental latency. Proc. Natl. Acad. Sci. USA 2017, 114, E10586–E10595. [Google Scholar] [CrossRef] [Green Version]
- Shnayder, M.; Nachshon, A.; Krishna, B.; Poole, E.; Boshkov, A.; Binyamin, A.; Maza, I.; Sinclair, J.; Schwartz, M.; Stern-Ginossar, N. Defining the Transcriptional Landscape during Cytomegalovirus Latency with Single-Cell RNA Sequencing. MBio 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Grinde, B. Herpesviruses: Latency and reactivation—Viral strategies and host response. J. Oral. Microbiol. 2013, 5. [Google Scholar] [CrossRef] [Green Version]
- Collin, V.; Flamand, L. HHV-6A/B Integration and the Pathogenesis Associated with the Reactivation of Chromosomally Integrated HHV-6A/B. Viruses 2017, 9, 160. [Google Scholar] [CrossRef] [Green Version]
- Kaufer, B.B.; Flamand, L. Chromosomally integrated HHV-6: Impact on virus, cell and organismal biology. Curr Opin Virol 2014, 9, 111–118. [Google Scholar] [CrossRef]
- Finkel, Y.; Schmiedel, D.; Tai-Schmiedel, J.; Nachshon, A.; Winkler, R.; Dobesova, M.; Schwartz, M.; Mandelboim, O.; Stern-Ginossar, N. Comprehensive annotations of human herpesvirus 6A and 6B genomes reveal novel and conserved genomic features. Elife 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Kondo, K.; Shimada, K.; Sashihara, J.; Tanaka-Taya, K.; Yamanishi, K. Identification of human herpesvirus 6 latency-associated transcripts. J. Virol. 2002, 76, 4145–4151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondo, K.; Sashihara, J.; Shimada, K.; Takemoto, M.; Amo, K.; Miyagawa, H.; Yamanishi, K. Recognition of a novel stage of betaherpesvirus latency in human herpesvirus 6. J. Virol. 2003, 77, 2258–2264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rotola, A.; Ravaioli, T.; Gonelli, A.; Dewhurst, S.; Cassai, E.; Di Luca, D. U94 of human herpesvirus 6 is expressed in latently infected peripheral blood mononuclear cells and blocks viral gene expression in transformed lymphocytes in culture. Proc. Natl. Acad. Sci. USA 1998, 95, 13911–13916. [Google Scholar] [CrossRef] [Green Version]
- Caselli, E.; Bracci, A.; Galvan, M.; Boni, M.; Rotola, A.; Bergamini, C.; Cermelli, C.; Dal Monte, P.; Gompels, U.A.; Cassai, E.; et al. Human herpesvirus 6 (HHV-6) U94/REP protein inhibits betaherpesvirus replication. Virology 2006, 346, 402–414. [Google Scholar] [CrossRef] [Green Version]
- Saviola, A.J.; Zimmermann, C.; Mariani, M.P.; Signorelli, S.A.; Gerrard, D.L.; Boyd, J.R.; Wight, D.J.; Morissette, G.; Gravel, A.; Dubuc, I.; et al. Chromatin Profiles of Chromosomally Integrated Human Herpesvirus-6A. Front. Microbiol. 2019, 10, 1408. [Google Scholar] [CrossRef]
- Piedade, D.; Azevedo-Pereira, J.M. The Role of microRNAs in the Pathogenesis of Herpesvirus Infection. Viruses 2016, 8, 156. [Google Scholar] [CrossRef] [Green Version]
- Skalsky, R.L.; Cullen, B.R. Viruses, microRNAs, and host interactions. Annu. Rev. Microbiol. 2010, 64, 123–141. [Google Scholar] [CrossRef] [Green Version]
- Prusty, B.K.; Gulve, N.; Chowdhury, S.R.; Schuster, M.; Strempel, S.; Descamps, V.; Rudel, T. HHV-6 encoded small non-coding RNAs define an intermediate and early stage in viral reactivation. NPJ Genom. Med. 2018, 3, 25. [Google Scholar] [CrossRef]
- Nukui, M.; Mori, Y.; Murphy, E.A. A human herpesvirus 6A-encoded microRNA: Role in viral lytic replication. J. Virol. 2015, 89, 2615–2627. [Google Scholar] [CrossRef] [Green Version]
- Stevens, J.G.; Wagner, E.K.; Devi-Rao, G.B.; Cook, M.L.; Feldman, L.T. RNA complementary to a herpesvirus alpha gene mRNA is prominent in latently infected neurons. Science 1987, 235, 1056–1059. [Google Scholar] [CrossRef]
- Wagner, E.K.; Bloom, D.C. Experimental investigation of herpes simplex virus latency. Clin. Microbiol. Rev. 1997, 10, 419–443. [Google Scholar] [CrossRef]
- Depledge, D.P.; Sadaoka, T.; Ouwendijk, W.J.D. Molecular Aspects of Varicella-Zoster Virus Latency. Viruses 2018, 10. [Google Scholar] [CrossRef] [Green Version]
- Javier, R.T.; Stevens, J.G.; Dissette, V.B.; Wagner, E.K. A herpes simplex virus transcript abundant in latently infected neurons is dispensable for establishment of the latent state. Virology 1988, 166, 254–257. [Google Scholar] [CrossRef]
- Sedarati, F.; Izumi, K.M.; Wagner, E.K.; Stevens, J.G. Herpes simplex virus type 1 latency-associated transcription plays no role in establishment or maintenance of a latent infection in murine sensory neurons. J. Virol. 1989, 63, 4455–4458. [Google Scholar] [CrossRef] [Green Version]
- Steiner, I.; Spivack, J.G.; Lirette, R.P.; Brown, S.M.; MacLean, A.R.; Subak-Sharpe, J.H.; Fraser, N.W. Herpes simplex virus type 1 latency-associated transcripts are evidently not essential for latent infection. EMBO J. 1989, 8, 505–511. [Google Scholar] [CrossRef]
- Amelio, A.L.; Giordani, N.V.; Kubat, N.J.; O’neil, J.E.; Bloom, D.C. Deacetylation of the herpes simplex virus type 1 latency-associated transcript (LAT) enhancer and a decrease in LAT abundance precede an increase in ICP0 transcriptional permissiveness at early times postexplant. J. Virol. 2006, 80, 2063–2068. [Google Scholar] [CrossRef] [Green Version]
- Cliffe, A.R.; Garber, D.A.; Knipe, D.M. Transcription of the herpes simplex virus latency-associated transcript promotes the formation of facultative heterochromatin on lytic promoters. J. Virol. 2009, 83, 8182–8190. [Google Scholar] [CrossRef] [Green Version]
- Kwiatkowski, D.L.; Thompson, H.W.; Bloom, D.C. The polycomb group protein Bmi1 binds to the herpes simplex virus 1 latent genome and maintains repressive histone marks during latency. J. Virol. 2009, 83, 8173–8181. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.Y.; Zhou, C.; Johnson, K.E.; Colgrove, R.C.; Coen, D.M.; Knipe, D.M. Herpesviral latency-associated transcript gene promotes assembly of heterochromatin on viral lytic-gene promoters in latent infection. Proc. Natl. Acad. Sci. USA 2005, 102, 16055–16059. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, M.; Lock, M.; Miller, C.G.; Fraser, N.W. Regions of the herpes simplex virus type 1 latency-associated transcript that protect cells from apoptosis in vitro and protect neuronal cells in vivo. J. Virol. 2002, 76, 717–729. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.; Chentoufi, A.A.; Hsiang, C.; Carpenter, D.; Osorio, N.; BenMohamed, L.; Fraser, N.W.; Jones, C.; Wechsler, S.L. The herpes simplex virus type 1 latency-associated transcript can protect neuron-derived C1300 and Neuro2A cells from granzyme B-induced apoptosis and CD8 T-cell killing. J. Virol. 2011, 85, 2325–2332. [Google Scholar] [CrossRef] [Green Version]
- Perng, G.C.; Jones, C.; Ciacci-Zanella, J.; Stone, M.; Henderson, G.; Yukht, A.; Slanina, S.M.; Hofman, F.M.; Ghiasi, H.; Nesburn, A.B.; et al. Virus-induced neuronal apoptosis blocked by the herpes simplex virus latency-associated transcript. Science 2000, 287, 1500–1503. [Google Scholar] [CrossRef] [Green Version]
- Held, K.; Junker, A.; Dornmair, K.; Meinl, E.; Sinicina, I.; Brandt, T.; Theil, D.; Derfuss, T. Expression of herpes simplex virus 1-encoded microRNAs in human trigeminal ganglia and their relation to local T-cell infiltrates. J. Virol. 2011, 85, 9680–9685. [Google Scholar] [CrossRef] [Green Version]
- Jurak, I.; Kramer, M.F.; Mellor, J.C.; van Lint, A.L.; Roth, F.P.; Knipe, D.M.; Coen, D.M. Numerous conserved and divergent microRNAs expressed by herpes simplex viruses 1 and 2. J. Virol. 2010, 84, 4659–4672. [Google Scholar] [CrossRef] [Green Version]
- Kramer, M.F.; Jurak, I.; Pesola, J.M.; Boissel, S.; Knipe, D.M.; Coen, D.M. Herpes simplex virus 1 microRNAs expressed abundantly during latent infection are not essential for latency in mouse trigeminal ganglia. Virology 2011, 417, 239–247. [Google Scholar] [CrossRef] [Green Version]
- Umbach, J.L.; Kramer, M.F.; Jurak, I.; Karnowski, H.W.; Coen, D.M.; Cullen, B.R. MicroRNAs expressed by herpes simplex virus 1 during latent infection regulate viral mRNAs. Nature 2008, 454, 780–783. [Google Scholar] [CrossRef] [Green Version]
- Flores, O.; Nakayama, S.; Whisnant, A.W.; Javanbakht, H.; Cullen, B.R.; Bloom, D.C. Mutational inactivation of herpes simplex virus 1 microRNAs identifies viral mRNA targets and reveals phenotypic effects in culture. J. Virol. 2013, 87, 6589–6603. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.H.; Lee, L.Y.; Garber, D.A.; Schaffer, P.A.; Knipe, D.M.; Coen, D.M. Neither LAT nor open reading frame P mutations increase expression of spliced or intron-containing ICP0 transcripts in mouse ganglia latently infected with herpes simplex virus. J. Virol. 2002, 76, 4764–4772. [Google Scholar] [CrossRef] [Green Version]
- Feldman, L.T.; Ellison, A.R.; Voytek, C.C.; Yang, L.; Krause, P.; Margolis, T.P. Spontaneous molecular reactivation of herpes simplex virus type 1 latency in mice. Proc. Natl. Acad. Sci. USA 2002, 99, 978–983. [Google Scholar] [CrossRef] [Green Version]
- Giordani, N.V.; Neumann, D.M.; Kwiatkowski, D.L.; Bhattacharjee, P.S.; McAnany, P.K.; Hill, J.M.; Bloom, D.C. During herpes simplex virus type 1 infection of rabbits, the ability to express the latency-associated transcript increases latent-phase transcription of lytic genes. J. Virol. 2008, 82, 6056–6060. [Google Scholar] [CrossRef] [Green Version]
- Green, M.T.; Rosborough, J.P.; Dunkel, E.C. In vivo reactivation of herpes simplex virus in rabbit trigeminal ganglia: Electrode model. Infect. Immun. 1981, 34, 69–74. [Google Scholar] [CrossRef] [Green Version]
- Kramer, M.F.; Coen, D.M. Quantification of transcripts from the ICP4 and thymidine kinase genes in mouse ganglia latently infected with herpes simplex virus. J. Virol. 1995, 69, 1389–1399. [Google Scholar] [CrossRef] [Green Version]
- Kramer, M.F.; Chen, S.H.; Knipe, D.M.; Coen, D.M. Accumulation of viral transcripts and DNA during establishment of latency by herpes simplex virus. J. Virol. 1998, 72, 1177–1185. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.S.; Raja, P.; Pan, D.; Pesola, J.M.; Coen, D.M.; Knipe, D.M. CCCTC-Binding Factor Acts as a Heterochromatin Barrier on Herpes Simplex Viral Latent Chromatin and Contributes to Poised Latent Infection. MBio 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Maillet, S.; Naas, T.; Crepin, S.; Roque-Afonso, A.M.; Lafay, F.; Efstathiou, S.; Labetoulle, M. Herpes simplex virus type 1 latently infected neurons differentially express latency-associated and ICP0 transcripts. J. Virol. 2006, 80, 9310–9321. [Google Scholar] [CrossRef] [Green Version]
- Nicoll, M.P.; Hann, W.; Shivkumar, M.; Harman, L.E.; Connor, V.; Coleman, H.M.; Proença, J.T.; Efstathiou, S. The HSV-1 Latency-Associated Transcript Functions to Repress Latent Phase Lytic Gene Expression and Suppress Virus Reactivation from Latently Infected Neurons. PLoS Pathog. 2016, 12, e1005539. [Google Scholar] [CrossRef]
- Pesola, J.M.; Zhu, J.; Knipe, D.M.; Coen, D.M. Herpes simplex virus 1 immediate-early and early gene expression during reactivation from latency under conditions that prevent infectious virus production. J. Virol. 2005, 79, 14516–14525. [Google Scholar] [CrossRef] [Green Version]
- Raja, P.; Lee, J.S.; Pan, D.; Pesola, J.M.; Coen, D.M.; Knipe, D.M. A Herpesviral Lytic Protein Regulates the Structure of Latent Viral Chromatin. MBio 2016, 7. [Google Scholar] [CrossRef] [Green Version]
- Russell, T.A.; Tscharke, D.C. Lytic Promoters Express Protein during Herpes Simplex Virus Latency. PLoS Pathog. 2016, 12, e1005729. [Google Scholar] [CrossRef]
- Tal-Singer, R.; Lasner, T.M.; Podrzucki, W.; Skokotas, A.; Leary, J.J.; Berger, S.L.; Fraser, N.W. Gene expression during reactivation of herpes simplex virus type 1 from latency in the peripheral nervous system is different from that during lytic infection of tissue cultures. J. Virol. 1997, 71, 5268–5276. [Google Scholar] [CrossRef] [Green Version]
- Singh, N.; Tscharke, D.C. Herpes simplex virus latency is noisier the closer we look. J. Virol. 2019. [Google Scholar] [CrossRef] [Green Version]
- Miller, C.S.; Danaher, R.J. Asymptomatic shedding of herpes simplex virus (HSV) in the oral cavity. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endod. 2008, 105, 43–50. [Google Scholar] [CrossRef]
- Ramchandani, M.S.; Jing, L.; Russell, R.M.; Tran, T.; Laing, K.J.; Magaret, A.S.; Selke, S.; Cheng, A.; Huang, M.L.; Xie, H.; et al. Viral Genetics Modulate Orolabial Herpes Simplex Virus Type 1 Shedding in Humans. J. Infect. Dis. 2019, 219, 1058–1066. [Google Scholar] [CrossRef]
- Chen, X.P.; Mata, M.; Kelley, M.; Glorioso, J.C.; Fink, D.J. The relationship of herpes simplex virus latency associated transcript expression to genome copy number: A quantitative study using laser capture microdissection. J. Neurovirol. 2002, 8, 204–210. [Google Scholar] [CrossRef]
- Maggioncalda, J.; Mehta, A.; Su, Y.H.; Fraser, N.W.; Block, T.M. Correlation between herpes simplex virus type 1 rate of reactivation from latent infection and the number of infected neurons in trigeminal ganglia. Virology 1996, 225, 72–81. [Google Scholar] [CrossRef] [Green Version]
- Mehta, A.; Maggioncalda, J.; Bagasra, O.; Thikkavarapu, S.; Saikumari, P.; Valyi-Nagy, T.; Fraser, N.W.; Block, T.M. In situ DNA PCR and RNA hybridization detection of herpes simplex virus sequences in trigeminal ganglia of latently infected mice. Virology 1995, 206, 633–640. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Lau, T.Y.; Morales, M.; Mont, E.K.; Straus, S.E. Laser-capture microdissection: Refining estimates of the quantity and distribution of latent herpes simplex virus 1 and varicella-zoster virus DNA in human trigeminal Ganglia at the single-cell level. J. Virol. 2005, 79, 14079–14087. [Google Scholar] [CrossRef] [Green Version]
- Cliffe, A.R.; Wilson, A.C. Restarting Lytic Gene Transcription at the Onset of Herpes Simplex Virus Reactivation. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [Green Version]
- Bloom, D.C. Alphaherpesvirus Latency: A Dynamic State of Transcription and Reactivation. Adv. Virus Res. 2016, 94, 53–80. [Google Scholar] [CrossRef]
- Nicoll, M.P.; Proença, J.T.; Efstathiou, S. The molecular basis of herpes simplex virus latency. FEMS Microbiol. Rev. 2012, 36, 684–705. [Google Scholar] [CrossRef]
- Cohrs, R.J.; Barbour, M.; Gilden, D.H. Varicella-zoster virus (VZV) transcription during latency in human ganglia: Detection of transcripts mapping to genes 21, 29, 62, and 63 in a cDNA library enriched for VZV RNA. J. Virol. 1996, 70, 2789–2796. [Google Scholar] [CrossRef] [Green Version]
- Cohrs, R.J.; Gilden, D.H.; Kinchington, P.R.; Grinfeld, E.; Kennedy, P.G. Varicella-zoster virus gene 66 transcription and translation in latently infected human Ganglia. J. Virol. 2003, 77, 6660–6665. [Google Scholar] [CrossRef] [Green Version]
- Cohrs, R.J.; Gilden, D.H. Prevalence and abundance of latently transcribed varicella-zoster virus genes in human ganglia. J. Virol. 2007, 81, 2950–2956. [Google Scholar] [CrossRef] [Green Version]
- Mahalingam, R.; Wellish, M.; Cohrs, R.; Debrus, S.; Piette, J.; Rentier, B.; Gilden, D.H. Expression of protein encoded by varicella-zoster virus open reading frame 63 in latently infected human ganglionic neurons. Proc. Natl. Acad. Sci. USA 1996, 93, 2122–2124. [Google Scholar] [CrossRef] [Green Version]
- Cohen, J.I.; Cox, E.; Pesnicak, L.; Srinivas, S.; Krogmann, T. The varicella-zoster virus open reading frame 63 latency-associated protein is critical for establishment of latency. J. Virol. 2004, 78, 11833–11840. [Google Scholar] [CrossRef] [Green Version]
- Hood, C.; Cunningham, A.L.; Slobedman, B.; Arvin, A.M.; Sommer, M.H.; Kinchington, P.R.; Abendroth, A. Varicella-zoster virus ORF63 inhibits apoptosis of primary human neurons. J. Virol. 2006, 80, 1025–1031. [Google Scholar] [CrossRef] [Green Version]
- Cohen, J.I.; Krogmann, T.; Ross, J.P.; Pesnicak, L.; Prikhod’ko, E.A. Varicella-zoster virus ORF4 latency-associated protein is important for establishment of latency. J. Virol. 2005, 79, 6969–6975. [Google Scholar] [CrossRef] [Green Version]
- Depledge, D.P.; Ouwendijk, W.J.D.; Sadaoka, T.; Braspenning, S.E.; Mori, Y.; Cohrs, R.J.; Verjans, G.M.G.M.; Breuer, J. A spliced latency-associated VZV transcript maps antisense to the viral transactivator gene 61. Nat. Commun. 2018, 9, 1167. [Google Scholar] [CrossRef]
- Sadaoka, T.; Rajbhandari, L.; Shukla, P.; Jagdish, B.; Lee, H.; Lee, G.; Venkatesan, A. Human stem cell derived sensory neurons are positioned to support varicella zoster virus latency. bioRxiv 2020. [Google Scholar] [CrossRef]
- Tang, Q.; Qin, D.; Lv, Z.; Zhu, X.; Ma, X.; Yan, Q.; Zeng, Y.; Guo, Y.; Feng, N.; Lu, C. Herpes simplex virus type 2 triggers reactivation of Kaposi’s sarcoma-associated herpesvirus from latency and collaborates with HIV-1 Tat. PLoS ONE 2012, 7, e31652. [Google Scholar] [CrossRef]
- Ye, F.; Lei, X.; Gao, S.J. Mechanisms of Kaposi’s Sarcoma-Associated Herpesvirus Latency and Reactivation. Adv. Virol. 2011, 2011. [Google Scholar] [CrossRef]
- Purushothaman, P.; Uppal, T.; Verma, S.C. Molecular biology of KSHV lytic reactivation. Viruses 2015, 7, 116–153. [Google Scholar] [CrossRef] [Green Version]
- Aneja, K.K.; Yuan, Y. Reactivation and Lytic Replication of Kaposi’s Sarcoma-Associated Herpesvirus: An Update. Front. Microbiol. 2017, 8, 613. [Google Scholar] [CrossRef]
- Flamand, L.; Stefanescu, I.; Ablashi, D.V.; Menezes, J. Activation of the Epstein–Barr virus replicative cycle by human herpesvirus 6. J. Virol. 1993, 67, 6768–6777. [Google Scholar] [CrossRef] [Green Version]
- Reynaldi, A.; Schlub, T.E.; Chelimo, K.; Sumba, P.O.; Piriou, E.; Ogolla, S.; Moormann, A.M.; Rochford, R.; Davenport, M.P. Impact of Plasmodium falciparum Coinfection on Longitudinal Epstein–Barr Virus Kinetics in Kenyan Children. J. Infect. Dis. 2016, 213, 985–991. [Google Scholar] [CrossRef] [Green Version]
- Karrasch, M.; Herfurth, K.; Kläver, M.; Miethke, J.; Mayer-Scholl, A.; Luge, E.; Straube, E.; Busch, M. Severe leptospirosis complicated by Epstein–Barr Virus reactivation. Infection 2015, 43, 763–769. [Google Scholar] [CrossRef]
- Ueda, S.; Uchiyama, S.; Azzi, T.; Gysin, C.; Berger, C.; Bernasconi, M.; Harabuchi, Y.; Zinkernagel, A.S.; Nadal, D. Oropharyngeal group A streptococcal colonization disrupts latent Epstein–Barr virus infection. J. Infect. Dis. 2014, 209, 255–264. [Google Scholar] [CrossRef] [Green Version]
- Byrne, C.M.; Johnston, C.; Orem, J.; Okuku, F.; Huang, M.-L.; Selke, S.; Wald, A.; Corey, L.; Schiffer, J.; Casper, T.C.; et al. Increased oral Epstein–Barr virus shedding with HIV-1 co-infection is due to a combination of B cell activation and impaired cellular immune control. bioRxiv 2019. [Google Scholar] [CrossRef]
- Makielski, K.R.; Lee, D.; Lorenz, L.D.; Nawandar, D.M.; Chiu, Y.F.; Kenney, S.C.; Lambert, P.F. Human papillomavirus promotes Epstein–Barr virus maintenance and lytic reactivation in immortalized oral keratinocytes. Virology 2016, 495, 52–62. [Google Scholar] [CrossRef]
- Hirsiger, J.R.; Fuchs, P.S.; Häusermann, P.; Müller-Durovic, B.; Daikeler, T.; Recher, M.; Hirsch, H.H.; Terracciano, L.; Berger, C.T. Syphilis Reactivates Latent Epstein–Barr Virus Reservoir via Toll-Like Receptor 2 and B-Cell Receptor Activation. Open Forum Infect. Dis. 2019, 6, ofz317. [Google Scholar] [CrossRef]
- Murata, T.; Tsurumi, T. Switching of EBV cycles between latent and lytic states. Rev. Med. Virol. 2014, 24, 142–153. [Google Scholar] [CrossRef]
- Mercader, M.; Taddeo, B.; Panella, J.R.; Chandran, B.; Nickoloff, B.J.; Foreman, K.E. Induction of HHV-8 lytic cycle replication by inflammatory cytokines produced by HIV-1-infected T cells. Am. J. Pathol. 2000, 156, 1961–1971. [Google Scholar] [CrossRef] [Green Version]
- Chang, J.; Renne, R.; Dittmer, D.; Ganem, D. Inflammatory cytokines and the reactivation of Kaposi’s sarcoma-associated herpesvirus lytic replication. Virology 2000, 266, 17–25. [Google Scholar] [CrossRef] [Green Version]
- Blackbourn, D.J.; Fujimura, S.; Kutzkey, T.; Levy, J.A. Induction of human herpesvirus-8 gene expression by recombinant interferon gamma. AIDS 2000, 14, 98–99. [Google Scholar] [CrossRef]
- Gandhi, J.; Gaur, N.; Khera, L.; Kaul, R.; Robertson, E.S. COX-2 induces lytic reactivation of EBV through PGE2 by modulating the EP receptor signaling pathway. Virology 2015, 484, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Davis, D.A.; Rinderknecht, A.S.; Zoeteweij, J.P.; Aoki, Y.; Read-Connole, E.L.; Tosato, G.; Blauvelt, A.; Yarchoan, R. Hypoxia induces lytic replication of Kaposi sarcoma-associated herpesvirus. Blood 2001, 97, 3244–3250. [Google Scholar] [CrossRef] [Green Version]
- Jiang, J.H.; Wang, N.; Li, A.; Liao, W.T.; Pan, Z.G.; Mai, S.J.; Li, D.J.; Zeng, M.S.; Wen, J.M.; Zeng, Y.X. Hypoxia can contribute to the induction of the Epstein–Barr virus (EBV) lytic cycle. J. Clin. Virol. 2006, 37, 98–103. [Google Scholar] [CrossRef]
- Kraus, R.J.; Yu, X.; Cordes, B.A.; Sathiamoorthi, S.; Iempridee, T.; Nawandar, D.M.; Ma, S.; Romero-Masters, J.C.; McChesney, K.G.; Lin, Z.; et al. Hypoxia-inducible factor-1α plays roles in Epstein–Barr virus’s natural life cycle and tumorigenesis by inducing lytic infection through direct binding to the immediate-early BZLF1 gene promoter. PLoS Pathog. 2017, 13, e1006404. [Google Scholar] [CrossRef]
- Ye, F.; Zhou, F.; Bedolla, R.G.; Jones, T.; Lei, X.; Kang, T.; Guadalupe, M.; Gao, S.J. Reactive oxygen species hydrogen peroxide mediates Kaposi’s sarcoma-associated herpesvirus reactivation from latency. PLoS Pathog. 2011, 7, e1002054. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Feng, J.; Sun, R. Oxidative stress induces reactivation of Kaposi’s sarcoma-associated herpesvirus and death of primary effusion lymphoma cells. J. Virol. 2011, 85, 715–724. [Google Scholar] [CrossRef] [Green Version]
- Coskun, O.; Sener, K.; Kilic, S.; Erdem, H.; Yaman, H.; Besirbellioglu, A.B.; Gul, H.C.; Eyigun, C.P. Stress-related Epstein–Barr virus reactivation. Clin. Exp. Med. 2010, 10, 15–20. [Google Scholar] [CrossRef]
- Mehta, S.K.; Laudenslager, M.L.; Stowe, R.P.; Crucian, B.E.; Sams, C.F.; Pierson, D.L. Multiple latent viruses reactivate in astronauts during Space Shuttle missions. Brain Behav. Immun. 2014, 41, 210–217. [Google Scholar] [CrossRef]
- Mehta, S.K.; Laudenslager, M.L.; Stowe, R.P.; Crucian, B.E.; Feiveson, A.H.; Sams, C.F.; Pierson, D.L. Latent virus reactivation in astronauts on the international space station. NPJ Microgravity 2017, 3, 11. [Google Scholar] [CrossRef] [Green Version]
- Pierson, D.L.; Stowe, R.P.; Phillips, T.M.; Lugg, D.J.; Mehta, S.K. Epstein–Barr virus shedding by astronauts during space flight. Brain Behav. Immun. 2005, 19, 235–242. [Google Scholar] [CrossRef]
- Rooney, B.V.; Crucian, B.E.; Pierson, D.L.; Laudenslager, M.L.; Mehta, S.K. Herpes Virus Reactivation in Astronauts during Spaceflight and Its Application on Earth. Front. Microbiol. 2019, 10, 16. [Google Scholar] [CrossRef]
- Feng, W.H.; Israel, B.; Raab-Traub, N.; Busson, P.; Kenney, S.C. Chemotherapy induces lytic EBV replication and confers ganciclovir susceptibility to EBV-positive epithelial cell tumors. Cancer Res. 2002, 62, 1920–1926. [Google Scholar]
- Westphal, E.M.; Blackstock, W.; Feng, W.; Israel, B.; Kenney, S.C. Activation of lytic Epstein–Barr virus (EBV) infection by radiation and sodium butyrate in vitro and in vivo: A potential method for treating EBV-positive malignancies. Cancer Res. 2000, 60, 5781–5788. [Google Scholar]
- Davies, A.H.; Grand, R.J.; Evans, F.J.; Rickinson, A.B. Induction of Epstein–Barr virus lytic cycle by tumor-promoting and non-tumor-promoting phorbol esters requires active protein kinase C. J. Virol. 1991, 65, 6838–6844. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Ikuta, K.; Tajima, M.; Sairenji, T. 12-O-tetradecanoylphorbol-13-acetate induces Epstein–Barr virus reactivation via NF-kappaB and AP-1 as regulated by protein kinase C and mitogen-activated protein kinase. Virology 2001, 286, 91–99. [Google Scholar] [CrossRef] [Green Version]
- Gorres, K.L.; Daigle, D.; Mohanram, S.; Miller, G. Activation and repression of Epstein–Barr Virus and Kaposi’s sarcoma-associated herpesvirus lytic cycles by short- and medium-chain fatty acids. J. Virol. 2014, 88, 8028–8044. [Google Scholar] [CrossRef] [Green Version]
- Renne, R.; Zhong, W.; Herndier, B.; McGrath, M.; Abbey, N.; Kedes, D.; Ganem, D. Lytic growth of Kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) in culture. Nat. Med. 1996, 2, 342–346. [Google Scholar] [CrossRef]
- Guito, J.; Lukac, D.M. KSHV Rta Promoter Specification and Viral Reactivation. Front. Microbiol. 2012, 3, 30. [Google Scholar] [CrossRef] [Green Version]
- Reddehase, M.J.; Lemmermann, N.A.W. Cellular reservoirs of latent cytomegaloviruses. Med. Microbiol. Immunol. 2019, 208, 391–403. [Google Scholar] [CrossRef]
- Zhu, D.; Pan, C.; Sheng, J.; Liang, H.; Bian, Z.; Liu, Y.; Trang, P.; Wu, J.; Liu, F.; Zhang, C.Y.; et al. Human cytomegalovirus reprograms haematopoietic progenitor cells into immunosuppressive monocytes to achieve latency. Nat. Microbiol. 2018, 3, 503–513. [Google Scholar] [CrossRef]
- Söderberg-Nauclér, C.; Fish, K.N.; Nelson, J.A. Reactivation of latent human cytomegalovirus by allogeneic stimulation of blood cells from healthy donors. Cell 1997, 91, 119–126. [Google Scholar] [CrossRef] [Green Version]
- Lautenschlager, I.; Lappalainen, M.; Linnavuori, K.; Suni, J.; Höckerstedt, K. CMV infection is usually associated with concurrent HHV-6 and HHV-7 antigenemia in liver transplant patients. J. Clin. Virol. 2002, 25 (Suppl. 2), S57–S61. [Google Scholar] [CrossRef]
- Gerdemann, U.; Keukens, L.; Keirnan, J.M.; Katari, U.L.; Nguyen, C.T.; de Pagter, A.P.; Ramos, C.A.; Kennedy-Nasser, A.; Gottschalk, S.M.; Heslop, H.E.; et al. Immunotherapeutic strategies to prevent and treat human herpesvirus 6 reactivation after allogeneic stem cell transplantation. Blood 2013, 121, 207–218. [Google Scholar] [CrossRef] [Green Version]
- Mendez, J.C.; Dockrell, D.H.; Espy, M.J.; Smith, T.F.; Wilson, J.A.; Harmsen, W.S.; Ilstrup, D.; Paya, C.V. Human beta-herpesvirus interactions in solid organ transplant recipients. J. Infect. Dis. 2001, 183, 179–184. [Google Scholar] [CrossRef]
- Fan, J.; Jing, M.; Yang, M.; Xu, L.; Liang, H.; Huang, Y.; Yang, R.; Gui, G.; Wang, H.; Gong, S.; et al. Herpesvirus infections in hematopoietic stem cell transplant recipients seropositive for human cytomegalovirus before transplantation. Int. J. Infect. Dis. 2016, 46, 89–93. [Google Scholar] [CrossRef] [Green Version]
- Shams El-Din, A.A.; El-Desoukey, N.A.; Amin Tawadrous, D.G.; Fouad, N.M.B.E.; Abdel-Mooti, M.; Hotar, S.F. The potential association of CMV-specific CD8+ T lymphocyte reconstitution with the risk of CMV reactivation and persistency in post allogeneic stem cell transplant patients. Hematology 2018, 23, 463–469. [Google Scholar] [CrossRef] [Green Version]
- Sinclair, J.; Sissons, P. Latency and reactivation of human cytomegalovirus. J. Gen. Virol. 2006, 87, 1763–1779. [Google Scholar] [CrossRef]
- Chapenko, S.; Folkmane, I.; Tomsone, V.; Amerika, D.; Rozentals, R.; Murovska, M. Co-infection of two beta-herpesviruses (CMV and HHV-7) as an increased risk factor for ‘CMV disease’ in patients undergoing renal transplantation. Clin. Transplant. 2000, 14, 486–492. [Google Scholar] [CrossRef]
- Mehta, S.K.; Stowe, R.P.; Feiveson, A.H.; Tyring, S.K.; Pierson, D.L. Reactivation and shedding of cytomegalovirus in astronauts during spaceflight. J. Infect. Dis. 2000, 182, 1761–1764. [Google Scholar] [CrossRef] [Green Version]
- D’Aiuto, L.; Di Maio, R.; Heath, B.; Raimondi, G.; Milosevic, J.; Watson, A.M.; Bamne, M.; Parks, W.T.; Yang, L.; Lin, B.; et al. Human induced pluripotent stem cell-derived models to investigate human cytomegalovirus infection in neural cells. PLoS ONE 2012, 7, e49700. [Google Scholar] [CrossRef] [Green Version]
- Luo, M.H.; Schwartz, P.H.; Fortunato, E.A. Neonatal neural progenitor cells and their neuronal and glial cell derivatives are fully permissive for human cytomegalovirus infection. J. Virol. 2008, 82, 9994–10007. [Google Scholar] [CrossRef] [Green Version]
- Luo, M.H.; Hannemann, H.; Kulkarni, A.S.; Schwartz, P.H.; O’Dowd, J.M.; Fortunato, E.A. Human cytomegalovirus infection causes premature and abnormal differentiation of human neural progenitor cells. J. Virol. 2010, 84, 3528–3541. [Google Scholar] [CrossRef] [Green Version]
- Odeberg, J.; Wolmer, N.; Falci, S.; Westgren, M.; Seiger, A.; Söderberg-Nauclér, C. Human cytomegalovirus inhibits neuronal differentiation and induces apoptosis in human neural precursor cells. J. Virol. 2006, 80, 8929–8939. [Google Scholar] [CrossRef] [Green Version]
- Taylor-Wiedeman, J.; Sissons, P.; Sinclair, J. Induction of endogenous human cytomegalovirus gene expression after differentiation of monocytes from healthy carriers. J. Virol. 1994, 68, 1597–1604. [Google Scholar] [CrossRef] [Green Version]
- Reeves, M.B.; MacAry, P.A.; Lehner, P.J.; Sissons, J.G.; Sinclair, J.H. Latency, chromatin remodeling, and reactivation of human cytomegalovirus in the dendritic cells of healthy carriers. Proc. Natl. Acad. Sci. USA 2005, 102, 4140–4145. [Google Scholar] [CrossRef] [Green Version]
- O’Connor, C.M.; Murphy, E.A. A myeloid progenitor cell line capable of supporting human cytomegalovirus latency and reactivation, resulting in infectious progeny. J. Virol. 2012, 86, 9854–9865. [Google Scholar] [CrossRef] [Green Version]
- Yasukawa, M.; Ohminami, H.; Sada, E.; Yakushijin, Y.; Kaneko, M.; Yanagisawa, K.; Kohno, H.; Bando, S.; Fujita, S. Latent infection and reactivation of human herpesvirus 6 in two novel myeloid cell lines. Blood 1999, 93, 991–999. [Google Scholar] [CrossRef]
- Ahlqvist, J.; Fotheringham, J.; Akhyani, N.; Yao, K.; Fogdell-Hahn, A.; Jacobson, S. Differential tropism of human herpesvirus 6 (HHV-6) variants and induction of latency by HHV-6A in oligodendrocytes. J. Neurovirol. 2005, 11, 384–394. [Google Scholar] [CrossRef]
- Yoshikawa, T.; Asano, Y.; Akimoto, S.; Ozaki, T.; Iwasaki, T.; Kurata, T.; Goshima, F.; Nishiyama, Y. Latent infection of human herpesvirus 6 in astrocytoma cell line and alteration of cytokine synthesis. J. Med. Virol. 2002, 66, 497–505. [Google Scholar] [CrossRef]
- Katsafanas, G.C.; Schirmer, E.C.; Wyatt, L.S.; Frenkel, N. In vitro activation of human herpesviruses 6 and 7 from latency. Proc. Natl. Acad. Sci. USA 1996, 93, 9788–9792. [Google Scholar] [CrossRef] [Green Version]
- Kondo, K.; Kondo, T.; Okuno, T.; Takahashi, M.; Yamanishi, K. Latent human herpesvirus 6 infection of human monocytes/macrophages. J. Gen. Virol. 1991, 72 Pt 6, 1401–1408. [Google Scholar] [CrossRef]
- Prasad, A.; Remick, J.; Zeichner, S.L. Activation of human herpesvirus replication by apoptosis. J. Virol. 2013, 87, 10641–10650. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, P.G.; Rovnak, J.; Badani, H.; Cohrs, R.J. A comparison of herpes simplex virus type 1 and varicella-zoster virus latency and reactivation. J. Gen. Virol. 2015, 96, 1581–1602. [Google Scholar] [CrossRef]
- Wilson, A.C.; Mohr, I. A cultured affair: HSV latency and reactivation in neurons. Trends Microbiol 2012, 20, 604–611. [Google Scholar] [CrossRef] [Green Version]
- Mazzarello, V.; Ferrari, M.; Decandia, S.; Sotgiu, M.A. Sunlight and Herpes Virus. In Human Herpesvirus Infection—Biological Features, Transmission, Symptoms, Diagnosis and Treatment; Thomasini, R.L., Ed.; IntechOpen: London, UK, 2018. [Google Scholar]
- Zak-Prelich, M.; Borkowski, J.L.; Alexander, F.; Norval, M. The role of solar ultraviolet irradiation in zoster. Epidemiol Infect 2002, 129, 593–597. [Google Scholar] [CrossRef]
- Nussbaum, R. Theories on Varicella Zoster Virus Reactivation Based on Shingles Patterns. Sci. J. Lander Coll. Arts Sci. 2014, 8, 10. [Google Scholar]
- Fatahzadeh, M.; Schwartz, R.A. Human herpes simplex virus infections: Epidemiology, pathogenesis, symptomatology, diagnosis, and management. J. Am. Acad. Dermatol. 2007, 57, 737–763. [Google Scholar] [CrossRef]
- Black, B.J.; Atmaramani, R.; Kumaraju, R.; Plagens, S.; Romero-Ortega, M.; Dussor, G.; Price, T.J.; Campbell, Z.T.; Pancrazio, J.J. Adult mouse sensory neurons on microelectrode arrays exhibit increased spontaneous and stimulus-evoked activity in the presence of interleukin-6. J. Neurophysiol. 2018, 120, 1374–1385. [Google Scholar] [CrossRef]
- Poon, D.C.; Ho, Y.S.; Chiu, K.; Wong, H.L.; Chang, R.C. Sickness: From the focus on cytokines, prostaglandins, and complement factors to the perspectives of neurons. Neurosci. Biobehav. Rev. 2015, 57, 30–45. [Google Scholar] [CrossRef]
- Harbour, D.A.; Blyth, W.A.; Hill, T.J. Prostanglandins enhance spread of herpes simplex virus in cell cultures. J. Gen. Virol. 1978, 41, 87–95. [Google Scholar] [CrossRef]
- Freeman, M.L.; Sheridan, B.S.; Bonneau, R.H.; Hendricks, R.L. Psychological stress compromises CD8+ T cell control of latent herpes simplex virus type 1 infections. J. Immunol. 2007, 179, 322–328. [Google Scholar] [CrossRef] [Green Version]
- Schmader, K.; Studenski, S.; MacMillan, J.; Grufferman, S.; Cohen, H.J. Are stressful life events risk factors for herpes zoster? J. Am. Geriatr. Soc. 1990, 38, 1188–1194. [Google Scholar] [CrossRef]
- Schmader, K.; George, L.K.; Burchett, B.M.; Hamilton, J.D.; Pieper, C.F. Race and stress in the incidence of herpes zoster in older adults. J. Am. Geriatr. Soc. 1998, 46, 973–977. [Google Scholar] [CrossRef]
- Lasserre, A.; Blaizeau, F.; Gorwood, P.; Bloch, K.; Chauvin, P.; Liard, F.; Blanchon, T.; Hanslik, T. Herpes zoster: Family history and psychological stress-case-control study. J. Clin. Virol. 2012, 55, 153–157. [Google Scholar] [CrossRef]
- Hsia, S.C.; Bedadala, G.R.; Balish, M.D. Effects of thyroid hormone on HSV-1 gene regulation: Implications in the control of viral latency and reactivation. Cell Biosci. 2011, 1, 24. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, M.; Wilson, A.C.; Chao, M.V.; Mohr, I. Control of viral latency in neurons by axonal mTOR signaling and the 4E-BP translation repressor. Genes Dev. 2012, 26, 1527–1532. [Google Scholar] [CrossRef] [Green Version]
- Yanez, A.A.; Harrell, T.; Sriranganathan, H.J.; Ives, A.M.; Bertke, A.S. Neurotrophic Factors NGF, GDNF and NTN Selectively Modulate HSV1 and HSV2 Lytic Infection and Reactivation in Primary Adult Sensory and Autonomic Neurons. Pathogens 2017, 6, 5. [Google Scholar] [CrossRef]
- Camarena, V.; Kobayashi, M.; Kim, J.Y.; Roehm, P.; Perez, R.; Gardner, J.; Wilson, A.C.; Mohr, I.; Chao, M.V. Nature and duration of growth factor signaling through receptor tyrosine kinases regulates HSV-1 latency in neurons. Cell Host Microbe 2010, 8, 320–330. [Google Scholar] [CrossRef] [Green Version]
- Wilcox, C.L.; Johnson, E.M. Nerve growth factor deprivation results in the reactivation of latent herpes simplex virus in vitro. J. Virol. 1987, 61, 2311–2315. [Google Scholar] [CrossRef] [Green Version]
- Cohrs, R.J.; Badani, H.; Baird, N.L.; White, T.M.; Sanford, B.; Gilden, D. Induction of varicella zoster virus DNA replication in dissociated human trigeminal ganglia. J. Neurovirol. 2017, 23, 152–157. [Google Scholar] [CrossRef] [Green Version]
- Pourchet, A.; Modrek, A.S.; Placantonakis, D.G.; Mohr, I.; Wilson, A.C. Modeling HSV-1 Latency in Human Embryonic Stem Cell-Derived Neurons. Pathogens 2017, 6, 24. [Google Scholar] [CrossRef] [Green Version]
- Laemmle, L.; Goldstein, R.S.; Kinchington, P.R. Modeling Varicella Zoster Virus Persistence and Reactivation—Closer to Resolving a Perplexing Persistent State. Front. Microbiol. 2019, 10, 1634. [Google Scholar] [CrossRef] [Green Version]
- Cliffe, A.R.; Arbuckle, J.H.; Vogel, J.L.; Geden, M.J.; Rothbart, S.B.; Cusack, C.L.; Strahl, B.D.; Kristie, T.M.; Deshmukh, M. Neuronal Stress Pathway Mediating a Histone Methyl/Phospho Switch Is Required for Herpes Simplex Virus Reactivation. Cell Host Microbe 2015, 18, 649–658. [Google Scholar] [CrossRef] [Green Version]
- Du, T.; Zhou, G.; Roizman, B. Induction of apoptosis accelerates reactivation of latent HSV-1 in ganglionic organ cultures and replication in cell cultures. Proc. Natl. Acad. Sci. USA 2012, 109, 14616–14621. [Google Scholar] [CrossRef] [Green Version]
- Alkharsah, K.R.; Dedicoat, M.; Blasczyk, R.; Newton, R.; Schulz, T.F. Influence of HLA alleles on shedding of Kaposi sarcoma-associated herpesvirus in saliva in an African population. J. Infect. Dis. 2007, 195, 809–816. [Google Scholar] [CrossRef]
- Alkharsah, K.R.; Alzahrani, A.J.; Obeid, O.E.; El-Harith, E.H.; Guella, A.; Mohamed, E.A.; Haykal, A.H.; Stuhrmann, M.; Al-Ali, A.K. Vascular endothelial growth factor A polymorphism and risk of Kaposi’s sarcoma herpesvirus viremia in kidney allograft recipients. Transpl. Infect. Dis. 2014, 16, 783–789. [Google Scholar] [CrossRef]
- Houldcroft, C.J.; Kellam, P. Host genetics of Epstein–Barr virus infection, latency and disease. Rev. Med. Virol. 2015, 25, 71–84. [Google Scholar] [CrossRef] [Green Version]
- Sezgin, E.; An, P.; Winkler, C.A. Host Genetics of Cytomegalovirus Pathogenesis. Front. Genet. 2019, 10, 616. [Google Scholar] [CrossRef] [Green Version]
- Moraru, M.; Cisneros, E.; Gómez-Lozano, N.; de Pablo, R.; Portero, F.; Cañizares, M.; Vaquero, M.; Roustán, G.; Millán, I.; López-Botet, M.; et al. Host genetic factors in susceptibility to herpes simplex type 1 virus infection: Contribution of polymorphic genes at the interface of innate and adaptive immunity. J. Immunol. 2012, 188, 4412–4420. [Google Scholar] [CrossRef] [Green Version]
- Kriesel, J.D.; Bhatia, A.; Thomas, A. Cold sore susceptibility gene-1 genotypes affect the expression of herpes labialis in unrelated human subjects. Hum. Genome Var. 2014, 1, 14024. [Google Scholar] [CrossRef] [Green Version]
- Crosslin, D.R.; Carrell, D.S.; Burt, A.; Kim, D.S.; Underwood, J.G.; Hanna, D.S.; Comstock, B.A.; Baldwin, E.; de Andrade, M.; Kullo, I.J.; et al. Genetic variation in the HLA region is associated with susceptibility to herpes zoster. Genes Immun. 2015, 16, 1–7. [Google Scholar] [CrossRef] [Green Version]
Figure 1. Latency establishment and lytic reactivation of herpesviruses. Following an initial lytic infection, herpesviruses establish latency mostly in dividing cells (except alphaherpesviruses), during which the viral genome is maintained as an episome and is tethered to cellular chromatin; lytic reactivation of gammaherpesviruses and betaherpesviruses may be cell differentiation dependent and occurs upon receiving the stimulus for terminal differentiation. EBV, a gammaherpesvirus, infects naïve B cells and remains latent throughout their differentiation until memory B cells carrying latent virus become activated and differentiate into plasma cells, which induces lytic reactivation. HCMV, a betaherpesvirus, establishes latency in cells of the myeloid lineage, its lytic cycle is activated upon differentiation into macrophages and dendritic cells. Additionally, HCMV can also establish latency in neural lineage and undergo lytic cycle in neurons, although the details of HCMV neural infection need to be studied further to resolve observed contradictions. In contrast HSV-1, an alphaherpesvirus establishes latency in sensory neurons and the lytic reactivation is induced upon receiving a specific stimulus. Viral genomes are symbolized by a black circle in the nucleus.
Figure 1. Latency establishment and lytic reactivation of herpesviruses. Following an initial lytic infection, herpesviruses establish latency mostly in dividing cells (except alphaherpesviruses), during which the viral genome is maintained as an episome and is tethered to cellular chromatin; lytic reactivation of gammaherpesviruses and betaherpesviruses may be cell differentiation dependent and occurs upon receiving the stimulus for terminal differentiation. EBV, a gammaherpesvirus, infects naïve B cells and remains latent throughout their differentiation until memory B cells carrying latent virus become activated and differentiate into plasma cells, which induces lytic reactivation. HCMV, a betaherpesvirus, establishes latency in cells of the myeloid lineage, its lytic cycle is activated upon differentiation into macrophages and dendritic cells. Additionally, HCMV can also establish latency in neural lineage and undergo lytic cycle in neurons, although the details of HCMV neural infection need to be studied further to resolve observed contradictions. In contrast HSV-1, an alphaherpesvirus establishes latency in sensory neurons and the lytic reactivation is induced upon receiving a specific stimulus. Viral genomes are symbolized by a black circle in the nucleus.


{kind=link}
{kind=link}
Table 1. Comparison of herpesviral latency reservoirs, latent transcripts, and inducers of lytic reactivation.
Table 1. Comparison of herpesviral latency reservoirs, latent transcripts, and inducers of lytic reactivation.
| Sub Family | Virus | Cell Tropism | Latent Reservoir | Major Latent Transcripts | Inducers of Lytic Cycle |
|---|---|---|---|---|---|
| Alphaherpesvirinae | HSV1 HSV2 |
neurons fibroblasts |
neurons | LATs: sRNA1 and 2, lncRNAs, miRNAs |
immunosuppression, stress, UV hypoxia local tissue trauma hypothermia and hyperthermia cytokines and prostaglandins * hormonal changes NGF withdrawal Dexamethasone, NaBu |
| VZV | neurons | neurons | ORF63 VLT |
immunosuppression, stress, UV NGF withdrawal PI3K inhibitor, NaBu |
|
| Betaherpesvirinae | HCMV | fibroblasts PBMCs macrophages dendritic cells endothelial cells |
CD34+ HSCs CD33+ myeloid progenitors CD14+ monocytes NSCs |
UL138, UL111A LUNA US28 IE1x4 lncRNA2.7 and 4.9 UL144 |
immunosuppression proinflammatory cytokines * differentiation of cells allogeneic T-cell activation TPA |
| HHV-6 HHV-7 |
T cells | CD34+ HSCs PBMCs |
HHV-6: H6LTs:ORF99, ORF142, ORF145 U94 miR-U86 HHV-7: no data available |
immunosuppression infection with other pathogen * (HHV-6B) T-cell activation (HHV-7) TPA (HHV-6A/6B, HHV-7), TSA (HHV-6A) |
|
| Gammaherpesvirinae | EBV | B cells epithelial cells |
B cells | Latency I: EBNA1 Latency II: EBNA1 and LMP1 and/or 2 Latency III: EBNA1, 2, 3a, 3c, 3b, LP and LMP1, 2a and 2b |
immunosuppression, stress, chemotherapy, radiotherapy, hypoxia infection with other pathogen * prostaglandins * BCR crosslinking, TPA, NaBu, TSA |
| KSHV | B cells endothelial cells |
B cells | LANA (ORF73) vCyc (ORF72) vFLIP (ORF71) Kaposins vIRF3 (PEL and MCD) miRNAs |
immunosuppression, UV proinflammatory cytokines * hypoxia infection with other pathogen * TPA, NaBu, TSA, VPA |
* For more detailed description please see text Section 4. Virus reactivation. HSV—herpes simplex virus; VZV—varicella zoster virus; HCMV—human cytomegalovirus; HHV—human herpesvirus; EBV—Epstein–Barr virus; KSHV—Kaposi sarcoma-associated herpesvirus; PBMCs—peripheral blood mononuclear cells; HSCs—haematopoietic stem cells; NSCs—neural stem cells; NPCs—neural progenitor cells; NGF—nerve growth factor; NaBu—sodium butyrate; PI3K—phosphoinositide 3-kinase; TPA-12-O—tetradecanoylphorbol-13-acetate; TSA—trichostatin A; BCR—B-cell receptor; VPA—valproic acid; LATs—latency associated transcripts; ORF—open reading frame; VLT—VZV latency-associated transcript; LUNA—latency unique nuclear antigen; EBNA—Epstein–Barr nuclear antigen; LMP—latency-associated membrane proteins; LANA—latency-associated nuclear antigen; vFLIP—viral FLICE inhibitory protein; vCyc—viral cyclin; PEL—primary effusion lymphoma; MCD—multicentric Castleman’s disease.
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Weidner-Glunde, M.; Kruminis-Kaszkiel, E.; Savanagouder, M. Herpesviral Latency—Common Themes. Pathogens 2020, 9, 125. https://doi.org/10.3390/pathogens9020125
AMA Style
Weidner-Glunde M, Kruminis-Kaszkiel E, Savanagouder M. Herpesviral Latency—Common Themes. Pathogens. 2020; 9(2):125. https://doi.org/10.3390/pathogens9020125
Chicago/Turabian StyleWeidner-Glunde, Magdalena, Ewa Kruminis-Kaszkiel, and Mamata Savanagouder. 2020. "Herpesviral Latency—Common Themes" Pathogens 9, no. 2: 125. https://doi.org/10.3390/pathogens9020125
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.