RNA Virus Reverse Genetics and Vaccine Design

1
Department of Pediatrics, Emory University School of Medicine, Atlanta, GA 30322, USA
2
Children's Healthcare of Atlanta, Atlanta, GA 30322, USA
*
Author to whom correspondence should be addressed.
Viruses 2014, 6(7), 2531-2550; https://doi.org/10.3390/v6072531
Submission received: 30 April 2014 / Revised: 18 June 2014 / Accepted: 19 June 2014 / Published: 25 June 2014
(This article belongs to the Special Issue Virus-based Vaccines)
Abstract
:
RNA viruses are capable of rapid spread and severe or potentially lethal disease in both animals and humans. The development of reverse genetics systems for manipulation and study of RNA virus genomes has provided platforms for designing and optimizing viral mutants for vaccine development. Here, we review the impact of RNA virus reverse genetics systems on past and current efforts to design effective and safe viral therapeutics and vaccines.
1. Introduction
Vaccines remain one of the greatest accomplishments of human ingenuity, scientific endeavor, and the combined global efforts of the public health community. The rates of incidence and mortality associated with infection by RNA viruses such as polio, measles, mumps and rubella have declined by greater than 95% compared to pre-vaccination rates [1]. Though highly successful in the past, conventional approaches to RNA virus vaccine development, such as live-attenuation through passaging (forward genetics) or inactivation, may be less efficient for generating good candidates than rational targeted mutagenesis (reverse genetics). Advancements in recombinant DNA technology and virus reverse genetics have provided key critical insights into the replication and pathogenesis of RNA viruses and facilitate vaccine development through targeted modifications and directed attenuation. The advent of reverse genetics and molecular engineering of viruses has transformed the field of virology by permitting study of targeted genetic changes in virus genomes. In 1981, the first infectious RNA virus clone was isolated from cDNA to generate poliovirus [2]. Since then, reverse genetics technology and recombinant virus design has been employed to generate reverse genetic clones representing all major virus families. In addition, these techniques and approaches have now become the focus of new efforts to design vaccines that incorporate specific changes in either component-based or virus-based systems to induce lasting immunity in the host without health risks or deleterious effects.
Since the development of effective live-attenuated vaccines, new vaccine preparations utilizing well-established vectors, expression of specific viral proteins or components (subunit vaccines), and development of virus-like particles (VLPs), have continued to shape the domain of vaccine discovery and development. The challenge of establishing a safe, immunogenic platform, which induces lasting immunity in the context of a wide variety of viral systems, has resulted in remarkable creativity and variability in the approaches employed. Use of replicating viruses in vaccines, such as live-attenuated or chimeric vector-based platforms, have the benefits of high immunogenicity, lower costs, and ease to transport and administer. Yet, these viruses have the potential to revert to more pathogenic phenotypes and may be under-attenuated in immunocompromised hosts. Conversely, component, subunit, or killed pathogen vaccines have the benefits of generally being safer and can be used to display the most immunogenic antigens. However, the costs, time of development, and weaker induced immune responses present their own challenges to design and implementation. In this review, we describe RNA virus reverse genetics systems and provide an overview of current efforts to use reverse genetics technology in the development of safe and effective vaccines.
2. RNA Virus Biology and Reverse Genetic Infectious Clone Design
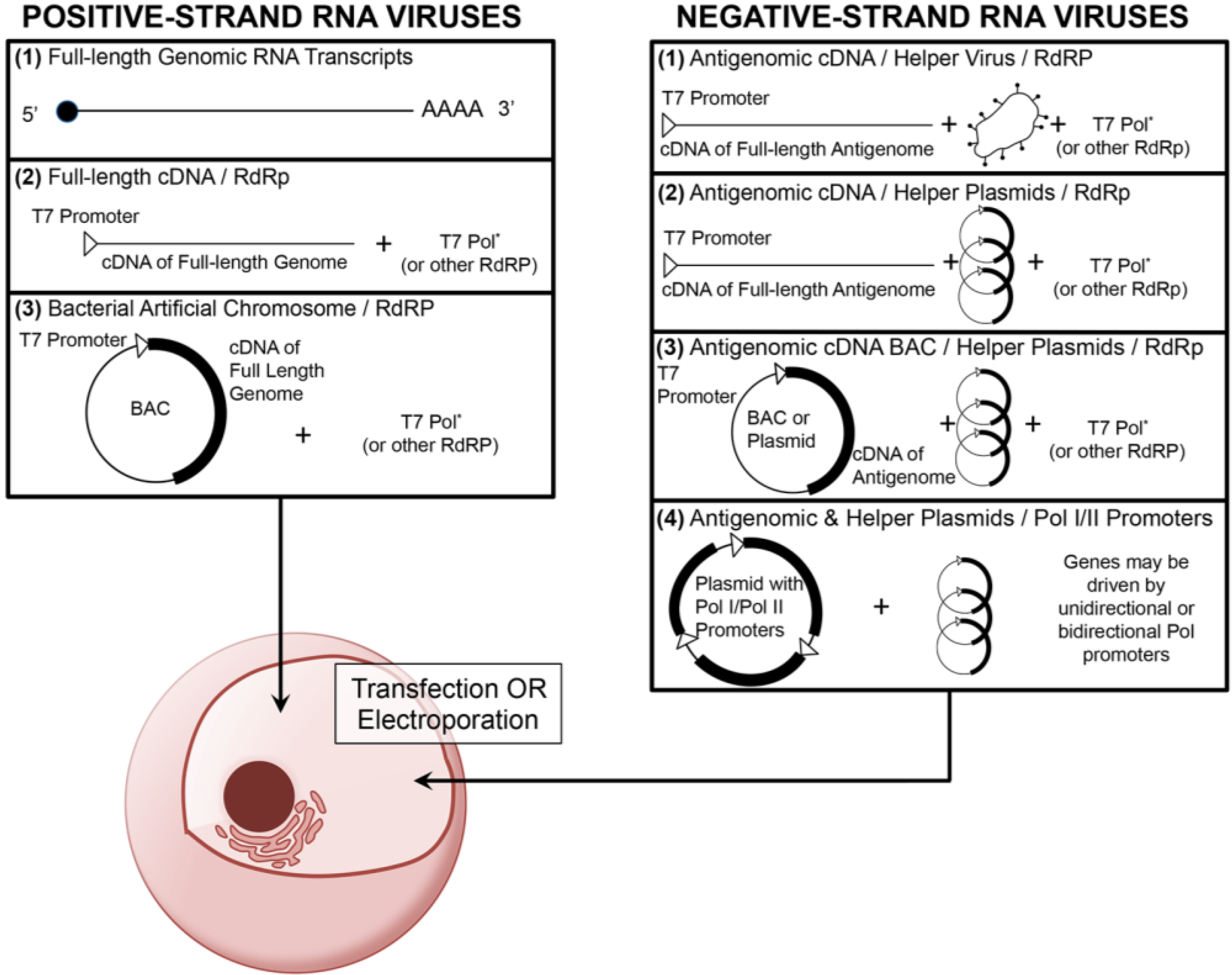
Current RNA virus reverse genetics systems make use of multiple common features of RNA virus biology. First, RNA viruses generate genomic copies through the activity of a viral RNA-dependent RNA polymerase (RdRp). In addition, nearly all RNA virus replication strategies are independent from the host cell nucleus and instead reside in the cytoplasm. For positive-strand RNA viruses, such as poliovirus, immediately after entry and uncoating, the genomic RNA is directly translated by host ribosomes to generate viral protein products. Since the virus rarely needs to package additional non‑structural proteins in the virion, most positive-sense RNA virus reverse genetics systems largely focus on delivery of either transcribed genomic RNA into the cell cytoplasm or delivery of cDNA under the control of a viral transcription promoter such as T7 or CMV (Figure 1) [3,4,5,6,7,8,9,10]. However, negative-strand and double-strand RNA virus reverse genetic systems often require the use of additional helper constructs to introduce the RdRp and other essential proteins to initiate genomic replication. A recent alternative approach, which has been employed in the field of influenza research, is to synthesize viral RNA and drive mRNA production through the activity of the host polymerases such as Pol I and Pol II [11,12]. This approach has simplified the logistics of plasmid transfection and increased the efficacy of recombinant virus recovery. A similar approach has been successfully employed for recovery of an arenavirus, lymphocytic choriomenigitis virus (LCMV) [13]. One additional platform that has been employed for RNA virus infectious clone generation is the bacterial artificial chromosome (BAC). BAC constructs are single-copy DNA plasmids based on the F-plasmid of bacteria, which are genetically stable in E. coli and permit the insertion of large DNA fragments to be transcribed under the control of a transcriptional promoter such as T7. BAC constructs have been employed for many years in the recovery of large DNA viruses. However, the genetic stability of these constructs has led to their use in recombinant RNA virus platforms. Several positive-strand RNA viruses and one negative-strand RNA virus have used BACs as a platform for reverse genetic design (Table 1). Since most RNA virus replication strategies are segregated from the host genome and replication machinery, few RNA viruses modify host gene expression and cause oncogenesis. As a result, RNA reverse genetic systems do not typically have to account for the potential transformation of host cells. Lastly, most RNA viruses are limited to small genome sizes with the majority smaller than 15 kb due to reduced genomic stability and the lowered fidelity of the viral RdRp. Subsequently, reverse genetic approaches often employ the use of cDNA genetic clones for greater versatility in manipulation and modification of the virus genome.
Figure 1. An overview of common reverse genetic platforms for the recovery of positive- and negative-strand RNA viruses. Most positive-strand RNA virus reverse genetic platforms consist of either direct introduction of full-length copies of the viral genome (which have been transcribed in vitro) or introduction of either a linear or plasmid‑associated (bacterial artificial chromosome, BAC) cDNA of the full-length genome in combination with an RdRp (such as T7 polymerase). Negative-strand RNA virus reverse genetic platforms often involve transfection or electroporation of genomic or more commonly subgenomic cDNA into permissive cells in combination with either a helper virus or helper plasmids, all of which driven by a RdRp. Some negative-strand RNA systems employ host polymerase I (Pol I) and II (Pol II) promoters to drive viral RNA synthesis and mRNA production. In both positive- and negative-strand reverse genetics systems, the RdRp (*) is typically constitutively or transiently expressed in the permissive cell type.
Figure 1. An overview of common reverse genetic platforms for the recovery of positive- and negative-strand RNA viruses. Most positive-strand RNA virus reverse genetic platforms consist of either direct introduction of full-length copies of the viral genome (which have been transcribed in vitro) or introduction of either a linear or plasmid‑associated (bacterial artificial chromosome, BAC) cDNA of the full-length genome in combination with an RdRp (such as T7 polymerase). Negative-strand RNA virus reverse genetic platforms often involve transfection or electroporation of genomic or more commonly subgenomic cDNA into permissive cells in combination with either a helper virus or helper plasmids, all of which driven by a RdRp. Some negative-strand RNA systems employ host polymerase I (Pol I) and II (Pol II) promoters to drive viral RNA synthesis and mRNA production. In both positive- and negative-strand reverse genetics systems, the RdRp (*) is typically constitutively or transiently expressed in the permissive cell type.


{kind=link}
| Virus | Family | Genome | Genome Size | Year | Reference |
|---|---|---|---|---|---|
| Transmissible gastroenteritis virus (TGEV) | Coronaviridae | +ssRNA | 28 kb | 2000 | [4] |
| Japanese encephalitis virus (JEV) | Flaviviridae | +ssRNA | 11 kb | 2003 | [14] |
| Porcine reproductive and respiratory syndrome virus (PRRSV) | Arteriviridae | +ssRNA | 15 kb | 2006 | [15] |
| Human coronavirus OC43 | Coronaviridae | +ssRNA | 31 kb | 2006 | [16] |
| Severe acute respiratory syndrome coronavirus (SARS-CoV) | Coronaviridae | +ssRNA | 30 kb | 2007 | [17] |
| Dengue virus type 1 | Flaviviridae | +ssRNA | 11 kb | 2007 | [18] |
| Bovine viral diarrheal virus (BVDV) | Flaviviridae | +ssRNA | 12 kb | 2008 | [19] |
| Border disease virus (BDV) | Flaviviridae | +ssRNA | 12 kb | 2010 | [20] |
| Classical swine fever virus (CSFV) | Flaviviridae | +ssRNA | 12 kb | 2010 | [20] |
| Respiratory syncytial virus (RSV) | Paramyxoviridae | -ssRNA | 15 kb | 2012 | [21] |
| Feline infectious peritonitis virus (FIPV) | Coronaviridae | +ssRNA | 29 kb | 2012 | [22] |
| Middle East respiratory syndrome coronavirus (MERS-CoV) | Coronaviridae | +ssRNA | 30 kb | 2013 | [3] |
| Dengue virus type 2 | Flaviviridae | +ssRNA | 11 kb | 2014 | [23] |
3. Positive-Strand RNA Virus Biology, Reverse Genetics, and Vaccine Design
Positive-strand RNA viruses have genomes which are infectious upon entry into host cells. Upon entry, host ribosomes translate the viral RNA into one or more polyproteins, which require either host or viral proteases for processing. Although many different reverse genetics platforms have been used to generate positive-strand RNA virus clones, nearly all share the common goal to introduce either sense genomic RNA transcripts directly or cDNA to be transcribed by an included RdRp (Figure 1). Below we describe approaches that have been employed to generate infectious clones of picornaviruses, coronaviruses, and flaviviruses and how these strategies have been and are currently being applied to develop vaccines.
3.3. Flaviviruses: Yellow Fever, Dengue, and West Nile Viruses
Flaviviruses are small, enveloped, positive-strand RNA viruses that infect a wide range of hosts. Flavivirus genomes are markedly smaller than coronaviruses at approximately 10–12 kb in size, and the transmission of most flaviviruses is dependent upon an arthropod vector (hence their common name, arboviruses). Flavivirus diseases range from asymptomatic to severe neurological disease such as encephalitis, meningitis, and myelitis [53]. Yellow fever virus (YFV), a deadly flavivirus associated with over 30,000 deaths annually (WHO), was identified in 1901 by Walter Reed and was the first human viral pathogen ever discovered [54,55,56]. Outbreaks and disease connected to flaviviruses have stressed the importance of developing reverse genetic platforms and efficacious vaccines.
Similar to picornaviruses, nearly all flavivirus reverse genetics platforms involve either in vitro transcription of full-length or ligated cDNA fragments of the genome or use a bacterial artificial chromosome (BAC) platform (Table 1) and are subsequently transfected or electroporated into competent cells. Reverse genetics platforms have been developed for a wide range of flaviviruses including YFV [57,58], Dengue Types 1–4 [59,60,61,62,63,64,65,66], JEV [67], Kunjin virus [68], Tick-borne encephalitis virus (TBEV) [69,70,71], Murray Valley encephalitis virus [72], Langat virus [73], West Nile virus (WNV) [6,74], and Omsk hemorrhagic fever virus [75]. Advances in the recombinant flavivirus approaches have been instrumental in new vaccine design efforts.
Some of the first vaccine efforts were directed at generating inactivated or live-attenuated strains for vaccinations against flaviviruses. In 1937, Max Theiler developed a safe YFV live-attenuated vaccine called 17D through the use of a serial passaged virus originally isolated from an African patient [76]. The vaccine composition used today remains largely the same as that first developed over 70 years ago and provides protective immunity for over 30 years [77,78,79]. JEV was first isolated and studied in the 1930’s and an inactivated vaccine derived from a mouse brain was first developed in Japan in 1954 [53,80]. The current formulations of the JEV vaccines include inactivated Beijing-1 strain or live‑attenuated strains, which elicit greater immunogenicity and broader protection than the original Nakayama strain [53]. Despite the success of vaccine development for YFV and JEV, unique challenges have been presented in developing vaccines to some well-known flaviviruses (such as dengue virus) and new emerging flaviviruses (such as WNV).
Dengue virus is endemic to tropical and subtropical locations worldwide. To date, five distinct serotypes of dengue virus are known, including a new serotype identified in 2013 [53,81]. Infection with one serotype increases the severity of disease upon a secondary infection with a different serotype [82]. Consequently, any dengue virus vaccine must either provide protection to all extent serotypes to prevent priming for increased disease by a heterotypic infection or remove the immunogenic components of the virus that cause increased disease severity upon heterotypic infections. Efforts to develop stable chimeric platforms for the development of dengue virus vaccines have recently been focused on expressing dengue virus surface proteins in chimeric viruses with other more stable flaviviruses such as YFV [83,84,85]. One vaccine candidate is a tetravalent vaccine from Sanofi Pasteur, which involves the formation of a chimeric YFV strain 17D virus containing the prM/E genes of each dengue serotype [85]. This approach has also recently been adapted to develop similar vaccine candidates to the recently identified WNV [85,86]. Despite several commercially available WNV vaccines for veterinary purposes, there remains no approved WNV vaccine for humans. The future licensure of dengue and WNV vaccines will likely continue to focus on development of live‑attenuated or inactivated virus models of vaccination due to the high immunogenicity of flavivirus infections.
4. Negative-Strand RNA Virus Biology, Reverse Genetics, and Vaccine Design
Negative-strand RNA virus genomes and antigenomes cannot act as an mRNA. To be a substrate of RdRp, they must be encapsidated with nucleocapsid and form ribonucleoprotein complexes (RNPs). The antigenomes of negative-strand RNA viruses are introduced either as a linearized cDNA or as a plasmid under a T7 promoter and are normally co-transfected with one or more plasmids encoding the nucleocapsid and replicase machinery. Due to these biological requirements to initiate infection, development of negative-strand RNA virus reverse genetics systems has been slower than positive‑strand virus systems. Initial recovery efforts to generate infectious clones involved recovery of viruses using helper viruses, which could supply the viral genes and proteins necessary for replication. However, these approaches made it difficult to isolate the mutants of interest. The first recovery of a negative‑strand RNA virus completely from cDNA was achieved in 1994 for rabies virus [87,88]. Despite the biological limitations and challenges to working with negative-strand RNA viruses, several effective negative-strand RNA virus vaccines have been successfully introduced and many more are currently in varying phases of clinical trials. Below we describe approaches that have been employed to generate infectious clones and vaccines in paramyxoviruses and orthomyxoviruses.
4.1. Paramyxoviruses: Measles Virus and Respiratory Syncytial Viruses
Paramyxoviruses are enveloped, negative-sense, single-strand RNA viruses which are responsible for a variety of human and animal diseases. Human paramyxoviruses have been identified which are responsible for diseases including measles, mumps, pneumonia, and the common cold. Paramyxoviruses carry a single copy of their genome, which is typically 15 to 19 kb in length. Like other negative-strand RNA viruses, paramyxoviruses must incorporate their replication machinery, including the RDRP, into the virion during assembly. Paramyxovirus reverse genetic systems employ very similar mechanisms. First, the full-length genome or antigenome and helper plasmids expressing nucleocapsid and polymerase proteins are cloned as cDNA and are under transcriptional control by a promoter such as T7 RNA polymerase. The plasmids are co-transfected into permissive cell lines. Earlier reverse genetics systems utilized a co-infection approach with a vaccinia virus expressing T7, however most modern reverse genetics systems use a T7 cell line or transfect a T7 plasmid. Recently, the first BAC-based reverse genetics system for a negative-strand RNA virus was developed for respiratory syncytial virus (RSV) [21]. Reverse genetics systems for many paramyxoviruses have been generated including measles virus [89], mumps virus [90], Hendra virus [91], Nipah virus [92], RSV [21,93]. Despite the availability of reverse genetics systems, development of paramyxovirus vaccines has been met with variable success.
The first paramyxovirus vaccine was developed during the 1950’s. John Enders was able to develop a cultivation system for measles virus and cultured an attenuated measles virus called the Edmonston strain (named after the child from which it was isolated). The Edmonston strain, though initially under‑attenuated, was later adapted and led to the successfully license of a measles vaccine in 1963 [94]. Maurice Hilleman was able to build on the success of the live-attenuated measles vaccine and cultured and adapted by passage in fertilized hen’s eggs, a strain of mumps called Jeryl Lynn (named after his daughter, from whom it was isolated) [95]. Hilleman was later instrumental in the development of the MMR vaccine combining live-attenuated strains of measles, mumps, rubella (Wistar RA 27/3 strain), which was first licensed for use in 1971 [96]. The early success of the measles and mumps vaccines prompted renewed efforts to develop vaccines to other pathogenic RNA viruses. One of the greatest challenges to viral vaccine design remains the development of a vaccine for respiratory syncytial virus (RSV).
Respiratory syncytial virus (RSV) was first isolated in 1955 from a chimpanzee displaying upper respiratory illness [97]. Since its identification, RSV has become recognized as the leading cause of infant mortality by a virus worldwide [98,99]. In the United States alone, RSV upper and lower respiratory infections have led to over 100,000 hospitalizations annually [98,99,100]. Despite a high clinical burden, no licensed RSV vaccines are available and current treatments are cost prohibitive while only providing passive immunity by administration of prophylactic antibodies [101,102,103]. The most susceptible population for RSV infection is young infants [98,99,100]. Consequently, the ideal RSV vaccine must be immunogenic, genetically stable, and safe for vaccination in infants. However, an early tragic failure during the 1960’s of a formalin-inactivated RSV vaccine has dampened efforts to develop and implement new vaccines [104,105]. To date, the most clinically advanced RSV vaccine candidates have been live-attenuated viruses developed through virus passage and reconstitution by reverse genetics [106,107,108]. One of the more promising of these candidates to date has been MEDI‑559, which includes many introduced mutations which render the virus temperature-sensitive and provides some level of protection [106,107]. However, these candidates have not been able to achieve the level of protection and genetic stability necessary for implementation [109]. Ongoing studies continue to evaluate new targets for attenuation; however successful development of an RSV vaccine will require finding the proper balance of attenuation and immunogenicity.
4.2. Orthomyxoviruses: Influenza virus
Orthomyxoviruses are enveloped, negative-sense RNA viruses whose genomes consist of multiple linear segments and are approximately 12 to 15 kb in size. Similar to paramyxoviruses, many orthomyxoviruses cause respiratory illnesses in humans, most notable is influenza (flu). A key hallmark of orthoymyxovirus evolution is the reassortment of the virus genomes through co-infection of a cell. In order to be transcriptionally active, influenza viruses require a functionally active viral ribonucleoprotein complex (RNP) which consists of the viral genomic RNA, nucleoprotein (NP), and the viral RdRp (comprised of PB1, PB2, and PA proteins) [110]. The first reverse genetics approaches developed for orthomyxoviruses were generated for influenza A virus (IAV), however these systems utilized helper viruses that had to be selected against to recover recombinants [111,112]. The first helper virus-free systems were developed around the turn of the millennium [113,114]. These first systems required the co-transfection of four or more plasmids under the control of a Pol II promoter as well as eight plasmids expressing the eight viral RNA segments. The number of plasmids co‑transfected for recovery varied greatly depending upon the number of viral genomic segments and the organization of the helper protein constructs, ranging from 10 (which expressed two of the helper proteins on each of two plasmids using the Pol II promoter and an IRES) to as many as 17 [113,114,115,116,117,118]. However, recent advances in the reverse genetics platforms have reduced the number of total plasmids needed to 8 or less using a bidirectional expression system [11,12]. In this system, a human Pol I promoter coupled with either a murine PolI terminator drives viral RNA synthesis and a CMV Pol II promoter is responsible for viral mRNA synthesis [11]. This improved production of IAV, however, generation of recombinant virus for vaccines is limited to a select number of mammalian cell lines such as African green monkey kidney epithelial cells (Vero) or Madin Darby canine kidney (MDCK) cells [119,120]. However, these cell lines had limited transfectability and differences in Pol I and Pol II compatibility have hindered the efficacy of these uni- and bidirectional approaches. Incorporation of species-specific polymerase promoters has provided improved efficacy of recovery in several of these cell lines [119,120]. A recent advancement has been the combination of up to 8 Pol I driven IAV genes on a single plasmid and up to 3 Pol II drive genes on an additional plasmid, this approach improves the probability of a single cell receiving all necessary plasmids during recovery [121]. The availability of applicable reverse genetics systems for studying and identifying the structure and function of influenza proteins has revolutionized influenza vaccination strategies.
For many years, the trivalent vaccine that is currently provided consisted of three separated strains (2 A strains and 1 B strain), which were selected based on the WHO recommendations prior to the next flu season. However, these vaccines were made using either live-attenuated (through cold-adaptation) or inactivated virus grown in fertilized chicken eggs. Next generation vaccination strategies include tetravalent or quadrivalent vaccines, which may be grown in animal cell cultures (rather than chicken eggs) or virus-like particles (VLPs) in cultures of S. frugiperda insect (Sf9) cells [122,123,124,125,126,127,128,129]. In 2013, the FDA approved a seasonal influenza vaccine comprised of purified HA proteins prepared using a baculovirus-expression system [130]. These recombinant vaccines were successfully used to vaccinate individuals between 18 and 49 years of age and represents a major step in influenza vaccine design and implementation because this system reduces production time compared to the conventional egg-based approach [131].
5. Synthetic Biology and the Future of RNA Virus Vaccine Design
5.1. Limitations and Challenges to the Application of Reverse Genetics to Vaccine Design
A major frontier of synthetic biology is the development of new vaccines and therapeutics and the improvement of the implementation and efficacy of those already on the market. One major complication to the successful implementation of vaccines currently is the limitations in time between vaccine design and production. Influenza viruses require seasonal vaccinations and determination of the proper formulations leaves little time for development and implementation. New advances in reverse genetics technology are reducing the potential time of recovery and production from months to weeks. For instance, Dormitzer et al. have developed an improved platform approach and demonstrated that with current reverse genetics technology, it is feasible to generate a recombinant influenza virus from new HA and NA sequences within 5 days [132]. As new emerging viruses continue to appear and pandemics continue to occur, rapid approaches to recover and adapt viruses will be instrumental in the public health response. Development of common chimeric reverse genetic platforms for rapid cloning and expression of surface antigens from emergent pathogens will help increase the efficacy and timing of delivery during epidemic and pandemic outbreaks.
One key limitation to the development of effective live-attenuated vaccines is genetic stability. RNA viruses generally exhibit high mutation rates due to decreased fidelity of the RDRP. Yet, recent studies have shown that codon-usage bias can be used to alter the translation and consequently, replication of viruses. Several groups have shown that substituting non-preferred codons based on host cell codon usage bias, a process referred to as codon-deoptimization, into the genome of poliovirus resulted in reduction of plaque areas and virus yields [133,134]. By changing codons rather than amino acids, the amount of viral protein may be modulated without impairment in its function. More importantly, the potential for reversion is greatly limited due to the sheer number of codon changes introduced into the coding sequence. This approach represents a promising new avenue to develop attenuated, but genetically stable vaccine platforms.
5.2. Current and Future Directions to Vaccine Design
Despite the presence of reverse genetics systems for many human pathogens, not all have resulted in successful vaccine platforms. Advances in biotechnology and key discoveries have led to novel reverse genetic approaches, which may be employed in new generation vaccines. Structural vaccinology or structure-based antigen design has become a common practice for optimizing antigens for display in vaccines [135]. As mentioned previously, RSV remains a key hurdle to reducing viral childhood morbidity. Since the discovery and implementation of palivizumab, currently the only licensed prophalytic inhibitory measure to RSV infection, considerable energy has been devoted to evaluating the antigenic sites of the RSV fusion (F) protein and optimizing F expression constructs for higher immunogenicity [105]. Current structural vaccinology efforts to evaluate the pre-fusion and post-fusion antigenic forms of RSV F protein have led to the induction and identification of potent neutralizing antibodies with higher neutralizing potencies than palivizumab [136,137]. The availability of new structures and sequences has made predictive structural modeling and structure-based antigen design viable options for renewed efforts at design of vaccines, for which no current vaccines exist.
Advances in biotechnology have shaped the field of virology as much as any other field of science. The capacity to engineer reverse genetics platforms for the study and manipulation of RNA virus genomes has revolutionized the field of vaccine design. We have described here RNA virus reverse genetic systems and past and current efforts to develop vaccines to provide immunity to several human RNA virus pathogens. The incorporation of new advances in reverse genetics technology, adjuvants, non-human models of infection, and surveillance will continue to drive the development of next generation vaccines to pathogens for which we already have vaccination strategies as well as those that we currently do not. Ongoing changes in human demographics and accessibility to health care are likely to modify cost-benefit analyses and provoke allocation of new resources for scientific study and development of vaccines. Despite the ongoing evolution of vaccine design and implementation, the hallmarks of an effective viral vaccine will remain the same: high efficacy, safety, and stability.
Acknowledgments
The authors thank Sujin Lee and Anne Hotard for their advice on formatting and manuscript organization. This work was supported by NIH grants R01AI087798 and U19AI095227.
Author Contributions
C.S. organized and drafted the manuscript. C.S. and M.M. reviewed and edited the manuscript. Both authors read and approved the manuscript.
Conflicts of Interest
MLM and Emory University are entitled to licensing fees derived from various agreements Emory has entered into non-exclusive licensing agreements related to some of the technologies described in this paper. The terms of this arrangement have been reviewed and approved by Emory University in accordance with its conflict of interest policies.
References and Notes
- Roush, S.W.; Murphy, T.V.; Vaccine-Preventable Disease Table Working Group. Historical comparisons of morbidity and mortality for vaccine-preventable diseases in the united states. JAMA 2007, 298, 2155–2163. [Google Scholar] [CrossRef]
- Racaniello, V.R.; Baltimore, D. Cloned poliovirus complementary DNA is infectious in mammalian cells. Science 1981, 214, 916–919. [Google Scholar]
- Almazan, F.; DeDiego, M.L.; Sola, I.; Zuniga, S.; Nieto-Torres, J.L.; Marquez-Jurado, S.; Andres, G.; Enjuanes, L. Engineering a replication-competent, propagation-defective middle east respiratory syndrome coronavirus as a vaccine candidate. mBio 2013, 4, e00650–e00613. [Google Scholar]
- Almazan, F.; Gonzalez, J.M.; Penzes, Z.; Izeta, A.; Calvo, E.; Plana-Duran, J.; Enjuanes, L. Engineering the largest rna virus genome as an infectious bacterial artificial chromosome. Proc. Natl. Acad. Sci. USA 2000, 97, 5516–5521. [Google Scholar]
- Scobey, T.; Yount, B.L.; Sims, A.C.; Donaldson, E.F.; Agnihothram, S.S.; Menachery, V.D.; Graham, R.L.; Swanstrom, J.; Bove, P.F.; Kim, J.D.; et al. Reverse genetics with a full-length infectious cdna of the middle east respiratory syndrome coronavirus. Proc. Natl. Acad. Sci. USA 2013, 110, 16157–16162. [Google Scholar] [CrossRef]
- Shi, P.Y.; Tilgner, M.; Lo, M.K.; Kent, K.A.; Bernard, K.A. Infectious cdna clone of the epidemic west nile virus from new york city. J. Virol. 2002, 76, 5847–5856. [Google Scholar] [CrossRef]
- Thiel, V.; Herold, J.; Schelle, B.; Siddell, S.G. Infectious rna transcribed in vitro from a cdna copy of the human coronavirus genome cloned in vaccinia virus. J. Gen. Virol. 2001, 82, 1273–1281. [Google Scholar]
- Yount, B.; Curtis, K.M.; Baric, R.S. Strategy for systematic assembly of large rna and DNA genomes: Transmissible gastroenteritis virus model. J. Virol. 2000, 74, 10600–10611. [Google Scholar] [CrossRef]
- Yount, B.; Curtis, K.M.; Fritz, E.A.; Hensley, L.E.; Jahrling, P.B.; Prentice, E.; Denison, M.R.; Geisbert, T.W.; Baric, R.S. Reverse genetics with a full-length infectious cdna of severe acute respiratory syndrome coronavirus. Proc. Natl. Acad. Sci. USA 2003, 100, 12995–13000. [Google Scholar] [CrossRef]
- Yount, B.; Denison, M.R.; Weiss, S.R.; Baric, R.S. Systematic assembly of a full-length infectious cdna of mouse hepatitis virus strain a59. J. Virol. 2002, 76, 11065–11078. [Google Scholar] [CrossRef]
- Hoffmann, E.; Neumann, G.; Hobom, G.; Webster, R.G.; Kawaoka, Y. "Ambisense" approach for the generation of influenza a virus: Vrna and mrna synthesis from one template. Virology 2000, 267, 310–317. [Google Scholar] [CrossRef]
- Hoffmann, E.; Webster, R.G. Unidirectional rna polymerase i-polymerase ii transcription system for the generation of influenza a virus from eight plasmids. J. Gen. Virol. 2000, 81, 2843–2847. [Google Scholar]
- Flatz, L.; Bergthaler, A.; de la Torre, J.C.; Pinschewer, D.D. Recovery of an arenavirus entirely from rna polymerase i/ii-driven cdna. Proc. Natl. Acad. Sci. USA 2006, 103, 4663–4668. [Google Scholar] [CrossRef]
- Yun, S.I.; Kim, S.Y.; Rice, C.M.; Lee, Y.M. Development and application of a reverse genetics system for japanese encephalitis virus. J. Virol. 2003, 77, 6450–6465. [Google Scholar] [CrossRef]
- Choi, Y.J.; Yun, S.I.; Kang, S.Y.; Lee, Y.M. Identification of 5' and 3' cis-acting elements of the porcine reproductive and respiratory syndrome virus: Acquisition of novel 5' au-rich sequences restored replication of a 5'-proximal 7-nucleotide deletion mutant. J. Virol. 2006, 80, 723–736. [Google Scholar] [CrossRef]
- St-Jean, J.R.; Desforges, M.; Almazan, F.; Jacomy, H.; Enjuanes, L.; Talbot, P.J. Recovery of a neurovirulent human coronavirus oc43 from an infectious cdna clone. J. Virol. 2006, 80, 3670–3674. [Google Scholar] [CrossRef]
- DeDiego, M.L.; Alvarez, E.; Almazan, F.; Rejas, M.T.; Lamirande, E.; Roberts, A.; Shieh, W.J.; Zaki, S.R.; Subbarao, K.; Enjuanes, L. A severe acute respiratory syndrome coronavirus that lacks the e gene is attenuated in vitro and in vivo. J. Virol. 2007, 81, 1701–1713. [Google Scholar] [CrossRef]
- Suzuki, R.; de Borba, L.; Duarte dos Santos, C.N.; Mason, P.W. Construction of an infectious cdna clone for a brazilian prototype strain of dengue virus type 1: Characterization of a temperature-sensitive mutation in ns1. Virology 2007, 362, 374–383. [Google Scholar] [CrossRef]
- Fan, Z.C.; Bird, R.C. An improved reverse genetics system for generation of bovine viral diarrhea virus as a bac cdna. J. Virol. Methods. 2008, 149, 309–315. [Google Scholar]
- Rasmussen, T.B.; Reimann, I.; Uttenthal, A.; Leifer, I.; Depner, K.; Schirrmeier, H.; Beer, M. Generation of recombinant pestiviruses using a full-genome amplification strategy. Vet. Microbiol. 2010, 142, 13–17. [Google Scholar] [CrossRef]
- Hotard, A.L.; Shaikh, F.Y.; Lee, S.; Yan, D.; Teng, M.N.; Plemper, R.K.; Crowe, J.E., Jr.; Moore, M.L. A stabilized respiratory syncytial virus reverse genetics system amenable to recombination-mediated mutagenesis. Virology 2012, 434, 129–136. [Google Scholar]
- Balint, A.; Farsang, A.; Zadori, Z.; Hornyak, A.; Dencso, L.; Almazan, F.; Enjuanes, L.; Belak, S. Molecular characterization of feline infectious peritonitis virus strain df-2 and studies of the role of orf3abc in viral cell tropism. J. Virol. 2012, 86, 6258–6267. [Google Scholar] [CrossRef]
- Usme-Ciro, J.A.; Lopera, J.A.; Enjuanes, L.; Almazan, F.; Gallego-Gomez, J.C. Development of a novel DNA-launched dengue virus type 2 infectious clone assembled in a bacterial artificial chromosome. Virus Res. 2014, 180, 12–22. [Google Scholar] [CrossRef]
- Kandolf, R.; Hofschneider, P.H. Molecular cloning of the genome of a cardiotropic coxsackie b3 virus: Full-length reverse-transcribed recombinant cdna generates infectious virus in mammalian cells. Proc. Natl. Acad. Sci. USA 1985, 82, 4818–4822. [Google Scholar] [CrossRef]
- Mizutani, S.; Colonno, R.J. In vitro synthesis of an infectious rna from cdna clones of human rhinovirus type 14. J. Virol. 1985, 56, 628–632. [Google Scholar]
- Cohen, J.I.; Ticehurst, J.R.; Feinstone, S.M.; Rosenblum, B.; Purcell, R.H. Hepatitis a virus cdna and its rna transcripts are infectious in cell culture. J. Virol. 1987, 61, 3035–3039. [Google Scholar]
- Roos, R.P.; Stein, S.; Routbort, M.; Senkowski, A.; Bodwell, T.; Wollmann, R. Theiler's murine encephalomyelitis virus neutralization escape mutants have a change in disease phenotype. J. Virol. 1989, 63, 4469–4473. [Google Scholar]
- Zibert, A.; Maass, G.; Strebel, K.; Falk, M.M.; Beck, E. Infectious foot-and-mouth disease virus derived from a cloned full-length cdna. J. Virol. 1990, 64, 2467–2473. [Google Scholar]
- Inoue, T.; Yamaguchi, S.; Saeki, T.; Sekiguchi, K. Production of infectious swine vesicular disease virus from cloned cdna in mammalian cells. J. Gen. Virol. 1990, 71, 1835–1838. [Google Scholar] [CrossRef]
- Blackburn, R.V.; Racaniello, V.R.; Righthand, V.F. Construction of an infectious cdna clone of echovirus 6. Virus Res. 1992, 22, 71–78. [Google Scholar] [CrossRef]
- Zimmermann, H.; Eggers, H.J.; Zimmermann, A.; Kraus, W.; Nelsen-Salz, B. Complete nucleotide sequence and biological properties of an infectious clone of prototype echovirus 9. Virus Res. 1995, 39, 311–319. [Google Scholar] [CrossRef]
- Palmenberg, A.C.; Spiro, D.; Kuzmickas, R.; Wang, S.; Djikeng, A.; Rathe, J.A.; Fraser-Liggett, C.M.; Liggett, S.B. Sequencing and analyses of all known human rhinovirus genomes reveal structure and evolution. Science 2009, 324, 55–59. [Google Scholar] [CrossRef]
- Waman, V.P.; Kolekar, P.S.; Kale, M.M.; Kulkarni-Kale, U. Population structure and evolution of rhinoviruses. PLoS One 2014, 9, e88981. [Google Scholar]
- Glanville, N.; McLean, G.R.; Guy, B.; Lecouturier, V.; Berry, C.; Girerd, Y.; Gregoire, C.; Walton, R.P.; Pearson, R.M.; Kebadze, T.; et al. Cross-serotype immunity induced by immunization with a conserved rhinovirus capsid protein. PLoS Pathog. 2013, 9, e1003669. [Google Scholar] [CrossRef]
- Rossmann, M.G.; Arnold, E.; Erickson, J.W.; Frankenberger, E.A.; Griffith, J.P.; Hecht, H.J.; Johnson, J.E.; Kamer, G.; Luo, M.; Mosser, A.G.; et al. Structure of a human common cold virus and functional relationship to other picornaviruses. Nature 1985, 317, 145–153. [Google Scholar] [CrossRef]
- Edlmayr, J.; Niespodziana, K.; Popow-Kraupp, T.; Krzyzanek, V.; Focke-Tejkl, M.; Blaas, D.; Grote, M.; Valenta, R. Antibodies induced with recombinant vp1 from human rhinovirus exhibit cross-neutralisation. Euro. Respir. J. 2011, 37, 44–52. [Google Scholar] [CrossRef]
- Bartlett, N.W.; Walton, R.P.; Edwards, M.R.; Aniscenko, J.; Caramori, G.; Zhu, J.; Glanville, N.; Choy, K.J.; Jourdan, P.; Burnet, J.; et al. Mouse models of rhinovirus-induced disease and exacerbation of allergic airway inflammation. Nat. Med. 2008, 14, 199–204. [Google Scholar] [CrossRef]
- Dobrikova, E.Y.; Goetz, C.; Walters, R.W.; Lawson, S.K.; Peggins, J.O.; Muszynski, K.; Ruppel, S.; Poole, K.; Giardina, S.L.; Vela, E.M.; et al. Attenuation of neurovirulence, biodistribution, and shedding of a poliovirus:Rhinovirus chimera after intrathalamic inoculation in macaca fascicularis. J. Virol. 2012, 86, 2750–2759. [Google Scholar] [CrossRef]
- Jahan, N.; Wimmer, E.; Mueller, S. Polypyrimidine tract binding protein-1 (ptb1) is a determinant of the tissue and host tropism of a human rhinovirus/poliovirus chimera pv1(ripo). PLoS One 2013, 8, e60791. [Google Scholar] [CrossRef]
- Perlman, S.; Netland, J. Coronaviruses post-sars: Update on replication and pathogenesis. Nat. Rev. Microbiol. 2009, 7, 439–450. [Google Scholar] [CrossRef]
- Zaki, A.M.; van Boheemen, S.; Bestebroer, T.M.; Osterhaus, A.D.; Fouchier, R.A. Isolation of a novel coronavirus from a man with pneumonia in saudi arabia. New Engl. J. Med. 2012, 367, 1814–1820. [Google Scholar] [CrossRef]
- Donaldson, E.F.; Yount, B.; Sims, A.C.; Burkett, S.; Pickles, R.J.; Baric, R.S. Systematic assembly of a full-length infectious clone of human coronavirus nl63. J. Virol. 2008, 82, 11948–11957. [Google Scholar] [CrossRef]
- Becker, M.M.; Graham, R.L.; Donaldson, E.F.; Rockx, B.; Sims, A.C.; Sheahan, T.; Pickles, R.J.; Corti, D.; Johnston, R.E.; Baric, R.S.; et al. Synthetic recombinant bat sars-like coronavirus is infectious in cultured cells and in mice. Proc. Natl. Acad. Sci. USA 2008, 105, 19944–19949. [Google Scholar] [CrossRef]
- Eckerle, L.D.; Lu, X.; Sperry, S.M.; Choi, L.; Denison, M.R. High fidelity of murine hepatitis virus replication is decreased in nsp14 exoribonuclease mutants. J. Virol. 2007, 81, 12135–12144. [Google Scholar] [CrossRef]
- Smith, E.C.; Blanc, H.; Vignuzzi, M.; Denison, M.R. Coronaviruses lacking exoribonuclease activity are susceptible to lethal mutagenesis: Evidence for proofreading and potential therapeutics. PLoS Pathog. 2013, 9, e1003565. [Google Scholar] [CrossRef]
- Denison, M.R.; Graham, R.L.; Donaldson, E.F.; Eckerle, L.D.; Baric, R.S. Coronaviruses: An rna proofreading machine regulates replication fidelity and diversity. RNA Biol. 2011, 8, 270–279. [Google Scholar] [CrossRef]
- Eckerle, L.D.; Becker, M.M.; Halpin, R.A.; Li, K.; Venter, E.; Lu, X.; Scherbakova, S.; Graham, R.L.; Baric, R.S.; Stockwell, T.B.; et al. Infidelity of sars-cov nsp14-exonuclease mutant virus replication is revealed by complete genome sequencing. PLoS Pathog. 2010, 6, e1000896. [Google Scholar] [CrossRef]
- Graham, R.L.; Becker, M.M.; Eckerle, L.D.; Bolles, M.; Denison, M.R.; Baric, R.S. A live, impaired-fidelity coronavirus vaccine protects in an aged, immunocompromised mouse model of lethal disease. Nat. Med. 2012, 18, 1820–1826. [Google Scholar] [CrossRef]
- Hotez, P.J.; Bottazzi, M.E.; Tseng, C.T.; Zhan, B.; Lustigman, S.; Du, L.; Jiang, S. Calling for rapid development of a safe and effective mers vaccine. Microbes Infect. 2014. [Google Scholar] [CrossRef]
- Ying, T.; Du, L.; Ju, T.W.; Prabakaran, P.; Lau, C.C.; Lu, L.; Liu, Q.; Wang, L.; Feng, Y.; Wang, Y.; et al. Exceptionally potent neutralization of mers-cov by human monoclonal antibodies. J. Virol. 2014.
- Tang, X.C.; Agnihothram, S.S.; Jiao, Y.; Stanhope, J.; Graham, R.L.; Peterson, E.C.; Avnir, Y.; Tallarico, A.S.; Sheehan, J.; Zhu, Q.; et al. Identification of human neutralizing antibodies against mers-cov and their role in virus adaptive evolution. Proc. Natl. Acad. Sci. USA 2014, 111, E2018–E2026. [Google Scholar] [CrossRef]
- Ma, C.; Li, Y.; Wang, L.; Zhao, G.; Tao, X.; Tseng, C.T.; Zhou, Y.; Du, L.; Jiang, S. Intranasal vaccination with recombinant receptor-binding domain of mers-cov spike protein induces much stronger local mucosal immune responses than subcutaneous immunization: Implication for designing novel mucosal mers vaccines. Vaccine 2014, 32, 2100–2108. [Google Scholar] [CrossRef]
- Ishikawa, T.; Yamanaka, A.; Konishi, E. A review of successful flavivirus vaccines and the problems with those flaviviruses for which vaccines are not yet available. Vaccine 2014, 32, 1326–1337. [Google Scholar] [CrossRef]
- Reed, W.; Carroll, J. The prevention of yellow fever. Public Health Pap. Rep. 1901, 27, 113–129. [Google Scholar]
- Reed, W.; Carroll, J.; Agramonte, A. The etiology of yellow fever: An additional note. 1901. Mil. Med. 2001, 166, 44–53. [Google Scholar]
- Reed, W. Recent researches concerning the etiology, propagation, and prevention of yellow fever, by the united states army commission. J. Hyg. 1902, 2, 101–119. [Google Scholar] [CrossRef]
- Rice, C.M.; Grakoui, A.; Galler, R.; Chambers, T.J. Transcription of infectious yellow fever rna from full-length cdna templates produced by in vitro ligation. The New biologist 1989, 1, 285–296. [Google Scholar]
- Bredenbeek, P.J.; Kooi, E.A.; Lindenbach, B.; Huijkman, N.; Rice, C.M.; Spaan, W.J. A stable full-length yellow fever virus cdna clone and the role of conserved rna elements in flavivirus replication. J. Gen. Virol. 2003, 84, 1261–1268. [Google Scholar] [CrossRef]
- Kapoor, M.; Zhang, L.; Mohan, P.M.; Padmanabhan, R. Synthesis and characterization of an infectious dengue virus type-2 rna genome (new guinea c strain). Gene 1995, 162, 175–180. [Google Scholar] [CrossRef]
- Kinney, R.M.; Butrapet, S.; Chang, G.J.; Tsuchiya, K.R.; Roehrig, J.T.; Bhamarapravati, N.; Gubler, D.J. Construction of infectious cdna clones for dengue 2 virus: Strain 16681 and its attenuated vaccine derivative, strain pdk-53. Virology 1997, 230, 300–308. [Google Scholar] [CrossRef]
- Polo, S.; Ketner, G.; Levis, R.; Falgout, B. Infectious rna transcripts from full-length dengue virus type 2 cdna clones made in yeast. J. Virol. 1997, 71, 5366–5374. [Google Scholar]
- Puri, B.; Polo, S.; Hayes, C.G.; Falgout, B. Construction of a full length infectious clone for dengue-1 virus western pacific,74 strain. Virus Genes 2000, 20, 57–63. [Google Scholar] [CrossRef]
- Blaney, J.E., Jr.; Hanson, C.T.; Firestone, C.Y.; Hanley, K.A.; Murphy, B.R.; Whitehead, S.S. Genetically modified, live attenuated dengue virus type 3 vaccine candidates. Am. J. Trop. Med. Hyg. 2004, 71, 811–821. [Google Scholar]
- Blaney, J.E., Jr.; Hanson, C.T.; Hanley, K.A.; Murphy, B.R.; Whitehead, S.S. Vaccine candidates derived from a novel infectious cdna clone of an american genotype dengue virus type 2. BMC Infect. Dis. 2004, 4, 39. [Google Scholar] [CrossRef]
- Chen, W.; Kawano, H.; Men, R.; Clark, D.; Lai, C.J. Construction of intertypic chimeric dengue viruses exhibiting type 3 antigenicity and neurovirulence for mice. J. Virol. 1995, 69, 5186–5190. [Google Scholar]
- Lai, C.J.; Zhao, B.T.; Hori, H.; Bray, M. Infectious rna transcribed from stably cloned full-length cdna of dengue type 4 virus. Proc. Natl. Acad. Sci. USA 1991, 88, 5139–5143. [Google Scholar] [CrossRef]
- Sumiyoshi, H.; Hoke, C.H.; Trent, D.W. Infectious japanese encephalitis virus rna can be synthesized from in vitro-ligated cdna templates. J. Virol. 1992, 66, 5425–5431. [Google Scholar]
- Khromykh, A.A.; Westaway, E.G. Completion of kunjin virus rna sequence and recovery of an infectious rna transcribed from stably cloned full-length cdna. J. Virol. 1994, 68, 4580–4588. [Google Scholar]
- Gritsun, T.S.; Gould, E.A. Infectious transcripts of tick-borne encephalitis virus, generated in days by rt-pcr. Virology 1995, 214, 611–618. [Google Scholar] [CrossRef]
- Gritsun, T.S.; Gould, E.A. Development and analysis of a tick-borne encephalitis virus infectious clone using a novel and rapid strategy. J. Virol. Methods. 1998, 76, 109–120. [Google Scholar] [CrossRef]
- Mandl, C.W.; Ecker, M.; Holzmann, H.; Kunz, C.; Heinz, F.X. Infectious cdna clones of tick-borne encephalitis virus european subtype prototypic strain neudoerfl and high virulence strain hypr. J. Gen. Virol. 1997, 78, 1049–1057. [Google Scholar]
- Hurrelbrink, R.J.; Nestorowicz, A.; McMinn, P.C. Characterization of infectious murray valley encephalitis virus derived from a stably cloned genome-length cdna. J. Gen. Virol. 1999, 80, 3115–3125. [Google Scholar]
- Campbell, M.S.; Pletnev, A.G. Infectious cdna clones of langat tick-borne flavivirus that differ from their parent in peripheral neurovirulence. Virology 2000, 269, 225–237. [Google Scholar] [CrossRef]
- Yamshchikov, V.F.; Wengler, G.; Perelygin, A.A.; Brinton, M.A.; Compans, R.W. An infectious clone of the west nile flavivirus. Virology 2001, 281, 294–304. [Google Scholar] [CrossRef]
- Yoshii, K.; Igarashi, M.; Ito, K.; Kariwa, H.; Holbrook, M.R.; Takashima, I. Construction of an infectious cdna clone for omsk hemorrhagic fever virus, and characterization of mutations in ns2a and ns5. Virus Res. 2011, 155, 61–68. [Google Scholar] [CrossRef]
- Theiler, M.; Smith, H.H. The use of yellow fever virus modified by in vitro cultivation for human immunization. J. Exp. Med. 1937, 65, 787–800. [Google Scholar] [CrossRef]
- Bonaldo, M.C.; Sequeira, P.C.; Galler, R. The yellow fever 17d virus as a platform for new live attenuated vaccines. Hum. Vaccin. Immunother. 2014, 10, 1256–1265. [Google Scholar]
- Lefeuvre, A.; Marianneau, P.; Deubel, V. Current assessment of yellow fever and yellow fever vaccine. Curr. Infect. Dis. Rep. 2004, 6, 96–104. [Google Scholar] [CrossRef]
- Poland, J.D.; Calisher, C.H.; Monath, T.P.; Downs, W.G.; Murphy, K. Persistence of neutralizing antibody 30–35 years after immunization with 17d yellow fever vaccine. Bull. World Health Organ. 1981, 59, 895–900. [Google Scholar]
- Webster, L.T. Japanese b encephalitis virus: Its differentiation from st. Louis encephalitis virus and relationship to louping-ill virus. Science 1937, 86, 402–403. [Google Scholar]
- Normile, D. Tropical medicine. Surprising new dengue virus throws a spanner in disease control efforts. Science 2013, 342, 415. [Google Scholar] [CrossRef]
- Halstead, S.B. Immune enhancement of viral infection. Prog. Allergy 1982, 31, 301–364. [Google Scholar]
- Guirakhoo, F.; Arroyo, J.; Pugachev, K.V.; Miller, C.; Zhang, Z.X.; Weltzin, R.; Georgakopoulos, K.; Catalan, J.; Ocran, S.; Soike, K.; et al. Construction, safety, and immunogenicity in nonhuman primates of a chimeric yellow fever-dengue virus tetravalent vaccine. J. Virol. 2001, 75, 7290–7304. [Google Scholar] [CrossRef]
- Caufour, P.S.; Motta, M.C.; Yamamura, A.M.; Vazquez, S.; Ferreira, II; Jabor, A.V.; Bonaldo, M.C.; Freire, M.S.; Galler, R. Construction, characterization and immunogenicity of recombinant yellow fever 17d-dengue type 2 viruses. Virus Res. 2001, 79, 1–14. [Google Scholar] [CrossRef]
- Guy, B.; Guirakhoo, F.; Barban, V.; Higgs, S.; Monath, T.P.; Lang, J. Preclinical and clinical development of yfv 17d-based chimeric vaccines against dengue, west nile and japanese encephalitis viruses. Vaccine 2010, 28, 632–649. [Google Scholar] [CrossRef]
- Arroyo, J.; Miller, C.; Catalan, J.; Myers, G.A.; Ratterree, M.S.; Trent, D.W.; Monath, T.P. Chimerivax-west nile virus live-attenuated vaccine: Preclinical evaluation of safety, immunogenicity, and efficacy. J. Virol. 2004, 78, 12497–12507. [Google Scholar] [CrossRef]
- Schnell, M.J.; Mebatsion, T.; Conzelmann, K.K. Infectious rabies viruses from cloned cdna. EMBO J. 1994, 13, 4195–4203. [Google Scholar]
- Conzelmann, K.K.; Schnell, M. Rescue of synthetic genomic rna analogs of rabies virus by plasmid-encoded proteins. J. Virol. 1994, 68, 713–719. [Google Scholar]
- Radecke, F.; Spielhofer, P.; Schneider, H.; Kaelin, K.; Huber, M.; Dotsch, C.; Christiansen, G.; Billeter, M.A. Rescue of measles viruses from cloned DNA. EMBO J. 1995, 14, 5773–5784. [Google Scholar]
- Clarke, D.K.; Sidhu, M.S.; Johnson, J.E.; Udem, S.A. Rescue of mumps virus from cdna. J. Virol. 2000, 74, 4831–4838. [Google Scholar]
- Marsh, G.A.; Virtue, E.R.; Smith, I.; Todd, S.; Arkinstall, R.; Frazer, L.; Monaghan, P.; Smith, G.A.; Broder, C.C.; Middleton, D.; et al. Recombinant hendra viruses expressing a reporter gene retain pathogenicity in ferrets. Virol. J. 2013, 10, 95. [Google Scholar] [CrossRef]
- Yoneda, M.; Guillaume, V.; Ikeda, F.; Sakuma, Y.; Sato, H.; Wild, T.F.; Kai, C. Establishment of a nipah virus rescue system. Proc. Natl. Acad. Sci. USA 2006, 103, 16508–16513. [Google Scholar] [CrossRef]
- Collins, P.L.; Murphy, B.R. New generation live vaccines against human respiratory syncytial virus designed by reverse genetics. Proc. Am. Thorac. Soc. 2005, 2, 166–173. [Google Scholar] [CrossRef]
- Katz, S.L. John F. Enders and measles virus vaccine—A reminiscence. Curr. Top. Microbiol. Immunol. 2009, 329, 3–11. [Google Scholar]
- Buynak, E.B.; Hilleman, M.R. Live attenuated mumps virus vaccine. 1. Vaccine development. Exp. Biol. Med. 1966, 123, 768–775. [Google Scholar] [CrossRef]
- Stokes, J., Jr.; Weibel, R.E.; Villarejos, V.M.; Arguedas, J.A.; Buynak, E.B.; Hilleman, M.R. Trivalent combined measles-mumps-rubella vaccine. Findings in clinical-laboratory studies. JAMA 1971, 218, 57–61. [Google Scholar] [CrossRef]
- Blount, R.E., Jr.; Morris, J.A.; Savage, R.E. Recovery of cytopathogenic agent from chimpanzees with coryza. Exp. Biol. Med. 1956, 92, 544–549. [Google Scholar] [CrossRef]
- Hall, C.B.; Weinberg, G.A.; Iwane, M.K.; Blumkin, A.K.; Edwards, K.M.; Staat, M.A.; Auinger, P.; Griffin, M.R.; Poehling, K.A.; Erdman, D.; et al. The burden of respiratory syncytial virus infection in young children. New Engl. J. Med. 2009, 360, 588–598. [Google Scholar]
- Nair, H.; Nokes, D.J.; Gessner, B.D.; Dherani, M.; Madhi, S.A.; Singleton, R.J.; O'Brien, K.L.; Roca, A.; Wright, P.F.; Bruce, N.; et al. Global burden of acute lower respiratory infections due to respiratory syncytial virus in young children: A systematic review and meta-analysis. Lancet 2010, 375, 1545–1555. [Google Scholar]
- Glezen, W.P.; Taber, L.H.; Frank, A.L.; Kasel, J.A. Risk of primary infection and reinfection with respiratory syncytial virus. Am. J. Dis. Child. 1986, 140, 543–546. [Google Scholar]
- Subramanian, K.N.; Weisman, L.E.; Rhodes, T.; Ariagno, R.; Sanchez, P.J.; Steichen, J.; Givner, L.B.; Jennings, T.L.; Top, F.H., Jr.; Carlin, D.; et al. Safety, tolerance and pharmacokinetics of a humanized monoclonal antibody to respiratory syncytial virus in premature infants and infants with bronchopulmonary dysplasia. Medi-493 study group. J. Pediatr. Infect. Dis. 1998, 17, 110–115. [Google Scholar] [CrossRef]
- Abarca, K.; Jung, E.; Fernandez, P.; Zhao, L.; Harris, B.; Connor, E.M.; Losonsky, G.A.; Motavizumab Study, G. Safety, tolerability, pharmacokinetics, and immunogenicity of motavizumab, a humanized, enhanced-potency monoclonal antibody for the prevention of respiratory syncytial virus infection in at-risk children. J. Pediatr. Infect. Dis. 2009, 28, 267–272. [Google Scholar]
- Kamal-Bahl, S.; Doshi, J.; Campbell, J. Economic analyses of respiratory syncytial virus immunoprophylaxis in high-risk infants: A systematic review. Arch. Pediatr. Adolesc. Med. 2002, 156, 1034–1041. [Google Scholar] [CrossRef]
- Kim, H.W.; Leikin, S.L.; Arrobio, J.; Brandt, C.D.; Chanock, R.M.; Parrott, R.H. Cell-mediated immunity to respiratory syncytial virus induced by inactivated vaccine or by infection. Pediatr. Res. 1976, 10, 75–78. [Google Scholar] [CrossRef]
- Collins, P.L.; Melero, J.A. Progress in understanding and controlling respiratory syncytial virus: Still crazy after all these years. Virus Res. 2011, 162, 80–99. [Google Scholar] [CrossRef]
- Karron, R.A.; Wright, P.F.; Belshe, R.B.; Thumar, B.; Casey, R.; Newman, F.; Polack, F.P.; Randolph, V.B.; Deatly, A.; Hackell, J.; et al. Identification of a recombinant live attenuated respiratory syncytial virus vaccine candidate that is highly attenuated in infants. J. Infect. Dis. 2005, 191, 1093–1104. [Google Scholar] [CrossRef]
- Schickli, J.H.; Kaur, J.; Tang, R.S. Nonclinical phenotypic and genotypic analyses of a phase 1 pediatric respiratory syncytial virus vaccine candidate medi-559 (ra2cp248/404/1030deltash) at permissive and non-permissive temperatures. Virus Res. 2012, 169, 38–47. [Google Scholar] [CrossRef]
- Luongo, C.; Winter, C.C.; Collins, P.L.; Buchholz, U.J. Increased genetic and phenotypic stability of a promising live-attenuated respiratory syncytial virus vaccine candidate by reverse genetics. J. Virol. 2012, 86, 10792–10804. [Google Scholar] [CrossRef]
- Graham, B.S. Biological challenges and technological opportunities for respiratory syncytial virus vaccine development. Immunol. Rev. 2011, 239, 149–166. [Google Scholar] [CrossRef]
- Engelhardt, O.G. Many ways to make an influenza virus—Review of influenza virus reverse genetics methods. Influenza Other Respir. Viruses 2013, 7, 249–256. [Google Scholar] [CrossRef]
- Enami, M.; Luytjes, W.; Krystal, M.; Palese, P. Introduction of site-specific mutations into the genome of influenza virus. Proc. Natl. Acad. Sci. USA 1990, 87, 3802–3805. [Google Scholar] [CrossRef]
- Luytjes, W.; Krystal, M.; Enami, M.; Parvin, J.D.; Palese, P. Amplification, expression, and packaging of foreign gene by influenza virus. Cell 1989, 59, 1107–1113. [Google Scholar] [CrossRef]
- Fodor, E.; Devenish, L.; Engelhardt, O.G.; Palese, P.; Brownlee, G.G.; Garcia-Sastre, A. Rescue of influenza a virus from recombinant DNA. J. Virol. 1999, 73, 9679–9682. [Google Scholar]
- Neumann, G.; Kawaoka, Y. Generation of influenza a virus from cloned cdnas--historical perspective and outlook for the new millenium. Rev. Med. Virol. 2002, 12, 13–30. [Google Scholar] [CrossRef]
- Koudstaal, W.; Hartgroves, L.; Havenga, M.; Legastelois, I.; Ophorst, C.; Sieuwerts, M.; Zuijdgeest, D.; Vogels, R.; Custers, J.; de Boer-Luijtze, E.; et al. Suitability of per.C6 cells to generate epidemic and pandemic influenza vaccine strains by reverse genetics. Vaccine 2009, 27, 2588–2593. [Google Scholar] [CrossRef]
- de Wit, E.; Spronken, M.I.; Vervaet, G.; Rimmelzwaan, G.F.; Osterhaus, A.D.; Fouchier, R.A. A reverse-genetics system for influenza a virus using t7 rna polymerase. J. Gen. Virol. 2007, 88, 1281–1287. [Google Scholar] [CrossRef]
- Crescenzo-Chaigne, B.; van der Werf, S. Rescue of influenza c virus from recombinant DNA. J. Virol. 2007, 81, 11282–11289. [Google Scholar] [CrossRef]
- Muraki, Y.; Murata, T.; Takashita, E.; Matsuzaki, Y.; Sugawara, K.; Hongo, S. A mutation on influenza c virus m1 protein affects virion morphology by altering the membrane affinity of the protein. J. Virol. 2007, 81, 8766–8773. [Google Scholar] [CrossRef]
- Massin, P.; Rodrigues, P.; Marasescu, M.; van der Werf, S.; Naffakh, N. Cloning of the chicken rna polymerase i promoter and use for reverse genetics of influenza a viruses in avian cells. J. Virol. 2005, 79, 13811–13816. [Google Scholar] [CrossRef]
- Murakami, S.; Horimoto, T.; Yamada, S.; Kakugawa, S.; Goto, H.; Kawaoka, Y. Establishment of canine rna polymerase i-driven reverse genetics for influenza a virus: Its application for h5n1 vaccine production. J. Virol. 2008, 82, 1605–1609. [Google Scholar] [CrossRef]
- Neumann, G.; Fujii, K.; Kino, Y.; Kawaoka, Y. An improved reverse genetics system for influenza a virus generation and its implications for vaccine production. Proc. Natl. Acad. Sci. USA 2005, 102, 16825–16829. [Google Scholar] [CrossRef]
- Vinnemeier, C.D.; Fischer-Herr, J.; Meyer, S.; Liebig, K.; Theess, W.; Burchard, G.D.; Cramer, J.P. Immunogenicity and safety of an inactivated 2012/2013 trivalent influenza vaccine produced in mammalian cell culture (optaflu): An open label, uncontrolled study. Hum. Vaccin. Immunother. 2013, 10, 441–448. [Google Scholar]
- Doroshenko, A.; Halperin, S.A. Trivalent mdck cell culture-derived influenza vaccine optaflu (novartis vaccines). Expert Rev. Vaccin. 2009, 8, 679–688. [Google Scholar] [CrossRef]
- Bright, R.A.; Carter, D.M.; Daniluk, S.; Toapanta, F.R.; Ahmad, A.; Gavrilov, V.; Massare, M.; Pushko, P.; Mytle, N.; Rowe, T.; et al. Influenza virus-like particles elicit broader immune responses than whole virion inactivated influenza virus or recombinant hemagglutinin. Vaccine 2007, 25, 3871–3878. [Google Scholar] [CrossRef]
- Pushko, P.; Tumpey, T.M.; Van Hoeven, N.; Belser, J.A.; Robinson, R.; Nathan, M.; Smith, G.; Wright, D.C.; Bright, R.A. Evaluation of influenza virus-like particles and novasome adjuvant as candidate vaccine for avian influenza. Vaccine 2007, 25, 4283–4290. [Google Scholar] [CrossRef]
- Pushko, P.; Tumpey, T.M.; Bu, F.; Knell, J.; Robinson, R.; Smith, G. Influenza virus-like particles comprised of the ha, na, and m1 proteins of h9n2 influenza virus induce protective immune responses in balb/c mice. Vaccine 2005, 23, 5751–5759. [Google Scholar] [CrossRef]
- Galarza, J.M.; Latham, T.; Cupo, A. Virus-like particle vaccine conferred complete protection against a lethal influenza virus challenge. Viral Immunol. 2005, 18, 365–372. [Google Scholar] [CrossRef]
- Quan, F.S.; Huang, C.; Compans, R.W.; Kang, S.M. Virus-like particle vaccine induces protective immunity against homologous and heterologous strains of influenza virus. J. Virol. 2007, 81, 3514–3524. [Google Scholar] [CrossRef]
- Treanor, J.J.; Schiff, G.M.; Hayden, F.G.; Brady, R.C.; Hay, C.M.; Meyer, A.L.; Holden-Wiltse, J.; Liang, H.; Gilbert, A.; Cox, M. Safety and immunogenicity of a baculovirus-expressed hemagglutinin influenza vaccine: A randomized controlled trial. JAMA 2007, 297, 1577–1582. [Google Scholar] [CrossRef]
- Traynor, K. First recombinant flu vaccine approved. AJHP 2013, 70, 382. [Google Scholar]
- Yang, L.P. Recombinant trivalent influenza vaccine (flublok(®)): A review of its use in the prevention of seasonal influenza in adults. Drugs 2013, 73, 1357–1366. [Google Scholar] [CrossRef]
- Dormitzer, P.R.; Suphaphiphat, P.; Gibson, D.G.; Wentworth, D.E.; Stockwell, T.B.; Algire, M.A.; Alperovich, N.; Barro, M.; Brown, D.M.; Craig, S.; et al. Synthetic generation of influenza vaccine viruses for rapid response to pandemics. Sci. Transl. Med. 2013, 5, 185ra168. [Google Scholar]
- Burns, C.C.; Shaw, J.; Campagnoli, R.; Jorba, J.; Vincent, A.; Quay, J.; Kew, O. Modulation of poliovirus replicative fitness in hela cells by deoptimization of synonymous codon usage in the capsid region. J. Virol. 2006, 80, 3259–3272. [Google Scholar] [CrossRef]
- Mueller, S.; Papamichail, D.; Coleman, J.R.; Skiena, S.; Wimmer, E. Reduction of the rate of poliovirus protein synthesis through large-scale codon deoptimization causes attenuation of viral virulence by lowering specific infectivity. J. Virol. 2006, 80, 9687–9696. [Google Scholar] [CrossRef]
- Dormitzer, P.R.; Grandi, G.; Rappuoli, R. Structural vaccinology starts to deliver. Nat. Rev. Microbiol. 2012, 10, 807–813. [Google Scholar] [CrossRef]
- Correia, B.E.; Bates, J.T.; Loomis, R.J.; Baneyx, G.; Carrico, C.; Jardine, J.G.; Rupert, P.; Correnti, C.; Kalyuzhniy, O.; Vittal, V.; et al. Proof of principle for epitope-focused vaccine design. Nature 2014, 507, 201–206. [Google Scholar] [CrossRef]
- McLellan, J.S.; Chen, M.; Joyce, M.G.; Sastry, M.; Stewart-Jones, G.B.; Yang, Y.; Zhang, B.; Chen, L.; Srivatsan, S.; Zheng, A.; et al. Structure-based design of a fusion glycoprotein vaccine for respiratory syncytial virus. Science 2013, 342, 592–598. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
MDPI and ACS Style
Stobart, C.C.; Moore, M.L. RNA Virus Reverse Genetics and Vaccine Design. Viruses 2014, 6, 2531-2550. https://doi.org/10.3390/v6072531
AMA Style
Stobart CC, Moore ML. RNA Virus Reverse Genetics and Vaccine Design. Viruses. 2014; 6(7):2531-2550. https://doi.org/10.3390/v6072531
Chicago/Turabian StyleStobart, Christopher C., and Martin L. Moore. 2014. "RNA Virus Reverse Genetics and Vaccine Design" Viruses 6, no. 7: 2531-2550. https://doi.org/10.3390/v6072531