Background
Pelizaeus-Merzbacher disease (PMD) [1] is a congenital hypomyelination disorder caused by changes affecting the proteolipid protein 1 gene (PLP1) located on Xq22.2. Generally, patients with PLP1 missense mutations show the most severe form of PMD (connatal form); however, two-thirds of patients with PMD carry PLP1 duplications and present typical manifestations of the disorder, recognized as the classical form. Although Pelizaeus-Merzbacher disease and X-linked spastic paraplegia type 2 are nosologically distinguished, they are at opposite ends of a clinical spectrum of X-linked diseases caused by mutations of the same gene, the proteolipid protein 1 (PLP1) gene, and result in defective central nervous system (CNS) myelination (see the image below). (See Etiology.) This disease has a progressive and almost unvarying course, which is the clinical key to differentiating it from other entities such as infantile cerebral palsy, peripheral neuropathies or multiple sclerosis, etc. It is necessary to suspect this diagnosis and confirm alterations in the PLP1 gene with the aim of obtaining a real incidence of this entity, which is probably underestimated, like other leukodystrophies.
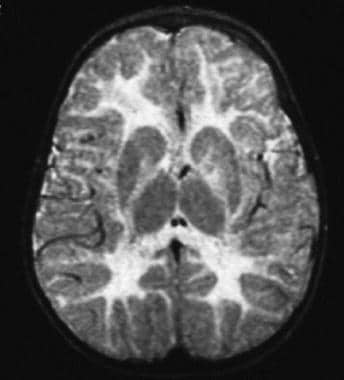 T2-weighted magnetic resonance imaging (MRI) scan of a child aged 10 months with duplication of the proteolipid protein (PLP) gene; note the high-intensity signal throughout the cerebral white matter.
T2-weighted magnetic resonance imaging (MRI) scan of a child aged 10 months with duplication of the proteolipid protein (PLP) gene; note the high-intensity signal throughout the cerebral white matter.
Clinical signs usually include some combination of nystagmus, stridor, spastic quadriparesis, hypotonia, cognitive impairment, ataxia, tremor, and diffuse leukoencephalopathy on magnetic resonance imaging (MRI). Seizures and perinatal stridor are rare signs and are typically seen only in the most severe cases. (See Presentation and Workup.)
Severe clinical syndromes (sometimes referred to as the connatal forms of Pelizaeus-Merzbacher disease) are typically caused by missense and other small mutations that affect critical positions in PLP1, whereas the milder spastic paraplegia syndrome is caused by mutations that presumably affect less critical regions of the protein. The most common mutations that cause Pelizaeus-Merzbacher disease are duplications of a region of the X chromosome that includes the entire PLP1 gene. (See Etiology.)
Severe Pelizaeus-Merzbacher disease is often fatal during the first decade of life, typically due to respiratory complications. (See Prognosis and Treatment.)
Etiology
In most cases, Pelizaeus-Merzbacher disease is caused by mutations of PLP1 on the long arm of the X chromosome (Xq22). Of note, the gene was previously termed PLP but is now designated as PLP1. PLP1 encodes 2 major products, PLP1 and a smaller protein, DM20, that results from alternative splicing. These proteins constitute about 50% of the mass of CNS white matter and are believed to serve an important structural function in compact myelin. [2, 3, 4, 5, 6]
Gene duplications
Approximately 60-70% of cases of Pelizaeus-Merzbacher disease result from duplications of the region of the X chromosome that contains PLP1 (caused, it has been proposed, by defective deoxyribonucleic acid [DNA] replication). The extent and breakpoints of duplications vary among different families. Inclusion of other genes in the duplicated region, or inclusion of aberrations of genes at the duplication endpoints, may potentially affect the phenotype.
Most individuals with PLP1 duplications present with classic Pelizaeus-Merzbacher disease, typified by nystagmus that begins in the first year of life, delayed motor and cognitive milestones, and ataxia. Most of these patients acquire some language function, which can be quite good (although slow).
Some patients with Pelizaeus-Merzbacher disease have been found to have 3 or more copies of the PLP1 gene. [7] These individuals have a more severe phenotype than most individuals with duplications.
Transgenic mice with extra copies of PLP1 develop a syndrome that effectively models the PLP1 duplication form of Pelizaeus-Merzbacher disease; this provides strong experimental support for the hypothesis that overexpression of PLP1 is deleterious to oligodendrocytes. [8, 9]
Point mutations
Approximately 15-20% of mutations in Pelizaeus-Merzbacher disease are point mutations or other small mutations that result in base substitutions, insertions, or deletions. Base substitutions usually result in missense mutations, but nonsense mutations (ie, substitution of an amino acid codon with a stop codon) and splicing mutations also occur. Splicing mutations are now recognized as quite common and may account for almost 20% of point mutations in the PLP1 gene.
The most severe form of Pelizaeus-Merzbacher disease, the so-called connatal form, usually results from missense substitutions. These severe mutations are believed to result in misfolding of the newly synthesized protein, which then accumulates in the endoplasmic reticulum and triggers apoptosis, or programmed cell death. Thus, oligodendrocyte numbers are severely reduced, and little (if any) myelin is made.
Mutations that prevent any PLP1 from being made result in a syndrome (PLP1 null syndrome) that is usually milder than classic Pelizaeus-Merzbacher disease. However, these mutations appear to cause a demyelinating peripheral neuropathy, although they do not result in oligodendrocyte cell death. Interestingly, mice that have been genetically engineered to prevent PLP1 expression develop a similar pathologic syndrome, characterized by severe, late-onset axonal degeneration.
Mutations that result in spastic paraplegia type 2 are generally missense mutations that do not prevent the processing of DM20, although they may interfere with the processing of PLP itself. These mutations do not appear to cause oligodendrocyte cell death.
Heterozygosity
Because females are mosaic with respect to X chromosome gene expression due to X inactivation, heterozygous females begin life with roughly equal proportions of oligodendrocytes that use the normal or mutated X chromosome. Females who are heterozygous for severe mutations are neurologically normal as adults, probably because the defective oligodendrocytes die, as described above, and are replaced by healthy ones. These females may have transient neurologic abnormalities during childhood. [10]
Females who are heterozygous for mutations that do not result in oligodendrocyte apoptosis (programmed cell death) continue to have oligodendrocytes that use the defective PLP1 and, therefore, are more likely to have detectable neurologic signs of Pelizaeus-Merzbacher disease.
Epidemiology
The frequency of Pelizaeus-Merzbacher disease in the United States is not known with certainty, but the estimated prevalence is at least 1 case per 500,000 population. However, this is a conservative estimate. Internationally, the frequency of the condition is estimated to be 1 case per 100,000-1,000,000 population.
Demographics
Pelizaeus-Merzbacher disease and spastic paraplegia type 2 are global syndromes and affect all major ethnic groups.
So far, no case reports of patients of African descent have been published; however, the author is aware of African Americans with Pelizaeus-Merzbacher disease. The disease has been reported in people of Asian, Middle Eastern, and European descent.
Pelizaeus-Merzbacher disease typically affects males, but female heterozygotes can be clinically affected, especially those who carry alleles that are relatively mild in males.
Pelizaeus-Merzbacher disease typically begins during infancy, but milder syndromes may not be recognized until early childhood. Although most heterozygous (ie, carrier) females are asymptomatic, young girls in families with severe to classic Pelizaeus-Merzbacher disease have reportedly developed classic Pelizaeus-Merzbacher disease that regresses as the child matures, followed by completely normal neurologic health.
Females who are heterozygous for the less severe alleles of PLP1 that are not believed to cause oligodendrocyte cell death or apoptosis may develop a more progressive and nonremitting syndrome, which usually begins during adulthood.
Prognosis
Individuals with connatal Pelizaeus-Merzbacher disease typically die of respiratory complications during childhood, but with attentive care, they can live into the third decade of life. Patients with classic Pelizaeus-Merzbacher disease (such as that caused by PLP1 gene duplications) can live into the fifth or sixth decade of life.
Patients with a predominantly spastic paraplegia phenotype have a normal life span and may even reproduce.
Morbidity
Each form of Pelizaeus-Merzbacher disease may have real or apparent intervals of stability, but the overall the trend is gradual progression. As discussed below, heterozygous females who carry a severe mutation are usually healthy, but those who carry a relatively mild mutation may develop neurologic signs, including spastic paraparesis and dementia, that typically manifest during adulthood.
Respiratory difficulty and stridor can be severe enough in infants with connatal disease to require the use of a tracheostomy or other airway protection. As the child grows older, the need for such measures may lessen.
Orthopedic complications are common in Pelizaeus-Merzbacher disease (PMD). Joint contractures are common in the legs and, to a lesser extent, the arms. Scoliosis can be severe enough to cause restrictive lung disease. Regular physical medicine evaluations, bracing, and physical therapy, as well as other treatments for spasticity, may reduce or delay the need for surgical therapy.
Dysphagia in Pelizaeus-Merzbacher disease can be severe enough to necessitate consideration of feeding tube placement.
Clinical course in heterozygous females
In heterozygous females with alleles that are severe in males, the defective oligodendrocytes die and are replaced by healthy oligodendrocytes, and neurologic function is maintained or improves with maturation. Females who are heterozygous for less severe alleles of PLP1 that are not believed to cause oligodendrocyte cell death or apoptosis may develop a more progressive and nonremitting syndrome, which usually begins during adulthood. [10]
Some females with Pelizaeus-Merzbacher disease (such as the original Pelizaeus-Merzbacher disease family) probably have a clinical course much like that of affected males, in which the symptoms do not remit and may be the result of skewed X inactivation (ie, most oligodendrocytes have inactivated the normal X chromosome, and insufficient healthy oligodendrocytes are available to effectively myelinate the CNS).
Patient Education
Families with confirmed Pelizaeus-Merzbacher disease must be referred to a geneticist or neurogeneticist for education about the disease and, especially, for genetic counseling. Pelizaeus-Merzbacher disease support online is available at http://www.pmdfoundation.org and http://groups.yahoo.com/group/PMDfamilysupport/join.
Information about Pelizaeus-Merzbacher disease is also available from the National Institutes of Health (NIH) at http://www.ninds.nih.gov/disorders/pelizaeus_merzbacher/pelizaeus_merzbacher.htm.
-
T2-weighted magnetic resonance imaging (MRI) scan of a child aged 10 months with duplication of the proteolipid protein (PLP) gene; note the high-intensity signal throughout the cerebral white matter.
-
T2-weighted magnetic resonance imaging (MRI) scan of a man aged 41 years with duplication of the proteolipid protein (PLP) gene; note the increased white matter signal, as well as diffuse atrophy.
-
T2-weighted magnetic resonance imaging (MRI) scan of a man aged 20 years with connatal Pelizaeus-Merzbacher disease due to a Pro14Leu mutation; note the severe reduction in white matter volume, as well as the increased white matter signal.
-
T2-weighted magnetic resonance imaging (MRI) scan of a boy aged 17 years with null mutation of the proteolipid protein (PLP) gene; note the more subtle increase in signal intensity relative to that seen in the previous images, and observe that the volume of white matter is normal.









