
Abstract
The kidney is the main organ that senses changes in systemic oxygen tension, but it is also the key detoxification, transit and excretion site of transition metals (TMs). Pivotal to oxygen sensing are prolyl-hydroxylases (PHDs), which hydroxylate specific residues in hypoxia-inducible factors (HIFs), key transcription factors that orchestrate responses to hypoxia, such as induction of erythropoietin (EPO). The essential TM ion Fe is a key component and regulator of the hypoxia–PHD–HIF–EPO (HPHE) signaling axis, which governs erythropoiesis, angiogenesis, anaerobic metabolism, adaptation, survival and proliferation, and hence cell and body homeostasis. However, inadequate concentrations of essential TMs or entry of non-essential TMs in organisms cause toxicity and disrupt health. Non-essential TMs are toxic because they enter cells and displace essential TMs by ionic and molecular mimicry, e. g. in metalloproteins. Here, we review the molecular mechanisms of HPHE interactions with TMs (Fe, Co, Ni, Cd, Cr, and Pt) as well as their implications in renal physiology, pathophysiology and toxicology. Some TMs, such as Fe and Co, may activate renal HPHE signaling, which may be beneficial under some circumstances, for example, by mitigating renal injuries from other causes, but may also promote pathologies, such as renal cancer development and metastasis. Yet some other TMs appear to disrupt renal HPHE signaling, contributing to the complex picture of TM (nephro-)toxicity. Strikingly, despite a wealth of literature on the topic, current knowledge lacks a deeper molecular understanding of TM interaction with HPHE signaling, in particular in the kidney. This precludes rationale preventive and therapeutic approaches to TM nephrotoxicity, although recently activators of HPHE signaling have become available for therapy.
Similar content being viewed by others

Introduction
Interactions between the environment and organisms are crucial for life. Careless and widespread use of pesticides, plastics, chemicals, and environmental pollutants in general, is one of the most serious problems affecting human health in the twenty-first century (Klaassen 2019). Metals and metal compounds disrupt the function of various organs, such as the central nervous system (CNS), the hematopoietic system, the liver, and the kidneys (Ufelle and Barchowsky 2019).The kidneys are complex organs that are vital for the maintenance of normal body homeostasis. A kidney contains over 1 million functional units called nephrons, each composed of a glomerulus and a tubule. Ultrafiltration of the blood occurs at the glomerulus, forming a primary filtrate that is free of cells and large proteins and enters the tubular lumen. Renal tubules are highly specialized in their various segments, producing the final urine through reabsorption and secretion of filtered solutes and water. Filtration, reabsorption, and secretion processes maintain homeostatic levels of water, minerals, electrolytes, and hydrogen ions as well as eliminating metabolic waste products and xenobiotics from the body (Yu et al. 2019). Moreover, the kidneys are important endocrine organs. They secrete humoral factors that regulate blood pressure (renin), blood calcium concentration (calcitriol) and red blood cell production (erythropoietin; EPO) (reviewed in Kurt and Kurtz 2015).
Renal handling of transition metals
The kidney is also a target organ of metal toxicity for its capability to filter, reabsorb, excrete and accumulate metal ions. Although, it has been assumed for a long time that the kidney plays no part in metal homeostasis, it is now established that the kidney is involved in transport of iron and other metal ions (Smith and Thévenod 2009; Thévenod and Wolff 2016; van Swelm et al. 2020). Several metal transport proteins have been identified in the kidney, including the multi-ligand receptor complex megalin:cubilin:amnionless (Christensen and Birn 2002) or the divalent metal transporter-1 (DMT-1; gene SLC11A2) that transports ferrous iron and a broad range of other divalent transition metals (TMs), such as cadmium, zinc, manganese, cobalt and nickel (Gunshin et al. 1997; Illing et al. 2012). Some TM ions are used as cofactors in enzymatic reactions, as they can readily transit between oxidized and reduced states, e.g. for nucleic acid and protein synthesis, enzymatic reactions, membrane stabilization, immune system, antioxidant defense, oxidative phosphorylation, etc. (Fraga 2005; Zoroddu et al. 2019), making them essential for cellular homeostasis.
One of the best studied examples of essential TM function is the role of iron in erythropoiesis in red bone marrow (Ganz 2013). Iron is required in sufficient amounts in erythroblasts where it is used for hemoglobin synthesis to maintain O2 transporting capacity and to prevent anemia. Iron is transported in the blood by transferrin (TF) and is then released to erythroblasts by interaction of di-ferric TF with the TF receptor (TFR). The TF-TFR endosomal cycle is indispensable for erythropoiesis, as erythroblasts do not have an alternative route for iron import. Additionally, iron affects erythropoiesis by contributing to the regulation of EPO production in the kidney through coordinated function of the iron-responsive element-binding protein 1 (IRP1) and hypoxia-inducible factors (HIFs). IRP1 binds to the iron-responsive element (IRE) present in several genes (Hentze et al. 2010).
In general, essential TMs are effective at very low concentrations, demanding tight regulation. Both deficiency and excess may cause severe illness or death (Bleackley and Macgillivray 2011; Crichton 2017). Transport and urinary excretion of essential metal ions by the kidney, together with the gastrointestinal absorption rates, contribute to keeping their plasma concentration low to prevent their accumulation in tissues and cells, which may result in organ damage and dysfunction, e.g. by causing cell death, inflammation and cancer. Non-essential metal ions, such as cadmium, lead and mercury serve no known purpose in the human body, but similar to essential metal ions, they are transported and excreted by the kidney and use the same transport pathways, hence they may accumulate and induce nephrotoxicity (Bridges and Zalups 2017; Satarug et al. 2020). When TMs, essential or non-essential, enter the body, the kidney is affected by uptake of the toxic metals into renal cells, which in turn leads to concomitant decrease of essential metal entry due to competition between the metal ions. The severity of renal damage depends on the metal concentration and duration of exposure. The sequels of acute intoxication will differ from those induced by chronic intoxication. Certain metals are known to generate free radicals either by their own redox activity or by interfering with reactive oxygen species (ROS) scavenging mechanisms, which then may lead to oxidative stress and cause cellular damage, resulting finally in cell death (Valko et al. 2005). Moreover, some metals are known to have carcinogenic effects. Several signaling proteins or cellular regulatory proteins that participate in apoptosis, cell cycle regulation, DNA repair, DNA methylation, cell growth, and differentiation are targets of metals (Chen et al. 2019a; Tokar et al. 2011). In recent years, mounting evidence from individual reports has revealed that TM exposure increases the risk of anemia (e.g. Ashley-Martin et al. 2021; Bayhan et al. 2017; Choi et al. 2017; Lopez-Rodriguez et al. 2017; Shen et al. 2019; Yadav et al. 2020).
The kidney as a sensor of hypoxia
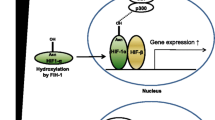
Hypoxia is a condition in which a cell or an organ has insufficient oxygen (O2) supply. Stimulation of red blood cell production is one of the classical physiological responses to systemic hypoxia during anemia. In the kidneys, branches of renal arteries and veins run in parallel over long distances in close contact with each other. This special architecture allows O2 to diffuse from the arterial system into the venous system before reaching the capillary bed (“arteriovenous O2 shunting”) and leads to relatively low O2 tensions in kidney tissues, despite high blood flow to the kidney (20% of cardiac output) (reviewed in Eckardt et al. 2005; Evans et al. 2008). O2 tension in the renal cortex is around 30–50 mmHg and does not rise above 10–25 mmHg in the renal medulla (Nangaku and Eckardt 2007) in comparison with blood (up to 100 mmHg) or well-oxygenated tissues like the intestine (up to 71 mmHg) (Carreau et al. 2011). Due to their high metabolic activity, renal tubules display high O2 consumption. Consequently, low O2 supply and high O2 demand make the kidneys particularly sensitive to changes in O2 delivery. When the kidneys sense hypoxia, peritubular fibroblasts-like interstitial cells (termed renal EPO-producing cells, REPCs) start to produce EPO. However, in rodents hypoxia-responsive Epo expression is limited to a small number of these fibroblasts in the cortico-medullary junction, that is the deep renal cortex (predominantly juxtamedullary region) and outer medulla (Bachmann et al. 1993; Eckardt et al. 1993; Koury et al. 1989; Maxwell et al. 1993; Paliege et al. 2010) (see Table 1 for a synopsis of EPO expression in renal structures). Although basal Epo expression was also seen in rodent tubular cells, these cells show almost no response to hypoxia (Nagai et al. 2014).The increased expression of rat Epo in the context of anemia involves progressive recruitment of additional REPCs situated more superficially in the kidney cortex (Eckardt et al. 1993). The origin of these REPCs is still under debate. REPCs have a unique morphology as they have dendrite-like processes and express the PDGF receptor beta (PDGFB) and ecto-5′-nucleotidase (CD73) (Asada et al. 2011; Bachmann et al. 1993). A number of studies, using genetic cell fate technologies, indicate that REPCs may originate from a distinct forkhead box protein D1 (Foxd1)-expressing subpopulation of migrating neural crest cells (Asada et al. 2011; Kobayashi et al. 2016; Souma et al. 2016; Yamazaki et al. 2013). The cell type-specific EPO gene expression may involve a GATA factor-binding motif (GATA box) that has been identified in the core promotor region of the EPO gene and acts as a negative regulatory element (Imagawa et al. 2002). This GATA-based repression seems to contribute to the switch of EPO production from liver to kidney during development (Dame et al. 2004), and may prevent EPO expression in epithelial cells, including nephron epithelia, despite hypoxic conditions (Kaneko et al. 2017; Obara et al. 2008). In other cell types or organs, EPO expression may be permanently silenced by epigenetic mechanisms, however, the exact mechanisms of organ and cell type-specific EPO production remain to be elucidated.
In renal disease, the hypoxic induction of EPO fails and anemia becomes more severe as the disease progresses without concomitant rise in EPO production (Erslev 1991). The main determinant of EPO production is the transcriptional activity of its gene, which is driven by O2 tension. Key mediators of this cellular adaptation to hypoxia are hypoxia-inducible factors (HIFs) (see “HIFs/PHDs: isoforms, regulation, tissue specific expression, mechanisms in the kidney”). Beside their role in EPO regulation, HIFs have different effects in the kidney. Chronic HIF activation may impair differentiation of renal progenitor cells, promote, or restrict cyst growth, and protect renal tubules in acute or chronic kidney injury (see “Impact of toxic metal ions on the renal HPHE signaling axis” and “Therapy of TM nephrotoxicity, controversies, outlook, and conclusions”). Furthermore, HIFs are involved in renal inflammation and fibrosis (for review see Schodel and Ratcliffe 2019). Whether TMs directly interfere with the renal hypoxia–PHD–HIF–EPO (HPHE) signaling axis remains unclear. Hence, in this review, we address the roles of O2 sensing and the HPHE signaling axis in the context of TM nephrotoxicity.
HIFs/PHDs: isoforms, regulation, tissue-specific expression, mechanisms in the kidney
Cellular responses to hypoxia involve increased glycolysis to compensate for energy loss due to reduced oxidative phosphorylation, and at the systemic level, promotion of erythrocytosis and angiogenesis to achieve efficient O2 utilization. Key to this adaptation mechanism is the rapid accumulation of HIFs. HIF transcription factors are heterodimers of an O2-regulated alpha-subunit (HIF-alpha) and a constitutively expressed beta-subunit (HIF1B, also known as aryl hydrocarbon receptor nuclear translocator, ARNT) (Wang and Semenza 1995). In humans, the HIF-alpha subunits consist of three isoforms, HIF1A, HIF2A (also known as Endothelial PAS Domain-Containing Protein 1; gene EPAS1), and HIF3A (Ema et al. 1997; Gu et al. 1998), and when complexed with HIF1B they are named HIF1, HIF2 and HIF3. All HIF subunits are members of the basic helix-loop-helix PER-ARNT-SIM (PAS) protein family (Brahimi-Horn et al. 2005). Whereas HIF1A and HIF2A are well studied, little is known regarding the biological functions of HIF3A. HIF3A shows complex cellular expression patterns with multiple splicing forms, several of these isoforms lack the transactivation domain found in the C-termini of HIF1A and HIF2A, suggesting a different role as a negative regulator of hypoxia-inducible gene expression (Duan 2016; Hara et al. 2001). Nevertheless, some HIF3 target genes have been identified and it was shown that HIF3A contributes to various diseases, such as idiopathic pulmonary fibrosis (Aquino-Galvez et al. 2019; Zhang et al. 2014). The HIF-alpha subunits are continuously transcribed and translated into protein and are maintained at low levels by O2-dependent hydroxylation on specific proline residues. Hydroxylated-alpha subunits are recognized by the von Hippel-Lindau (VHL) protein of the E3 ubiquitin ligase complex and are then rapidly degraded via the polyubiquitination/proteasomal pathway (Maxwell et al. 1999). Under normoxia, HIF-alpha proteins have a half-life of less than 5 min in human cell lines (Huang et al. 1996). As intracellular O2 concentration decreases, non-hydroxylated HIF-alpha accumulates and forms the functional transcription factor complexes HIF1 and HIF2 in the nucleus by heterodimerization with the HIF1B subunit.
Regulation of HIF-alpha levels by O 2 -sensing PHDs
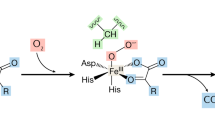
HIF1A/2A stability and abundance are regulated by prolyl-4-hydroxylases, known as prolyl-hydroxylase domain proteins (PHDs) (also known as Egl-9 Family Hypoxia Inducible Factors 1-3; genes EGLN1-3) that function as oxygen sensors. HIF-alpha contain a C- and an N-terminal O2-dependant degradation (ODD) domain and PHDs hydroxylate two specific proline residues (HIF1A: Pro402 and Pro564, HIF2A: Pro405 and Pro531) in the ODD domain of human HIF-alpha (Kaelin 2005). In rodents and humans three PHD isoforms have been identified, PHD1/EGLN2, PHD2/EGLN1 and PHD3/EGLN3 (Epstein et al. 2001; Ivan et al. 2001; Jaakkola et al. 2001). All three enzymes use oxygen and 2-oxoglutarate (2-OG) as co-substrates and ferrous iron (Fe2+) and ascorbate (AA) as cofactors. Catalytic Fe2+ is bound in a bi-dentate manner by a 2-histidine-1-aspartate triad of amino residues. In the catalytic cycle, oxidation of the prolyl residue in HIF-alpha is coupled to the oxidative decarboxylation of 2-OG in a redox cycle that involves the creation of a ferryl-oxo (FeIV = O) intermediate at the catalytic center. Binding of HIF weakens complexation of a water molecule to the iron, thereby opening a coordination site for oxygen binding. The reactive FeIV = O intermediate oxidizes HIF via a direct insertion into a C-H bond. Product dissociation completes the catalytic cycle (Loenarz and Schofield 2011). Replacing the 2-OG in PHDs by dimethyloxalylglycine (DMOG), which is intracellularly converted to N-oxalylglycine (a structural analog of 2-OG), results in normoxic accumulation of HIFs in human cells (Epstein et al. 2001). AA is required by PHDs for their full catalytic capacity, as it reduces the catalytic iron center following the oxidation that occurs during uncoupled catalytic cycles (Flashman et al. 2010). In vitro studies show that the binding affinity of PHD enzymes for Fe2+ and 2-OG is unusually strong compared to other 2-OG oxygenases, which may reflect the pivotal role of HIF hydroxylases in hypoxic signaling to ensure that in the presence of sufficient concentrations of Fe2+ and 2-OG the enzyme is maintained in a form ‘primed’ for catalysis (McNeill et al. 2005).
PHDs differ in their expression patterns, tissue distribution, subcellular localization, and their ability to hydroxylate HIF-alpha (see Table 1 for an overview of PHD expression in the kidney). Egln2 mRNA is highly expressed in murine testis, moderately in liver, and in low quantities in the heart, brain, and kidney (Lieb et al. 2002). PHD1 is a constitutively expressed protein with a nuclear localization sequence (Metzen et al. 2003; Steinhoff et al. 2009; Yasumoto et al. 2009), yet heterologously expressed human PHD1 shows no response to hypoxia (Metzen et al. 2003). Egln1 mRNA is highly expressed in the murine heart and testis, and moderately in the brain, liver, and kidney (Lieb et al. 2002). Human PHD2 protein is primarily localized in the cytosol but shuttles between the nucleus and cytoplasm (Steinhoff et al. 2009). The nuclear localization of PHDs indicates that they hydroxylate HIF-alpha proteins also in the nucleus (reviewed in Depping et al. 2015). PHD2 has the lowest O2 affinity among the PHDs and is the most active and most important O2 sensor (Berra et al. 2003). Human PHD3 is present in both the cytoplasmic and nuclear compartment (Metzen et al. 2003), and its mRNA is expressed highly in the heart and liver, and moderately in the brain and kidney of mice (Lieb et al. 2002). Although mRNA of all three Egln isoforms are expressed in the rodent kidney, Egln1 is most abundant in tubular cells, whereas Egln2 and Egln3 are predominantly expressed in interstitial fibroblasts of the kidney (Schodel et al. 2009). All human PHD proteins appear to be abundant in tubular segments of the inner medulla, where O2 tension is low (Soilleux et al. 2005). In human cells lines, all three PHDs can contribute to the regulation of both HIF1A and HIF2A, although PHD1 and PHD3 are more active on HIF2A than on HIF1A, whereas PHD2 hydroxylates HIF1A more efficiently (Appelhoff et al. 2004). Consistent with this study, PHD2 deficiency in mouse liver and kidney leads to the accumulation of nuclear HIF1A but not HIF2A (Takeda et al. 2007). In contrast, PHD1/PHD3 double deficiency in mice led to hepatic accumulation of HIF2A but not HIF1A (Takeda et al. 2008). Selectivity among HIF substrates is mediated by sequences contained within the mobile loop in the PHD polypeptide (Chowdhury et al. 2016; Villar et al. 2007). Strikingly, accumulation of HIF1A by hypoxia or inhibitors leads to feedback upregulation of human or rodent PHD2 and PHD3, but not PHD1, which prevents further accumulation of HIF1A (reviewed in Fong and Takeda 2008).
In addition to PHDs, factor inhibiting HIF (FIH) is a vital O2-sensitive enzyme for HIF regulation. FIH is an O2-dependent dioxygenase that, similarly to PHDs, requires Fe2+ and 2-OG. Human FIH hydroxylates an asparagine residue (HIF1A: Asn803, HIF2A: Asn847) in the C-terminal transactivation domain and prevents binding of the co-activators p300 and CREB-binding protein (CBP) to HIFs, which is required for full transcriptional activity (Lando et al. 2002a, b). Human FIH protein remains active at lower O2 concentrations than PHDs and dominates HIF activation during exposure to lower PO2 range (Stolze et al. 2004). FIH is expressed in distal tubules and podocytes in the kidney (Schodel et al. 2010). Interestingly, in human cells FIH is less effective on HIF2A than on HIF1A (Bracken et al. 2006).
Renal expression of HIF-alpha isoforms and regulation of HIF-target genes
Whereas HIF1A is ubiquitously expressed, HIF2A exhibits a more tissue-specific expression pattern. In human and rat kidney, HIF1A is the predominant isoform in tubular cells, whereas HIF2A is strongly expressed in interstitial cells, endothelial cells and the glomeruli, but mostly absent from tubular cells (Bernhardt et al. 2006b; Rosenberger et al. 2002) (see Table 1 for a summary of HIF expression in the kidney). Beside their different expression patterns, HIF1 and HIF2 transcription factors also differ in their transcriptional targets, activation kinetics and O2 dependency. In human cell lines, HIF1A rapidly accumulates during severe hypoxia (< 5% O2) and takes part in the initial adaptation process to this condition, but then declines to low levels after 24 h, whereas HIF2A accumulation occurs—in addition to severe hypoxia—under prolonged and less severe hypoxic conditions (Holmquist-Mengelbier et al. 2006; Wiesener et al. 1998). This difference in kinetics may be due to the specific action of a negative feedback loop via an HIF1A antisense transcript that negatively regulates human HIF1A, but not HIF2A (Rossignol et al. 2002).
When HIFs accumulate in the nucleus under hypoxia, they bind to hypoxia response elements (HRE) in the enhancer or promotor region of their target genes that contain the core sequence RCGTG (R = A or G), resulting in transcription (Wenger et al. 2005). HIF1 seems to predominantly regulate glycolytic genes, whereas HIF2 preferentially regulates erythropoiesis and angiogenesis via EPO and vascular endothelial growth factor A (VEGFA), respectively (Hu et al. 2006; Morita et al. 2003; Warnecke et al. 2004). HIF1 increases almost all enzymes in the glycolytic pathway, as well as the glucose transporters 1 and 3 (Chen et al. 2001; Wenger 2002). Without sufficient O2, cells need to switch to O2-independent glycolysis to meet their energy demand, as the O2-dependent tricarboxylic acid cycle (TCA) is no longer operative (Dang and Semenza 1999; Seagroves et al. 2001). As glycolysis only generates two molecules ATP from each glucose molecule instead of 34–38 ATP molecules that the TCA cycle provides, cells need to increase glucose uptake. More elaborate adaptive responses to hypoxia seem to depend on HIF2. Hence, in response to systemic hypoxia, murine HIF proteins binds to the HRE in the 3’ enhancer region of Epo resulting in the rapid production of EPO by interstitial fibroblast-like cells to promote erythropoiesis (Koury et al. 1989; Semenza and Wang 1992), and at least in mice conditional ablation of HIF2A in the murine kidney has established that hypoxic induction of EPO is completely dependent on HIF2, even under severe hypoxia, and not on HIF1 (Kapitsinou et al. 2010; see also Gruber et al. 2007; Rankin et al. 2007; Scortegagna et al. 2005). In addition, EPO can also protect against kidney injury by reducing apoptosis and inflammation, and increasing tubular cell proliferation (for review see Moore and Bellomo 2011). VEGFA plays a central role in angiogenesis by activating the receptor tyrosine kinases VEGFR-1, -2, and -3 (Forsythe et al. 1996; Shibuya 2013), which may be important in pathological angiogenesis, promoting tumor growth and metastasis. In the kidney, the function of the glomerulus is dependent on the special vasculature maintained by VEGFA, and dysregulation may lead to glomerulopathy and breakdown of the filtration barrier (Eremina et al. 2008, 2003; Keir et al. 2017).
Relevant to this review, hypoxic cancer tissue may trigger HPHE signaling with consequent activation of hypoxia-induced target genes, such as growth factors (including VEGFA, PDGFB, and TGFA) that can then bind to their respective receptors and induce angiogenesis and proliferation. HIF transcription factors also increase the expression of several genes that regulate glucose metabolism (such as SLC2A1, LDHA and PDK1), as well as EPO, all to promote cancer cell proliferation, survival, angiogenesis and metabolic reprogramming. Moreover, certain types of kidney cancer, in particular clear cell renal carcinoma, display mutations in genes associated with HPHE signaling, especially VHL (Huang 2003; Linehan and Ricketts 2013; Linehan and Rouault 2013; Linehan et al. 2019, 2010), resulting in a “pseudo-hypoxic state” accompanied by increased EPO (and its receptor) expression (reviewed in Morais et al. 2013).
VHL in renal cancer
Renal cancers have become increasingly prevalent, accounting for 2.2% of new adult malignancies and 1.8% of deaths. From the 36 cancer types that were reported, kidney cancer ranked 16th and is the 9th most common cancer type in males (Sung et al. 2021). Clear cell renal cell carcinoma (ccRCC) is the most common type of renal cancer and whilst it has been long known that it is a metabolic disease, comprehensive integrated molecular scrutinization utilizing whole genome sequencing, whole exome sequencing, RNA sequencing, array-based gene expression, copy number and DNA methylation analyses have now genetically and molecularly defined ccRCC (Cancer Genome Atlas Research et al. 2013; Sato et al. 2013). Up to 28 genes were identified to be significantly mutated in ccRCC with VHL, PBRM1, SETD2 and BAP1 belonging to the most significant mutated genes in both studies. Intriguingly, VHL is a key affected gene, occurring in approximately 60% of ccRCC, yet rarely in other renal cancer subtypes, such as papillary or chromophobe (Sukosd et al. 2003). VHL is located on chromosome 3p, which was deleted in more than 90% of patient samples (Zbar et al. 1987). Other key mutated genes, including the tumor suppressor gene PBRM1, are also located on the short arm of chromosome 3, which is frequently targeted in other cancer types, such as lung cancer (Zabarovsky et al. 2002). This leads to the question as to why chromosome 3p is susceptible to chromosomal aberrations and why VHL mutations are especially prevalent in ccRCC.
Due to the frequent alterations in chromosome 3p, it would be plausible to hypothesize susceptible regions are targeted during carcinogenesis. Common fragility sites (CFS) are chromosomal regions wherein breaks and gaps are often found, perturbing DNA replication and leading to replication stress (Glover et al. 2017). The structure of CFS—they are particularly AT-rich—may contribute to their sensitivity to damage and designation as “hotspots” of genomic instability. Interestingly, one of the most fragile sites has been localized to FRA3B region on chromosome 3p14 and is the top fragile locus in lymphoblasts (Hosseini et al. 2013; Huebner and Croce 2001). Furthermore, the tumor suppressor gene FHIT is localized to the FRA3B region and synergizes with VHL. However, a role for FRA3B/FHIT in ccRCC has been debated in conflicting studies. The low occurrence of terminal deletions encompassing the FRA3B region in nonpapillary RCC (3 from 100) (Bugert et al. 1997) or normal FHIT transcripts in renal cancer cell lines (van den Berg et al. 1997) lead to the conclusion that FRA3B/FHIT is not involved in the development of nonpapillary RCC. In a later study, a continuous deletion of a region of chromosome 3p, harboring VHL and FHIT, was found in 96% of ccRCC (Sukosd et al. 2003), which is supported by a number of earlier studies that speculate or imply involvement of the FRA3B region or FHIT in RCC (Hadaczek et al. 1998; Shridhar et al. 1997; Yamakawa et al. 1992). Decisively, the latest molecular analyses do not report FHIT among the significantly mutated genes in ccRCC (Cancer Genome Atlas Research et al. 2013; Sato et al. 2013), suggesting it does not drive ccRCC progression.
As a major site of excretion, the kidney comes into contact with numerous potentially toxic substances. Some of these substances are removed into the urine, yet others are retained in the kidney where they accumulate and may elicit stress and adaptive responses, culminating in cell death or survival. Risk factors for renal cancer include cigarette smoking, workplace exposures, obesity and hypertension. Could the VHL gene be directly targeted by potential carcinogens? Indeed, VHL mutations in renal tumors have been reported in nitrosamine-exposed Wistar rats (Shiao et al. 1998), potassium bromate-exposed F344 rats (Shiao et al. 2002) and individuals occupationally exposed to the industrial solvent and carcinogen trichloroethylene (Brauch et al. 2004). Furthermore, the impact of toxic substances and carcinogens on DNA damage (e.g. through oxidative stress), DNA replication, replication stress and DNA repair systems must be taken into consideration in the mutation of VHL and other tumor suppressor genes. To this end, transition metals are particularly relevant because of their ability to cause oxidative stress as well as their requirement for some enzymatic processes, such as DNA repair enzymes. Despite some indication of metals affecting HIFs and HIF-target genes (Li et al. 2006), the effect of transition metals on VHL mutation, expression and/or activity has remain largely unexplored.
Transition metals: environmental presence, exposure, general modes of toxicity in the kidney
According to the International Union of Pure and Applied Chemistry (IUPAC), a TM is defined as "an element whose atom has an incomplete d sub-shell, or which can give rise to cations with an incomplete d sub-shell" (Chemistry IUoPaA 1997). Most TMs are found in the so-called d-block in the periodic table in groups 3–12 and have common features, such as electricity and heat conduction, malleability and, of particular importance from a biological point of view, multiple oxidation states, which is a result of their valence electrons (used to combine with other elements) being present in more than one shell. TMs naturally occur in the Earth, usually as salts, and their distribution was determined by geological events, such as geotectonic/metamorphic, volcanic and oceanic, that occurred. From the 38 elements found in the d-block, we have selected to review TMs that pose a potential threat to human health, namely, cadmium, chromium, cobalt, iron, nickel, and platinum. Of these, cobalt, iron and chromium are essential trace elements that are required for chemical reactions and physiological processes in the human body. The TMs copper and mercury have not been included because of little evidence and reports in the literature in the context of hypoxia and HIFs. The abundance of the selected TMs in the Earth’s crust follows the sequence: iron (Fe) > chromium (Cr) > nickel (Ni) > cobalt (Co) > cadmium (Cd) > platinum (Pt) (Haynes et al. 2016). Furthermore, Cd is in the top ten of the Agency for Toxic Substances and Disease Registry (ATSDR) 2019 Substance Priority List, which is based on their frequency, toxicity and potential for human exposure (ATSDR 2019).
TMs are released into routes of human exposure either by natural weathering or following industrial activities that mine and extract ores for further processing (Smith and Huyck 1999). Both methods would result in excess levels of TMs entering the food chain and further contributing to human exposure to the general population not employed in the industries. For both industrial workers and the general population, the inhalational and ingestion routes are the major entry pathways of TMs into the body; though the skin is penetrated by some metals, it is unlikely to be in large quantities. Maladies of the lung, intestinal tract, liver and kidney are usually reported. For the general nonsmoking population, exposure to contaminated water and foodstuffs is the major entry route and metal accumulation may contribute to diseases of the digestive tract and detoxification organs. Airborne pollutants containing TMs, such as particulate matter, gaseous pollutants or tobacco smoke, may also pose a risk and cause injury to the lung, liver and kidneys (see Toxicological Profiles from Agency for Toxic Substances and Disease Registry (ATSDR 2021).
Quoting the Father of Toxicology, Paracelsus, “What is there that is not poison? All things are poison, and nothing is without poison. Solely the dose determines that a thing is not a poison”, this statement is especially relevant for TMs. Up until a certain dose, these metals can be tolerated and are even required by the human body. The kidney is often targeted by increased metal loading due to the plethora of membrane transporters available for crossing the lipid bilayer by molecular mimicry to access the intracellular space where their toxic effects can be unveiled (Bridges and Zalups 2005). Initially, the elevated influx of TMs can be buffered by renal cells through upregulation of chelating proteins, such as metallothionein, or efflux transporters. In addition, general cellular stress responses may be activated to counteract potential damage (Lynes et al. 2007; Weinhouse 2021). However, once the number of binding sites, reservoirs or capacity of storage compartments is exceeded, toxicity occurs. Based on the variable oxidation states and ease of electron donation of TMs, the generation of ROS is a common denominator across TMs and plays a pivotal role in the toxicity execution program (Sabolic 2006) (Fig. 1).

Possible targets of transition metals (TMs) on the HPHE signaling pathway. TMs may enter renal cells via transporters and channels for essential metal ions, such as Fe2+. One of these transporters is the divalent metal transporter-1 (DMT-1). TMs may compete with Fe2+ and thereby prevent entry of Fe2+ via DMT-1, resulting in depletion of intracellular iron needed as a cofactor for PHDs (1). TMs contribute to augmented generation of free radicals, either by interfering with reactive oxygen species (ROS) scavenging mechanisms (2), by their own redox activity (3), or by disrupting the mitochondrial electron transport chain (4). Increased ROS inhibit PHDs by oxidizing PHD bound Fe2+ to Fe3+. TMs may activate HIFs by substituting for Fe2+ in PHDs and inactivation of the enzyme (5). TMs may deplete intracellular ascorbate (AA) and consequent oxidation of Fe2+ to Fe3+ in the catalytic center of PHDs (6). TMs may stabilize HIF by occupying the VHL-binding domain thereby inhibiting the interaction between VHL protein and hydroxylated HIFs (7). For further details, please refer to the text
Free radicals: generation and eradication
The configuration of unpaired electrons in each outer orbital shell of diatomic O2 makes it particularly susceptible to free radical formation (Halliwell 1991). Reduction results in an anionic form of O2, the superoxide anion O2⋅−, which is short-lived (Hayyan et al. 2016). Other free radicals generated through reduction of O2 include the more reactive hydroxyl radicals (⋅OH) and (hydrogen) peroxide (H2(O2⋅−2)), henceforth referred to as H2O2 (Munro and Treberg 2017). Though H2O2 itself is not a free radical, oxidation of iron (Fe2+ to Fe3+) can generate ⋅OH through Haber–Weiss and Fenton chemistry (Halliwell 1991; Wardman and Candeias 1996). Naturally, iron can be substituted by other TMs. Moreover, superoxide can react with nitric oxide (NO⋅) to form peroxynitrite (ONOO−) and other reactive nitrogen species (RNS). Similar to ROS, RNS are highly reactive and can oxidize thiols and nitrate proteins (Adams et al. 2015).
To limit their damage potential, ROS are either metabolized or neutralized. Superoxide dismustases (SODs) drive the reaction of two superoxide anions with two H+ ions to generate the more stable H2O2 and water (Fukai and Ushio-Fukai 2011; Hayyan et al. 2016). Though H2O2 can still form ·OH radicals, it can be used as a substrate for several antioxidant enzyme systems, including catalase, glutathione peroxidase, and peroxiredoxins, so that it can be more efficiently removed than superoxide. ROS can be neutralized by glutathione, the most abundant antioxidant, or vitamins. Glutathione is synthesized through a two-enzyme reaction catalyzed by glutamate cysteine ligase and glutathione synthetase. It is a tripeptide nucleophile with thiol groups capable of accepting electrons in its reduced state (GSH), thereby becoming oxidized (GSSG), and rendering ROS to a lesser or non-reactive state (Diaz-Vivancos et al. 2015; Halliwell 1991; Scire et al. 2019; Wardman and Candeias 1996). Both water-soluble (AA, niacin, folic acid) and lipid-soluble (tocopherols) vitamins contribute to the cellular antioxidative capacity. The accumulation and incorporation of tocopherols, eight compounds under the umbrella term of vitamin E (Khadangi and Azzi 2019), into the lipid bilayer (with preference for polyunsaturated fatty acid chains) limits damaging lipid peroxidation.
In addition to modulation and inhibition of antioxidative enzymes and ROS-neutralizing substances, TMs increase ROS levels and oxidative stress by acting directly on mitochondria (Fig. 1). The mitochondrial electron transport chain (mETC) is the largest source of cellular ROS, which are rapidly removed by antioxidants in the mitochondrial matrix. Through the shunting of electrons along four multimeric protein complexes with TM (iron or copper) containing components, electrons can escape and contribute to ROS generation, specifically superoxide anions generated from complexes I and III (Drose and Brandt 2012). Impairment of electron shuttling by damaging complex proteins exposed to elevated levels of TMs culminates in increased ROS production and disruption of physiological ROS signals (e.g. Hosseini et al. 2014; Wang et al. 2004; Xiao et al. 2012).
ROS: physiological signaling messengers
Whilst unregulated ROS are damaging to cells, driving detrimental effects through unsolicited oxidation of encountered proteins and lipids, it is increasingly apparent that ROS also have physiological functions, such as signal transmission or monitoring of function. How can the cell differentiate between sub-toxic and toxic ROS signals? A key strategy is to compartmentalize ROS, to limit their levels by balancing generation with antioxidative mechanisms, and to control their diffusion capacity. TMs do not only participate in ROS generation as detailed above but can also influence antioxidative capacity, such as by exhausting glutathione supply, inhibiting or damaging antioxidative enzymes, altering protein structure and conformation, and influencing synthesis or regeneration of antioxidants (Dobritzsch et al. 2020; Limon-Pacheco and Gonsebatt 2009). Consequently, physiological ROS can be impacted through disruption, diversion or ablation of the signaling pathway.
Signaling through ROS make take several different routes depending on the TM, its concentration, its route of cellular uptake and intracellular metabolism (Villalpando-Rodriguez and Gibson 2021). Initial stress defense programs could be initiated through low levels of ROS as a sub-toxic load of TM slowly accumulates. This could include the heat shock protein response, prosurvival MAPK signaling, hypoxic response, antioxidative response, unfolded protein response or epigenetic response. Ultimately, toxicity will be elicited through engagement of a cell death program that results in renal injury. Though other forms of cell death may be utilized in specialized cases, such as ferroptosis in the case of iron or autophagic cell death, apoptosis or necrosis are the most likely forms of cell death to be encountered in TM toxicity. The reader is referred to excellent reviews concerning the molecular signaling pathways associated with these cell death forms (Galluzzi et al. 2016; Kalkavan and Green 2018; Lee and Thévenod 2020; Martinou and Youle 2011; Sano and Reed 2013).
Contribution of mitochondrial ROS to HPHE signaling
Increased production of ROS by cells in a hypoxic environment is counterintuitive and has, therefore, been debated (Clanton 2007). This argument was based on the observation that conditions of hyperoxia lead to elevations in ROS production in many tissues, especially that the amount of superoxide formed from mitochondria is directly proportional to the concentration of O2 (reviewed in Jamieson et al. 1986). Nonetheless, as one approaches anoxia, O2 availability can become critical for the production of ROS.
Cellular hypoxia is a state that is generally characterized by being in a more cellular reductive state and has been described as a form of “reductive stress” (Dawson et al. 1993). This condition is associated with elevations in reducing equivalents (mostly NADH and FADH2) that accumulate in the mitochondria, when there is not sufficient O2 is available for reduction by the mETC. The reducing equivalents also make electrons more available for reduction reactions, such as O2 to superoxide. Hence the conditions necessary for ROS formation in hypoxia comprise both high reductive capacity (e.g. high NADH/NAD+) and sufficient O2 available for reaction, whereas in hyperoxia, ROS formation occurs at the expense of low reducing capacity. This bimodal distribution of ROS formation as a function of PO2 is observed because both hypoxia and hyperoxia support elevations in ROS formation (Clanton 2007).
Regulation of HPHE signaling by hypoxia-induced mitochondrial ROS
The work of Chandel and Schumacker has provided a link between hypoxia-induced mitochondrial ROS formation and the HPHE signaling axis. Their experimental evidence suggested that the mETC is involved in O2 sensing and, therefore, responds to changes in O2 levels. In their initial study (Chandel et al. 1998), they used liver Hep3B cells that display transcriptional activation of EPO, glycolytic enzymes, and VEGFA during hypoxia or in response to cobalt chloride (CoCl2) that is a “chemical hypoxia mimic” and are, therefore, commonly used to investigate hypoxia signaling. They tested whether mitochondria act as O2 sensors during hypoxia in these cells and whether hypoxia and Co activate transcription by increasing ROS. Wild-type Hep3B cells increased ROS generation during hypoxia (1.5% O2) or Co (100 μM) incubation under normoxic conditions for 24 h, whereas Hep3B cells depleted of mitochondrial DNA (ρ0 cells), and, therefore, do not respire, failed to increase ROS and to induce mRNA for EPO, glycolytic enzymes, or VEGFA during hypoxia. In contrast, ρ0 cells increased ROS generation in response to Co and retained the ability to induce expression of these genes. Finally, antioxidants abolished transcriptional activation of these genes during hypoxia or Co in wild-type cells and abolished the response to Co in ρ0 cells. Thus, the authors concluded that hypoxia activates transcription via a mitochondria-dependent signaling process involving increased ROS, whereas Co activates transcription by stimulating ROS generation via a mitochondria-independent mechanism. In a subsequent study, Chandel et al. (2000) showed that hypoxia increases mitochondrial ROS generation at complex III of the mETC, which causes accumulation of HIF1A protein responsible for initiating expression of a hypoxia-inducible luciferase reporter construct. This response was lost in ρ0 cells. Overexpression of catalase abolished hypoxia-induced luciferase expression. Exogenous H2O2 stabilized HIF1A protein during normoxia and activated luciferase expression in wild-type and ρ0 cells. Moreover, isolated mitochondria increased ROS generation during hypoxia. Hence for the first time, these findings revealed that mitochondria-derived ROS are both required and sufficient to initiate HIF1A stabilization during hypoxia.
Mechanisms of stabilization of HIF-alpha by mitochondrial ROS
These groundbreaking and subsequent studies established that decreasing ROS levels using genetic or pharmacological tools during hypoxia diminishes HIF1A and HIF2A protein levels (Brunelle et al. 2005; Chandel et al. 1998, 2000; Guzy et al. 2005; Lin et al. 2008; Mansfield et al. 2005). However, the questions were still debated whether mitochondria contribute indirectly or directly to HIF-alpha protein stability and which function of the mETC is necessary for HIF-alpha protein stabilization. It had been suggested that under hypoxia, mitochondria, with their high O2 consumption, leave the rest of the cell ‘anoxic’. PHDs, would then be deprived of their cofactor, O2, leaving them unable to hydroxylate HIF-alpha protein to target it for degradation (Hagen et al. 2003). The second model implicated ROS generation by the mETC as a signaling molecule for HIF-alpha protein stabilization (Bell et al. 2005) and was supported by the above mentioned study (Chandel et al. 2000) as well as by experiments in which cells genetically depleted of cytochrome c or the Rieske-Fe-S protein also failed to increase production of ROS during hypoxia (Brunelle et al. 2005; Guzy et al. 2005; Mansfield et al. 2005).
Further studies conclusively identified the complex within the mETC responsible for hypoxic ROS generation and HIF-alpha protein stabilization. Pharmacological evidence indicated that the Qo site of complex III is a likely site of ROS generation during hypoxia (reviewed in Klimova and Chandel 2008). This was validated using cytochrome b mutant cybrids, generated by reconstituting 143B ρ0 cells with wild-type mitochondrial DNA or that containing a 4-bp deletion in the cytochrome b gene found in a patient suffering from parkinsonism (loss of cytochrome b renders these cells incapable of O2 consumption and unable to generate ROS at the Qi site specifically). However, although respiratory incompetent, these cytochrome b-deficient cells were still capable of upregulating hypoxic ROS and stabilizing HIF1A protein (Bell et al. 2007). The mitochondrial antioxidant MITOQ prevented the HIF1A protein stabilization. Furthermore, RNAi to knock down Rieske Fe-S protein to abolish ROS generation at the Qo site in mutant cytochrome b cybrids, prevented hypoxic ROS generation and HIF1A protein stabilization, implicating the Qo site of complex III as the key site in hypoxic ROS generation and HIF1A protein stabilization. These cells, however, retained the ability to stabilize HIF1A protein after direct PHD inhibition by DMOG, showing an otherwise intact HIF-signaling pathway. Furthermore, a link between hypoxic ROS generation and hydroxylation of HIF1A protein was established. Neutralizing the ROS with antioxidants allowed HIF1A protein to remain hydroxylated even under hypoxic conditions and, therefore, primed for degradation. On the contrary, increasing ROS levels under normoxia by overexpressing glucose oxidase prevented normoxic HIF1A protein hydroxylation. Altogether, these data demonstrated that the ROS generated by mitochondria under hypoxia prevent hydroxylation of HIF1A protein.
How mitochondrial ROS inactivate PHDs to stabilize the HIF-alpha protein subunit is not understood. One hypothesis is that mitochondrial ROS generated during hypoxia promote the oxidation of cysteine residues within PHD2, resulting in oxidative PHD2 homodimerization and inactivation and leading to HIF1A protein stabilization (Lee et al. 2016). Indeed, PHD2 has several reactive cysteine residues in its C-terminal catalytic domain that may be oxidized by ROS (Briggs et al. 2016; Lee et al. 2016). Interestingly, PHD2 activity requires high intracellular levels of free cysteine, which is regulated by cysteine dioxygenase (Briggs et al. 2016). Free intracellular cysteines may compete with the reactive cysteine residues of PHD2 for ROS-mediated oxidation. Thus, when free intracellular cysteine levels are high, PHD2 cysteine oxidation is prevented. PHD2 is then active, and HIF-alpha protein levels are low. By contrast, limiting the amount of free intracellular cysteine would trigger HIF-alpha protein accumulation. Currently, the significance of these PHD2-reactive cysteines in the stabilization of HIF-alpha under physiological hypoxic conditions remains to be clarified. Metabolites, such as succinate and fumarate, are a second input that inhibits PHD2 activity (reviewed in Lee et al. 2020). To date, no reports have been published that investigated the mechanisms of ROS-induced PHD1/3 inactivation.
In summary, a unifying model to explain HIF-alpha activation is the intrinsic decrease in PHD2 activity owing to declining O2 levels coupled with added inputs, such as ROS or metabolites that further diminish PHD2 activity to maximally increase HIF-alpha protein levels.
Impact of toxic metal ions on the renal HPHE signaling axis
General considerations
Once toxic metal ions have entered the body they disseminate and accumulate in organs via the blood circulation where they bind to blood cells as well as to various high- and low-molecular weight plasma proteins and peptides. The latter are filtered by the glomerulus and taken up by kidney tubules where they accumulate and damage renal tissue. After acute exposure to high concentrations of metal ions, other organs are damaged as well and kidney injury—likely necrosis—may be indirect and a sequel of pulmonary or cardiovascular problems. Chronic exposure to low concentrations of metals may also lead to renal damage, but the effect is protracted, and ultimately results in chronic kidney disease due to replacement of functional tissue by fibrotic material. The impact on the renal HPHE signaling axis may, therefore, differ between both durations of exposure.
TM ion concentration and time dependence of renal HPHE responses
Acute exposure to low TM ion concentrations may trigger activation of (protective) EPO signaling, e.g. by mimicking hypoxia (“chemical hypoxia”), and is either transient or long lasting depending on the exposure time. Protective effects of hypoxia—in addition to Fe, Co, Ni and pharmacological PHD inhibitors—on renal damage induced by various insults have been observed in many studies (Hou et al. 2013; Nath et al. 1992; Nezu et al. 2017; Shimizu et al. 2000; Zager et al. 2016, 2021, 1993). Yet protective effects will only be efficient when interventions occur before the damaging stress, and this strategy has been termed protective “preconditioning” (Wang et al. 2012b). This “(hypoxic) preconditioning” protects organs, including the kidney, against injury, and could be beneficial and improve renal function (reviewed in Bernhardt et al. 2007; Heyman et al. 2011; Shu et al. 2019); see also “Therapy of TM nephrotoxicity, controversies, outlook, and conclusions”). Indeed, the renal HPHE signaling can be stimulated in both acute (e.g. Bernhardt et al. 2006a; Conde et al. 2012; Kudo et al. 2005; Matsumoto et al. 2003; Schley et al. 2011; Schodel et al. 2009; Weidemann et al. 2008) and chronic (e.g. Schley et al. 2019; Tanaka et al. 2005b, c; Theilig et al. 2011) kidney injury, which induces the expression of a variety of tissue protective genes—in particular EPO but also HMOX1—for adaptation and repair.
Nevertheless, the underlying mechanism is very complex because several hundred genes are targeted by HIFs (Dengler et al. 2014), and time-, isoform- and compartment-specific actions of the HIF pathway seem of utmost importance. But whether activation of the HPHE pathway promotes or antagonizes renal fibrosis elicited by acute kidney injury (AKI), and particularly chronic kidney disease (CKD), is controversial and shows variable outcomes (reviewed in Faivre et al. 2021). Interestingly, a study using a remnant kidney model of CKD in rats has possibly shed some light on these divergent results: Administration of a small molecule inhibitor of PHD dioxygenases (see “Therapy of TM nephrotoxicity, controversies, outlook, and conclusions”) at an early stage accelerated renal fibrosis, whereas at a more advanced stage it decreased renal fibrosis (Yu et al. 2012). Strikingly, the inhibitor given at the early stage activated both HIF1A and HIF2A, whereas given at the later stage it only activated HIF2A with no effect on HIF1A. Consequently, whether HIFs are pro- or anti-fibrotic seems context- and HIF isoform-dependent. The role of the renal HPHE signaling in protection against ischemic kidney injury was investigated in more details using a genetic approach to dissect the contributions of endothelial HIF1A and HIF2A in murine models of hypoxic kidney injury induced by ischemia/reperfusion injury (IRI) or ureteral obstruction (Kapitsinou et al. 2014). In both models, inactivation of endothelial HIF2A, but not endothelial HIF1A, increased expression of renal injury markers and inflammatory cell infiltration in the post-ischemic kidney. Genetic or pharmacologic activation of HIF via HIF prolyl-hydroxylase inhibition protected wild-type animals from ischemic kidney injury and inflammation; however, these protective effects were not observed in HIF prolyl-hydroxylase inhibitor-treated animals lacking endothelial HIF2A. This indicated that endothelial HIF2 mediates protection and recovery from hypoxia-induced renal damage and represents a potential therapeutic target for renoprotection and prevention of fibrosis following acute ischemic injury.
In contrast, high TM ion concentrations may rapidly induce failure of the HPHE system due to cell death and disruption of mitochondrial function (see “Transition metals: environmental presence, exposure, general modes of toxicity in the kidney”; reviewed in Thévenod et al. 2020), thus abolishing physiological ROS signaling and HIF-alpha stabilization (see “Contribution of mitochondrial ROS to HPHE signaling”; reviewed in Lee et al. 2020) (Fig. 1). Chronic exposure to low TM ion concentrations could activate the renal HPHE signaling axis (“preconditioning”; see above) that may delay onset of failure, which occurs after longer periods of exposure and is paralleled by other signs of chronic renal dysfunction. This may occur in organisms, such as humans and experimental animals, but also in cell lines. Hence, seemingly disparate or equivocal observations can be reconciled if concentrations and exposure times are considered. Moreover, the impact of toxic metal ions on renal HPHE signaling may vary depending on whether it occurs in a hypoxic or normoxic environment, or because metal ions may differentially affect O2 binding to PHDs.
Iron (Fe)
Exposure and nephrotoxicity
Systemic Fe overload may occur in hereditary hemochromatosis or β-thalassemia, however, the major form of Fe overload is acquired by repeated blood transfusions (Siddique and Kowdley 2012). Systemic Fe overload diseases are associated with chronic damage to a variety of organs, including the heart, liver and endocrine glands. Excess Fe accumulation in these organs is associated with cellular toxicity and death because of its pro-oxidant effects but has also been associated with a number of diseases, and in particular the development of cancer (Toyokuni 2009), wherein excess Fe may cause DNA damage leading to persistent mutations. In addition, Fe is also essential for maintaining the rapid growth rate of cancer cells and may nurture the tumor microenvironment and metastasis. However, Fe can also contribute to cancer defense by inducing toxic ROS and/or initiating specific forms of cell death, including ferroptosis, necroptosis and pyroptosis. Not surprisingly, the carcinogenicity of Fe has been under debate for quite a while (see for instance Huang 2003; Thévenod 2018; Torti et al. 2018; Ying et al. 2021).
The kidney is rarely affected by systemic Fe overload but can be specifically targeted, e.g. in the context of hemoglobin (Hb)-induced AKI subsequent to hemolysis, IRI or due to proteinuria associated with chronic kidney diseases (reviewed in Scindia Ph et al. 2019; Van Avondt et al. 2019; van Swelm et al. 2020). When present in excess and in non-physiologic labile forms, Fe is toxic to the kidneys (as in Hb-associated AKI) and causes renal damage or aggravates AKI elicited by other insults (e.g. Moussavian et al. 2007; van Swelm et al. 2016; Zager and Gamelin 1989; reviewed in van Swelm et al. 2020). Labile (catalytic) Fe is a transitional pool of Fe (Leaf and Swinkels 2016; Slotki and Cabantchik 2015) that is readily available to participate in redox cycling and induces formation of ROS (Halliwell 1991). Through the Fenton reaction (see “Transition metals: environmental presence, exposure, general modes of toxicity in the kidney”) catalytic iron causes oxidative damage to cell membranes, proteins and DNA, which may trigger ER stress (van Raaij et al. 2018; van Swelm et al. 2018) and various forms of regulated cell death, such as ferroptosis (Linkermann et al. 2014) or necroptosis (van Swelm et al. 2018). Yet Fe-induced oxidative damage may be mitigated by nuclear factor erythroid 2-related factor 2 (NRF2)-mediated induction of heme oxygenase-1 (HO-1) (Adedoyin et al. 2018; Alam et al. 2003; Rubio-Navarro et al. 2019; reviewed in Lever et al. 2016; Nath and Agarwal 2020; Tracz et al. 2007). Still the role of Fe in this process appears to be more complex because apart from the obvious injuring effect of Fe on kidney tissue (see above), protective “preconditioning” (reviewed in Bernhardt et al. 2007; Heyman et al. 2011; Shu et al. 2019) by Fe may also occur (see “General considerations” and Hou et al. 2013; Nath et al. 1992; Zager et al. 2016; Zager et al. 2021; Zager et al. 1993).
Disruption of HPHE signaling by Fe
However, excess Fe represses the renal HPHE signaling (Oshima et al. 2017; Suzuki et al. 2018). Epo gene expression was suppressed in mice following Fe treatment (Oshima et al. 2017). HIF2A (but not HIF1A) was also diminished in the kidney of mice following Fe treatment (2 mg saccharated ferric oxide in a volume of 200 μl per 25 g mouse i.p. for five consecutive days). Moreover, anemia-induced increase in renal EPO and HIF2A expression were inhibited by Fe treatment. Additional cell culture experiments using EPO-producing HepG2 cells showed that Fe stimulation (50–200 µg/ml for 24 h) reduces the expression of the Epo gene, as well as HIF2A. Moreover, Fe treatment augmented oxidative stress, and Fe-induced reduction of Epo and HIF2A expression was restored by the antioxidant Tempol. HIF2 interaction with the Epo promoter was inhibited by Fe treatment and reversed by Tempol. Taken together, these findings suggested that Fe supplementation reduces Epo gene expression via an oxidative stress-HIF2A-dependent signaling pathway (Oshima et al. 2017). This was confirmed and extended in a mouse model of EPO-deficient anemia to show that during Fe overload renal interstitial fibroblasts accumulate Fe, and this impairs the hypoxia-driven transcription of the Epo gene via renal HIF2 (Suzuki et al. 2018). The authors used “ISAM” (“inherited super anemic mice”) mice, in which both alleles of the Epo gene are replaced with the green fluorescent protein (GFP) gene, resulting in constitutive activation of the mutant Epo-GFP gene in renal EPO-producing cells and hepatocytes due to chronic anemia conditions. By measuring EPO-GFP expression levels in ISAM mice without any specific treatment, the ability of these mice to produce EPO in vivo can be evaluated. Injection of Fe-dextran (10 mg Fe a day for 2 days i.p.) in ISAM mice caused severe Fe deposition in renal interstitial fibroblasts, the site of EPO production. Fe overload induced by either i.p. injection or feeding decreased activity of endogenous Epo gene expression by reducing levels of HIF2A. Administration of a Fe-deficient diet to the anemic mice reduced hyperferremia in ISAM mice to normal concentrations and enhanced the ability of renal EPO production. These results demonstrate that Fe overload due to EPO deficiency anemia attenuates endogenous Epo gene expression in the kidneys. Thus, iron suppresses EPO production by reducing HIF2A concentration in renal interstitial fibroblasts (Suzuki et al. 2018), indicating that EPO and Fe are in a “conflicted alliance” (Ganz 2018).
In summary, the effects of Fe on the kidney are complex and like a double-edged sword. Fe injures kidney tissues by inducing oxidative stress and various forms of cell death, in particular ferroptosis. At the same time Fe-induced ROS formation activates protective and adaptive signaling pathways, such as NRF2/HO-1 signaling. Whereas hypoxia and Fe may trigger activation of the renal HPHE signaling to protect against acute or chronic kidney injury (“hypoxic preconditioning”; see “General considerations”), excess Fe may also repress HPHE signaling (see Table 2) and thereby prevent induction of tissue protective hypoxia-induced gene products, such as EPO and HMOX1.
Cobalt (Co) and nickel (Ni)
Co exposure and nephrotoxicity
Co in hard metal production represents the main source of occupational exposure, e.g. as Co metal dust in the fabrication of tungsten carbide. Other less dominant sources are environmental, dietary, and medical (such as wear and tear of certain metal-on-metal hip prostheses) (reviewed in ATSDR 2004; Leyssens et al. 2017; Simonsen et al. 2012). The evidence for Co nephrotoxicity is weak although it accumulates in the kidney (ATSDR 2004; Simonsen et al. 2012). Rather, Co may be beneficial by attenuating kidney damage induced by various forms of renal insult (see “Iron (Fe)” and below). For instance, in an acute ischemic tubule-interstitial injury model of rats induced by 45-min clamping of renal pedicles with contralateral nephrectomy, elevation of serum creatinine and morphologic injury after the ischemic insult was improved by co-administration of cobalt chloride (2 mM in drinking water from day − 10 to day 3) associated with amelioration of tubulo-interstitial damage and reduction of macrophage infiltration (Matsumoto et al. 2003).
Renal HIF-alpha stabilization by Co
In the kidney of rats treated with Co, renal HIF1A protein was upregulated and mRNA or protein levels of several renoprotective genes, such as Hmox1, Epo, Slc2a1 and Vegfa, were increased before ischemic injury (Matsumoto et al. 2003). In a subsequent study (Tanaka et al. 2005c), the same group applied Co treatment (2.7 mg/kg, subcutaneously, once every 3 days for 3–5 weeks) to uninephrectomized Thy1 nephritis rats, a tubulo-interstitial renal injury model. Although Co did not change glomerular structural damage or urinary protein excretion rate, tubulo-interstitial damage was improved in Co-treated animals and was associated with upregulation of renoprotective HIF-regulated genes (Hmox1, Epo, Slc2a1, and Vegfa) as well as increased HIF1A, SLC2A1 and VEGFA proteins. TUNEL staining revealed that the number of apoptotic cells was reduced in the renal cortex by Co administration, suggesting that renoprotection was achieved partly through its antiapoptotic properties. In another study, gentamicin-induced AKI was established in rats by intramuscular injection of 80 mg/kg gentamicin once a day for 7 days (Ahn et al. 2012). Co was continuously infused (10 mg/kg/day for 7 days) into the rats to activate HIF. Co (or DMOG) significantly increased HIF1A expression as well as increased HIF-target gene Vegfa and reduced the number of gentamicin-induced apoptotic cells in rat kidneys. HIF activation ameliorated the extent of histologic injury, reduced macrophage infiltration into the tubular interstitium and improved creatinine clearance and proteinuria in gentamicin-induced AKI. An important study was performed by Rosenberger et al. (2002) who investigated the expression of HIF1A and -2A in nephron segments of rats exposed to Co (CoCl2 injected subcutaneously twice, at a dose of 30 mg/kg, with a dosing interval of 12 h and animals were euthanized 6 h after the second injection). Proximal tubular cells were negative but marked induction of HIF1A was observed in 70–80% of distal tubular cross-sections, mainly in distal tubules and collecting ducts. In contrast, expression of HIF2A was weak. Unfortunately, no such study was performed with Ni (see below). Interestingly, this nephron distribution of HIF1A matches apical expression of SLC11A2/DMT-1 in the rodent nephron (Ferguson et al. 2001), which transports divalent metal ions with a selectivity of Cd > Fe > Co >> Ni and shows competition of Co with Fe for uptake (Gunshin et al. 1997; Illing et al. 2012). This suggests that the effect of Co on HIF1A is limited by its uptake into nephron cells.
At the cellular level, HK-2 human renal cells were pre-treated for 24 h with Co (150 µM) or DMOG (1 mM) to activate HIF and were then exposed to the nephrotoxic compound gentamicin (3 mM) for another 24 h (Ahn et al. 2012). Co or DMOG significantly increased HIF1A expression in HK-2 cells and inhibited gentamicin-induced ROS formation. HIF1A also protected these cells from gentamycin-induced apoptosis by reducing caspase-3 activity and the amount of cleaved caspase-3, and -9 proteins.
Ni exposure and nephrotoxicity
Environmental pollution from Ni may be due to industry (e.g. for alloy production, electroplating, in the production of Ni–Cd batteries and as a catalyst in chemical and food industry), the use of liquid and solid fuels, as well as municipal and industrial waste. Ni-plated water taps may contaminate water and soil; mining and smelting may dump Ni into wastewater; Ni–steel alloy cookware and Ni-pigmented dishes may release Ni into food. Ni contact can cause a variety of side effects on human health, such as allergy, cardiovascular and kidney diseases, lung fibrosis, lung as well as nasal cancer (reviewed in ATSDR 2005; Denkhaus and Salnikow 2002; Genchi et al. 2020). Ni absorbed by humans is excreted by the kidney into the urine. Hence, Ni is not inevitably a cumulative toxin, but larger doses or chronic inhalatory exposure may be toxic, even carcinogenic, and constitute an occupational hazard. When Ni accumulates in the kidney it may or may not induce acute or chronic nephrotoxicity, depending on the dosage and duration of exposure (ATSDR 2005; Das et al. 2008; Denkhaus and Salnikow 2002). Horak and Sunderman exposed rats to inhalation of Ni-carbonyl (0.6 mg/l of air per 15 min), which led to acute toxicity and death of about 20–30% of the animals within 2 h whereas the remaining animals survived for at least 3 days and showed increased proteinuria and aminoaciduria (Horak and Sunderman 1980). Exposure of rats to Ni (44.7–223.5 mg/l via their drinking water for 13 weeks induced a significant decrease in urine volume and an increase in blood urea nitrogen at the highest dose group only, and both immune and pulmonary systems were more sensitive targets than the kidney (Obone et al. 1999). In another chronic nephrotoxicity study, rats were given 100 mg/l of Ni (as Ni–sulfate) in drinking water for 6 months which resulted in increased urinary excretion of albumin (but not N-acetyl-β-d-glucosaminidase (NAG) or β2-microglobulin), suggesting glomerular rather than tubular damage (Vyskocil et al. 1994).
Activation of renal HPHE signaling by Co and Ni through chemical hypoxia
When hypoxia causes increased ROS formation (Lee et al. 2020), this triggers HO-1 expression (see “Iron (Fe)”; also reviewed in Agarwal and Nick 2000; Gozzelino et al. 2010). Co and Ni rapidly induce renal microsomal HO-1 under normoxic conditions, similarly as other TM ions, such as Cd or Pt (see “Platinum (Pt)”; reviewed in Agarwal and Nick 2000; Sunderman 1987). Because HO-1 induction by metals was generally suppressed by treatments with SH compounds (for example, cysteine and glutathione) and enhanced by agents that deplete tissue SH levels (for example, diethyl maleate), Sunderman concluded that the induction mechanism may involve binding of metal ions to SH-containing regulatory molecules. However, oxidative stress induced by metal ions, which affects the redox signature of kidney tissue (Stohs and Bagchi 1995) and results in cytoprotective induction of HO-1, is the other side of the coin (reviewed in Agarwal and Nick 2000; Gozzelino et al. 2010), where increased HO-1 may be mediated by HIF induction as well (Lee et al. 1997). Cell culture experiments support oxidative stress induced by Co and Ni. Ni (1–500 µM for 12–72 h) increased the formation of ROS, lipid peroxidation, apoptosis and DNA damage in normal rat kidney (NRK) cells (Chen et al. 2010). Along the same lines, exposure of HK-2 cells to Ni (160–480 µM for 12–72 h) increased ROS, apoptosis, and DNA damage, which were prevented by pretreatment with N-acetylcysteine (Wang et al. 2012a).
Co and Ni caused EPO production in dog and rat kidneys after acute (4 h for Co) or chronic exposure (3 weeks for Ni), respectively (Fisher and Langston 1968; Hopfer et al. 1984). At the cellular level, most mechanistic studies were performed on the human hepatoma cell line Hep3B, which regulates its production of EPO in a physiologic manner. Other studies mainly used respiratory human non-cancerous or cancerous cell lines because of the entry route of Co and Ni by inhalation (ATSDR 2004, 2005). No studies have been performed on kidney cell lines so far. Therefore, it is difficult to estimate whether the observations made in liver and respiratory cells are valid for the kidney. Historically, evidence for a mechanism common to hypoxia, Co, and Ni was obtained by demonstrating increased EPO production in Hep3B cells (Goldberg et al. 1988). Inhibition of EPO production at low partial pressures of O2 by carbon monoxide indicated that a heme protein is integrally involved in the O2-sensing mechanism. Moreover, when heme synthesis was blocked, hypoxia-, Co- and Ni-induced EPO production were all inhibited. The authors concluded that EPO is induced by hypoxia or Fe depletion and that its induction by the TMs Co or Ni results from substitution of the Fe atom in an “O2 sensor” (Goldberg et al. 1988). The most likely explanation at that time (which turned out to be inaccurate) was that the O2 sensor is a heme protein in which Co and Ni can substitute for Fe in the porphyrin ring, whereby Fe-protoporphyrin would bind O2 with high affinity, Co-protoporphyrin with low affinity and Ni-protoporphyrin not at all (reviewed in Huang et al. 1997). EPO transcription is now known to be under the control of HIF proteins (Semenza and Wang 1992). In response to 75 µM CoCl2, HIF1A and HIF1B mRNAs of Hep3B cells peaked at 4 h, declined at 8 h, and increased again at 16 h (Wang et al. 1995). Exposure of human osteosarcoma (HOS) cells and a Ni-transformed derivative, SA-8, as well as MCF-7 and A549 human cancer cells to Co (0.2 mM) or Ni (1 mM) for 6–24 h has also been shown to induce HIF1A (Salnikow et al. 1999).
Maxwell and Salnikow (2004) have reviewed various mechanisms that could underlie “chemical hypoxia” associated induction of HIF-alpha elicited by Co and Ni. One means by which Co and Ni may activate HIF-alphas was that they substitute for Fe in regulatory HIF dioxygenases, the PHDs (see Epstein et al. 2001; Ivan et al. 2001; Jaakkola et al. 2001 and “HIFs/PHDs: isoforms, regulation, tissue specific expression, mechanisms in the kidney”), and this substitution inactivates the enzymes (see above), as already demonstrated with phthalate dioxygenase (see above and Batie et al. 1987). Another mechanism was proposed following in vitro experiments demonstrating that Co binds with high affinity to HIF2A via sites within the ODD domain in an O2-dependent manner and that the Co binding site overlaps with the VHL-binding site of HIF. Hence, Co and other TMs may disrupt the interaction between VHL protein and HIFs by directly binding to hydroxylated HIFs in vivo (Yuan et al. 2001). An alternative explanation to direct substitution for Fe in the regulatory dioxygenases was that Ni or (less strongly) Co could bind more tightly than Fe2+ to the membrane transporter SLC11A2/DMT-1 (Gunshin et al. 1997; Illing et al. 2012) and suppress delivery of ferrous iron into cells. Another option was that Co and Ni produce increased ROS levels in cells (see “Transition metals: environmental presence, exposure, general modes of toxicity in the kidney” and Fig. 1), which may stabilize HIF-alpha because (mitochondrial) ROS play an important role in HIF-alpha stabilization (see “Contribution of mitochondrial ROS to HPHE signaling”; reviewed in Lee et al. 2020). Oxidative stress would either inactivate the PHDs directly or indirectly by AA depletion, which is necessary for optimal function of the enzymes. Alternatively, direct interaction of metals with AA could prevent entry of AA into cells resulting in depletion of intracellular AA (reviewed in Maxwell and Salnikow 2004).
Mechanisms of PHD inhibition by Co and Ni
Most publications investigating the role of Co and Ni in HPHE signaling were performed in human airway normal or carcinoma cell lines, although the conclusions drawn from these studies may not be applicable to the kidney, but the main results are summarized as follows. Undoubtedly, Co and Ni are good inducers of HIF-alpha, and ROS are produced during the exposure of cells to these TMs. However, Salnikow and colleagues concluded that the formation of ROS is not involved in HIF stabilization or the activation of HIF1-dependent genes (Andrew et al. 2001; Salnikow et al. 2000a, b) (in contrast, see “Contribution of mitochondrial ROS to HPHE signaling” and Chandel et al. 1998 for different results obtained in Hep3B cells). One weakness of all these studies is that ROS formation was assayed using dichlorodihydrofluorescein diacetate, which cannot be reliably used to measure intracellular H2O2 and other ROS (see Dikalov and Harrison 2014). Hence, Salnikow et al. proposed that in addition to low oxygenation, Co, Ni (and possibly other TMs) deplete intracellular AA by excessive oxidation or insufficient supply of AA (due to inhibition of the AA transporter SVCT2) causing inhibition of PHDs, which are dependent on AA (Kaczmarek et al. 2007; Karaczyn et al. 2006; Salnikow et al. 2004). The role of AA is to reduce PHD enzyme-bound Fe, which is important for maintaining hydroxylase activity (Kaczmarek et al. 2009). The latter study indicated that the energy required for Fe substitution by Ni or Co in the enzyme is too high to be achieved in a biological system, thus contradicting the model favoring Fe replacement. Additionally, computer modeling identified a tridentate coordination of AA with the enzyme-bound Fe, which would explain the specific demand for AA as the Fe reductant. Thus, these data supported the hypothesis of Fe oxidation in the hydroxylases following exposure to TM ions (Kaczmarek et al. 2009). In contrast, using airway epithelial cell models as well the group of Costa and colleagues provided evidence that Ni and Co replace Fe in the hydroxylases and interfere with Fe uptake (Chen et al. 2005; Davidson et al. 2005, 2006; Li et al. 2006). Costa and coworkers (Davidson et al. 2006) demonstrated that Ni stabilizes HIF1A and decreases VHL binding to the ODD domain of HIF1A. Furthermore, Ni inhibited cellular PHDs as well as purified PHD2 in vitro through direct interference with the enzyme. Through theoretical calculations, the authors demonstrated that Ni may be able to replace the Fe in the active site of this enzyme. Hence, in contrast to the work of Salnikow et al. (Kaczmarek et al. 2009, 2007; Karaczyn et al. 2006; Salnikow et al. 2004) these authors concluded that Ni can interfere with PHDs directly and does not inhibit the enzyme by depleting cellular factors, such as Fe or AA (Davidson et al. 2006).
In summary, chronic Ni accumulation in the kidney may disrupt renal HPHE signaling axis by causing cell death and fibrosis, whereas Co may be beneficial by attenuating kidney damage induced by various forms of renal insults, which is consistent with the hypothesis of protective hypoxic "preconditioning". Whereas various animal studies have investigated Co effects on HPHE signaling, no such studies have been published for Ni (see Table 2 for a summary). At the cellular level, no studies have been performed on renal cell lines. Strikingly, there are few experimental data showing Co/Ni dependent stabilization of HIF2A. The underlying mechanism of HIF-alpha stabilization remains a matter of debate and may involve iron substitution of PHDs, which inactivates the enzymes, disruption of the interaction between VHL protein and HIFs by directly binding to the ODD domain of HIFs, or ROS formation with consequent AA depletion and subsequent iron oxidation (see Fig. 1). Considering the current literature, it is difficult to explain why Ni should be more nephrotoxic than Co only on the base of their interaction with the (nephroprotective) renal HPHE signaling axis. Hence other factors will have to be considered, including the distribution of putative transporters for Co and Ni in different nephron segments, the affinities of Co and Ni to those transporters, their role in iron depletion or displacement, their impact on the antioxidative status of tubule cells, etc., and all these factors may account for differential Co and Ni nephrotoxicities.
Cadmium (Cd)
Exposure and nephrotoxicity
Contamination of the environment by Cd may occur through anthropogenic and natural sources (WHO 1992, 2010). Food and drinking water are the main routes of exposure to Cd for the nonsmoking general population. Although the efficiency of Cd absorption through inhalation (25–50%) is much higher than that through ingestion (1–10%), concerns about airborne exposure is limited to special populations, including smokers, people living near smelters, and metal-processing workers. In contrast, dietary Cd intake is an important public health issue for the general world population despite the lower bioavailability of Cd through the gastrointestinal tract. The real challenge setting seems to be the chronic (i.e., over decades or even throughout life) low (i.e., in concentrations barely exceeding the “natural” environmental Cd concentrations) Cd exposure (CLCE) from dietary sources and cigarette smoking. Modern agriculture globally uses Cd-containing phosphate fertilizers to increase the efficiency of harvests. Plants, including tobacco, accumulate Cd, which is passed on to animals and man in the food chain (reviewed in Moulis and Thévenod 2010; Thévenod and Lee 2013). Cd in tobacco smoke takes a share in the development of smoking-associated chronic ailments, such as cardiovascular diseases or diabetes mellitus. Moreover, Cd is a class I carcinogen because it interacts indirectly with DNA consequent to elevated ROS levels, interferes with major DNA repair systems, as well as inactivates tumor suppressor functions by targeting proteins with Zn-binding structures. This may cause genomic instability and promote tumor initiation and progression. Cd is stored in various organs, and particularly in the kidney, with a half-life of several decades. Hence, CLCE damages multiple organs in humans and other mammalian organisms by causing nephrotoxicity, osteoporosis, neurotoxicity, genotoxicity, teratogenicity, or endocrine and reproductive defects (Thévenod 2003, 2009; Thévenod and Lee 2013).
Disruption of renal EPO production by Cd
An early study investigated nephrotoxicity and anemia in rats exposed to Cd (0.05 or 0.5 mg/kg b.w.) by intravenous application for 50 weeks (Hiratsuka et al. 1996). In the high Cd group, renal tubular disorders became marked at 16 weeks and cortical fibrosis with glomerular dysfunction appeared at 50 weeks. Anemia occurred at 12 weeks and worsened over time. Early on, EPO levels increased as the hemoglobin level decreased, indicating intact renal HPHE signaling axis. In contrast, EPO levels were not elevated at 50 weeks despite marked iron deficiency anemia, indicating that chronic renal damage and fibrosis disrupted the renal HPHE signaling axis (Hiratsuka et al. 1996). Concomitantly, another group confirmed these data (Horiguchi et al. 1996). Rats were exposed to 2.0 mg Cd/kg b.w. by subcutaneous injection once a week for 6 or 9 months and showed anemia with low levels of plasma EPO over time along with hypoinduction of Epo mRNA in the kidneys, which were accompanied by biochemical and histological renal tubular damage. The results indicated that chronic Cd-intoxication causes anemia by disturbing the EPO production capacity of renal cells (Horiguchi et al. 1996). In a subsequent study, the same group investigated the local relationship between hypoxia-induced EPO production and renal tubular injury in control rats and rats injected with Cd at 2 mg/kg twice a week for 8 months (Horiguchi et al. 2006). Anemia due to insufficient production of EPO was observed in chronic Cd-intoxication of rats. In situ hybridization detected Epo mRNA expression in proximal tubule (PT) cells of hypoxic control rats, but Cd-intoxicated rats showed atrophy of EPO expressing PT and tissue fibrosis (similar effects were observed a single dose of cisplatin at 8 mg/kg, which targets PT). The authors concluded that Cd (and cisplatin) induces anemia through direct damage of PT cells that mainly contribute to renal EPO production (peritubular fibroblasts-like interstitial cells did not express Epo mRNA in this study; however, see Nagai et al. 2014 for a reevaluation of EPO production by the nephron). Using the same Cd-intoxication protocol, these authors also investigated the role of Cd-induced hemolysis on renal damage in rats (Horiguchi et al. 2011). Renal iron overload was observed after 3 months of Cd application and was accompanied by increased urinary levels of NAG, glucose, transferrin, and hemoglobin that are all indicative of PT damage. Furthermore, increased renal expression of Il6 indicated increased inflammation of damaged renal tissue. The authors concluded that in addition to direct damaging effects of Cd on PT, Cd-induced hemolysis leads to Fe accumulation in the kidneys with resulting oxidative stress and cell death (see also “Iron (Fe)”), which aggravates renal damage and decreases EPO production, e.g. by destruction of EPO-producing cells (Horiguchi et al. 2011).
Roles of Cd in HPHE signaling
Cell culture studies have been performed in an attempt to determine mechanistically the interference of Cd with HPHE signaling (Chun et al. 2000; Gao et al. 2013; Horiguchi et al. 2000; Jing et al. 2012; Li et al. 2006; Obara et al. 2003), yet no study with renal cell lines has been published so far, which limits the conclusions drawn from these studies for the kidney (see also “Cobalt (Co) and nickel (Ni)”). Moreover, it is difficult to estimate whether the acute effects of exposure to submicromolar or low micromolar concentrations of Cd for up to 24 h in cell lines can be extrapolated to the more chronic effects observed after systemic application of Cd for weeks or months in vivo. Most importantly, only recently has an in vivo study determined the impact of chronic Cd exposure on several components of the renal HPHE signaling axis (Jacobo-Estrada et al. 2018) and was able to provide information at the same biochemical level as the cell culture studies. In most studies with Cd cited above, hypoxia was used a stimulus to activate HPHE signaling (Chun et al. 2000; Horiguchi et al. 2000; Obara et al. 2003), but some investigators triggered “chemical hypoxia” with Co (Gao et al. 2013; Horiguchi et al. 2000) or determined the effect of Cd under normoxic conditions (Jing et al. 2012; Li et al. 2006). Several cell culture studies with the human hepatoma cell line, Hep3B, found that Cd at concentrations and exposure times that do not affect cell viability (0.5–50 µM for 16–24 h) inhibits HIF1A DNA-binding activity (Chun et al. 2000; Horiguchi et al. 2000; Obara et al. 2003). This observation is relevant for the kidney because inhibition of HIF1A DNA-binding was recently confirmed in vivo in embryonic kidneys after exposure of pregnant rats to Cd (by inhalation for 2 h/day to a mist of a solution of 1 mg CdCl2/ml from gestational day 8–20) and removal of embryonic kidneys at day 21 (Jacobo-Estrada et al. 2018). Accordingly, expression of hypoxia-inducible target genes, such as Epo or Vegfa, or hypoxia-induced luciferase reporter gene activity were reduced by Cd (Chun et al. 2000; Horiguchi et al. 2000; Jacobo-Estrada et al. 2018; Obara et al. 2003). However, the effect of Cd on HIF1A mRNA and HIF1A protein is less consistent in the literature. Chun et al. (2000) observed no change of HIF1A mRNA but decreased HIF1A protein, which they attributed to Cd stimulation of proteasomal activity, whereas Jacobo-Estrada et al. (2018) did not observe any effect of Cd on mRNA or protein levels of HIF1A and PHD2. Interestingly, using a recombinant catalytic domain of human PHD2 and non-denaturing ionization electrospray mass spectrometry, Mecinovic et al. have demonstrated Cd binding to the apo-enzyme at the catalytic site plus at a novel binding site (as well as with Zn, Cu and Co, but not Fe or Ni) (Mecinovic et al. 2008). Cd could partially displace Fe from the PHD2 active site, suggesting Cd inhibition of PHD2. These in vitro data are in contrast to the above in vivo and cellular studies where Cd showed no increased HIF-alpha stabilization as well as to studies proposing AA depletion as the sole mechanism underlying PHD2 inhibition and excluding Fe displacement by other TMs (see “Cobalt (Co) and nickel (Ni)”; reviewed in Maxwell and Salnikow 2004).
Because Cd intake by organisms occurs through inhalation in addition to the oral route, Cd effects on HPHE signaling were also studied in airway epithelial cell lines. However, the results were variable, possibly because of differences in Cd concentrations, exposure times and cell lines used. Exposure of chemically hypoxic (Co treatment) rat lung fibroblasts (RFL6) to Cd resulted in inhibition of HIF1A DNA binding to the hypoxia-inducible gene lysyl oxidase Lox and reduced Hif1a mRNA expression (Gao et al. 2013). Other studies showed either no effect of Cd on HIF1A expression in normoxic cells (Li et al. 2006) or increased HIF1A and VEGFA expression under normoxia (Jing et al. 2012).
In summary, in renal tissue Cd may disrupt the renal HPHE signaling axis by interfering with HIF1A binding to DNA of hypoxia-inducible target genes and/or decrease Hif1a mRNA and protein expression, resulting in decreased renal EPO production and ensuing renal anemia, which may aggravate Cd-induced renal injury by disrupting nephroprotective HPHE signaling. To the best of our knowledge, no study has addressed the effect of Cd on (renal) HIF2A expression. Table 2 summarizes data from animal studies.
Chromium (Cr)
Exposure and nephrotoxicity
Hexavalent Cr (Cr6+) is often found in occupational settings as a by-product of various industrial processes, such as leather working, smelting, welding, and metal plating (ATSDR 2012). Cr6+ is also found in automobile exhaust and in tobacco products, such as traditional and electronic cigarettes and hookahs, (Williams et al. 2017) and is, therefore, a human respiratory carcinogen, and produces a variety of toxic effects through inhalation (ATSDR 2012). Systemic toxicity attributable to Cr6+ has been documented in the respiratory and pulmonary system, gastrointestinal tract, dermis, and renal system (ATSDR 2012). Contact dermatitis is frequently documented following exposure to chromate and dichromate (Lejding et al. 2018). Exposure to Cr6+ can also cause acute tubular necrosis, which is localized to the proximal convoluted tubules and may result in rapid onset of renal failure (Wedeen and Qian 1991). In addition, multiple mechanisms of Cr6+ carcinogenesis have been proposed involving oxidative stress, DNA damage and genomic instability, and epigenetic modulation (reviewed in Chen et al. 2019b). Cr6+ compounds, such as chromate and dichromate, are strong oxidizing agents at low or neutral pH. The product of Cr6+ reduction, Cr3+, is rather non-toxic and has even been considered an essential nutrient in humans for insulin, sugar, and lipid metabolism.
The mechanisms of Cr-associated nephrotoxicity are only partly elucidated. In rats, Cr selectively accumulates in the renal cortex at 6–20 times the level present in red blood cells or the liver (Weber 1983). Cr may induce nephrotoxicity in humans. However, tubular damage following occupational exposure is mostly due to acute absorption and transient in nature (Franchini and Mutti 1988; Sharma et al. 1978). Experimental studies in rats support acute Cr nephrotoxicity caused by oxidative stress (Gumbleton and Nicholls 1988; Ngaha 1981; Patlolla et al. 2009; Pedraza-Chaverri et al. 2005; Seiken et al. 1994; Zhou et al. 2008). The involvement of different nephron segments in Cr-induced oxidative stress and toxicity was assessed in AKI induced by K2Cr2O7 (Arreola-Mendoza et al. 2006). Rats received K2Cr2O7 (a single dose of 15 mg/kg, s.c.). Altered PT function, decreased glomerular filtration, and distal segment dysfunction were accompanied by oxidative damage 48 h after exposure to Cr. In α-tocopherol-treated animals (125 mg/kg by gavage 5 days before and during Cr exposure) proximal reabsorptive and secretory functions were preserved, implying that oxidative damage contributes to Cr toxicity. In contrast, glomerular or distal dysfunction were not prevented by α-tocopherol, suggesting nephron segment specificity of Cr-induced oxidative stress.
Chronic Cr nephrotoxicity has also been described in humans and experimental animals. Nuyts et al. (1995) observed that occupational chromium exposure increases the risk of chronic renal failure by about threefold in industrial areas, which was confirmed in subsequent studies (Tsai et al. 2017; Wang et al. 2011). Animal experiments are consistent with these observations and indicate chronic Cr nephrotoxicity induced by oxidative stress (Soudani et al. 2010). In another study, K2Cr2O7 administrated to female rats during late pregnancy and early postnatal periods provoked kidney damage mediated by oxidative stress in dams and their offspring (Soudani et al. 2011). Malondialdehyde (an indicator of lipid peroxidation), GSH and NO levels increased in kidneys of Cr-treated mothers and their suckling pups. Activities of SOD, CAT and glutathione peroxidase were increased in dams and decreased in their pups. Significant decrease in creatinine clearance was also found in treated mothers and in their progeny.
At the cellular level, a few studies have investigated mechanisms of acute Cr toxicity in cultured renal cells. Dartsch et al. (1998) compared opossum kidney and HepG2 liver cells because acute Cr nephrotoxicity is known to be more prominent than hepatotoxicity in vivo. Cr6+ (0.01 µM to 1 mM for 24 h), but not Cr3+, had a dose-dependent cytotoxic effect with loss of cell viability (EC50 ~ 5 µM for kidney and ~ 50 µM for liver epithelial cells). Chloride channel blockers did not inhibit cell damage, suggesting that the uptake of Cr6+ did not occur through anion transporters. In another study, the contribution of oxidative damage was investigated in HK-2 cells incubated with 10 μM K2Cr2O7 for 24 h (Lin et al. 2018). Supplementation with AA (30 μg/ml) inhibited damage to HK-2 cells, if incubated within 1–8 h of Cr toxicity by preventing ROS generation, apoptosis, and autophagy, as well as Cr entry into cells, possibly by reduction of Cr6+ to Cr3+.
Mechanisms of Cr6+-induced HIF-alpha stabilization
Salnikow and colleagues tested their hypothesis that TM ions can induce HIF-alpha by depleting intracellular AA via oxidation and by inhibiting AA uptake by cells (Karaczyn et al. 2006; Salnikow et al. 2004). Cr6+ is known to oxidize AA directly (Zhitkovich 2005) and is a principal AA oxidant in rat liver and kidneys (Standeven and Wetterhahn 1991). Salnikow and coworkers (Kaczmarek et al. 2007) compared the accumulation of HIF1A protein in human lung epithelial cells (1HAEo- and A549) following Ni (500 µM) or Cr6+ (5 µM) exposure. The experiments showed that there is a difference in the time course of this accumulation. Thus, HIF1A protein was induced only by Cr6+ at 1–8 h and disappeared after the latter was reduced to Cr3+. This correlated with cell-free experiments showing a rapid phase of AA oxidation by Cr6+, which ends after the completion of Cr6+ reduction to Cr3+. In contrast, extended stabilization of HIF1A was observed following acute exposure to Ni for up to 24 h. Ni was found to be a catalyst, which facilitates continuous oxidation of AA by ambient O2. A HIF1A-dependent reporter assay revealed that 20–24 h was required to fully develop HIF1 transcriptional response and the acute exposure to Ni, but not Cr, met this requirement. However, repeated (chronic) exposure to Cr6+ also led to extended stabilization of HIF1A. Thus, these data emphasized the important role of AA in regulation of HIF1 transcriptional activity in metal-exposed human lung cells. In a very recent study, another aspect of the regulation of HPHE signaling was investigated in HepG2 cells exposed to Cr (Nishimura et al. 2021). At variance with the report by Kaczmarek et al. (2007), the authors reported that Cr3+ (100 µM for 24 h) increases HIF1A protein, EPO mRNA expression and EPO protein levels in HepG2 cells. The effect of Cr on EPO production was abolished by co-incubation with the inhibitor of proliferator-activated receptor γ (PPARγ), SR-202 (100 µM), suggesting a different mode of action of Cr3+ on HIF1A protein levels that was mediated by PPARγ induction of HIF1A, and which is supported by other studies (Tsave et al. 2016; Urakami-Takebayashi et al. 2018; Zhou et al. 2009).
Epigenetic mechanisms in HPHE signaling affected by Cr6+
Hypoxia-induced HIF1A stabilization is known to regulate epigenetic mechanisms, e.g. by promoting histone demethylation (Beyer et al. 2008; Krieg et al. 2010; Pollard et al. 2008; Wellmann et al. 2008; Xia et al. 2009), which may act as signal amplifiers to facilitate hypoxic gene expression and ultimately enhance tumor growth (reviewed in Hancock et al. 2015; Watson et al. 2010). Costa and coworkers (reviewed in Chervona et al. 2012) proposed that Cr (as well as Ni) may induce post-translational histone modifications and affect the enzymes that modulate them, i.e., members of the Fe- and 2-oxoglutarate-dependent dioxygenase family, including PHD2, histone demethylases JHDM2A/JMJD1A, and DNA repair enzymes ABH3 and ABH2, as well as histone methyltransferases, G9a. Hence, given the effects that these metals may exert on the epigenome, their involvement in the dynamics of histone modifying enzymes could also account for their respective toxicities and carcinogenicities.
In summary, there is ample evidence that Cr6+ causes acute and chronic nephrotoxicity. Strikingly, no studies are available studying the impact of Cr6+ on HPHE signaling in the kidney or kidney cell lines. As a “chemical hypoxia mimetic” Cr6+ is a HIF1A stabilizer, but the underlying mechanism is not clear because only few studies have investigated this process, although it may not displace Fe from PHDs. The role of Cr6+ on other aspects of HPHE signaling (e.g. on HIF2A) and on nephroprotection remains to be investigated.
Platinum (Pt)
Exposure and nephrotoxicity
Platinum is mainly used in the automobile industry for autocatalysts, sensors and spark plugs. Other uses in industrial applications, such as in electronics, and jewelry make up the worldwide demand for this metal (Rauch and Morrison 2008). Interactions of platinum compounds with biological systems have been well reported in the literature. It was first observed that bacterial, viral and fungal pathogens could be rendered nonviable by platinum metals (Cochran and Maassab 1970; LeRoy 1975; Rosenberg et al. 1967; Shulman and Dwyer 1964). Based on these observations, platinum compounds were tested in leukemia and sarcoma cancers, which were implanted into mice. Of the four metal-amine complexes reported in the study, cis-Pt(II)(NH3)2Cl2, commonly known as cisplatin or Peyrone’s salt, was the most effective in reducing sarcoma tumor mass (by > 95%) or prolonging mean survival time (by > 80%) in the leukemic mouse model (Rosenberg et al. 1969). Moreover, the mice remained cancer-free for six months post-treatment. By 1972, phase I clinical trials with cisplatin were completed and in 1978, it was approved by the FDA for treatment of testicular, ovarian and bladder cancer. Cisplatin therapy is tied with toxicities predominantly in the kidney, intestinal tract and ear (Barabas et al. 2008; Karasawa and Steyger 2015; Qi et al. 2019), due to uptake of the drug by membrane transporters (Ciarimboli 2014), in particular organic cation transporters (Ciarimboli et al. 2005; Zhang et al. 2006), which are prevalent in these tissues (Lee et al. 2009). Nephrotoxicity seems to be the limiting factor in therapeutic dosing of cisplatin. Despite this and development of drug resistance, cisplatin is widely used to treat a number of solid tumors. A number of less toxic analogs have since been developed, including carboplatin and oxaliplatin (Sharma et al. 2022).
Cisplatin’s square-planar configuration predominantly undergoes nucleophilic substitution and, therefore, permitting its activation by displacement of the chloride ions by water molecules. Hydrolyzation of cisplatin results in a potent electrophile, which reacts with sulfhydryl groups on proteins and nitrogen donor atoms on nucleic acids (Dasari and Tchounwou 2014; Sharma et al. 2022). Hence, cisplatin’s antitumor action is not dependent on redox reactions of platinum itself. Rather, it attacks several intracellular targets, such as deoxyribonuclease I (Basnakian et al. 2005), involved in DNA and protein syntheses as well as intercalating into DNA, reducing Na+/K+-ATPase activity and total GSH levels (Courjault et al. 1993) to render its cytostatic actions. Of note, these aforementioned mechanisms appear to be specific to the antitumor action of cisplatin whereas the toxic effects are primarily consequent of oxidative stress due to mitochondrial dysfunction.
Role of cisplatin and HPHE signaling in renal injury
Hypoxia and HIFs have been implicated in both cisplatin-induced injury and advancement of drug resistance in the kidney (reviewed in Li et al. 2021). Following cellular uptake, cisplatin elicits an inflammatory response as well as mitochondrial dysfunction and decreasing antioxidative capacity, which culminates in increased levels of ROS and renal cell injury (reviewed in Holditch et al. 2019; McSweeney et al. 2021; Volarevic et al. 2019). The exact role of hypoxia and HIFs in renal damage elicited by cisplatin is hampered by conflicting data. In immortalized PTCs, hypoxia protected against cisplatin-induced apoptosis by attenuating Bax accumulation and cytochrome c release yet was independent of the activated HIF1A (Wang et al. 2006). Rather, the authors postulated a role for p53 activation by cisplatin that is suppressed by hypoxia, and inhibition of the mETC (Wang et al. 2006). Conversely, cisplatin-induced apoptosis was augmented by hypoxia in a cell line resembling principal cells of the renal collecting duct (Schwerdt et al. 2005). Cisplatin concentrations up to 30 µM did not induce proapoptotic caspase-3 activity whereas co-incubation with mETC inhibitors activated caspase-3 by up to 20-fold over controls and initiated DNA laddering. Inhibition of the mETC (to mimic hypoxic conditions), but not cisplatin per se, increased lactic acid thereby reducing intracellular pH and potentiated caspase-3 activation. Furthermore, caspase-3 activity was elevated by > twofold in cisplatin-treated cells under severe hypoxic conditions. These conflicting studies could be explained by the cell models (proximal tubule versus collecting duct) used wherein the HIF turnover is regulated differently based on the O2 tension. Due to physiological low O2 tension in the renal medulla, wherein collecting ducts are located, higher expression levels of PHD2 and PHD3 were detected (Schodel et al. 2009), presumably to maintain low HIF levels. Cisplatin treatment in Sprague–Dawley rats depreciated total PHD2 and PHD3 expression in parallel with increased blood creatinine, though in the absence of HIF induction, indicating HIFs are not essential to renal injury progression, which has been confirmed in resistant cell clones following repeated episodes of hypoxia (Brooks et al. 2007). Thus, whether hypoxia protects against or exacerbates cisplatin nephrotoxicity depends on the origin of the renal cells and does not seem to involve HPHE signaling.
The activation of HIFs following cisplatin is also unresolved. Tanaka et al. (2005a) observed increased HIF expression in the outer medulla three days after cisplatin administration to rats and Zhao et al. (2021) detected HIF1A in mouse renal tubules and primary and immortalized PTCs post-cisplatin treatment whereas Weidemann et al. (2008) did not find increased HIF1A in immortalized PTCs or rat renal tubules after exposure to cisplatin. Only hypoxic preconditioning with 0.1% carbon monoxide (without cisplatin) led to marked increase in HIF1A, in particular in the outer medulla (Weidemann et al. 2008), coincidently as observed by Tanaka et al. (2005a). In both studies, elevated HIF1A protected against cisplatin-induced tubular cell apoptosis and improved renal function compared to without hypoxic preconditioning (Weidemann et al. 2008) or expression of dominant negative HIF1A (Tanaka et al. 2005a).
HIF-alpha protection against cisplatin-induced renal injury
In contrast to the role of HIFs in cisplatin-induced injury, there is a general consensus that HIFs play a key role in conferring drug resistance through expression of drug transporters, such as ABCB1, detoxification of cisplatin, increased antioxidative capacity, disruption of apoptosis signaling or strengthened ability to repair DNA damage (reviewed in Belisario et al. 2020; Kim and Lee 2017; Shenoy et al. 2017). Indeed, it has been postulated that low HIF expression is decisive in determining the chemosensitivity of testicular cancer (Shenoy et al. 2017) and induction of pseudohypoxia in renal carcinoma cells culminates in augmented chemoresistance and cell motility, which is signaled through HIF-alpha (Liu et al. 2017). In agreement with the notion that HIF upregulation confers protection against cisplatin injury, enhanced HIF1A-induced mitochondrial autophagy (Li et al. 2020), increased HO-1 expression (Bolisetty et al. 2016; Shiraishi et al. 2000) and increased HO-1 activity induced by hemin (Al-Kahtani et al. 2014; Schaaf et al. 2002; Shiraishi et al. 2000) ameliorated oxidative stress, attenuated cisplatin-induced apoptosis of PTCs and increased antioxidative capacity, respectively, in both cell culture and animal models. Co-culture of genetically-engineered HIF1A-expressing stem cells with renal cells (Wang et al. 2014, 2015), administration of conditioned medium from mouse Hmox1+/+ stem cells (Zarjou et al. 2011) or implantation of EPO-secreting stromal cells (Eliopoulos et al. 2011) to cisplatin-treated mice reduced cisplatin-induced apoptosis and renal injury in a paracrine manner. Furthermore, infusion of rat fetal kidney stem cells ameliorates cisplatin-induced apoptosis with concomitant HIF1A, VEGFA and NOS upregulation, culminating in increased capillary density (Gupta et al. 2015). Finally, recombinant EPO conferred renoprotection against cisplatin-induced injury (Kong et al. 2013; Mohamed et al. 2013; Rjiba-Touati et al. 2011; Zafirov et al. 2008).
In summary, HIFs protect against cisplatin-induced injury to renal cells and fosters development of drug resistance whereas cisplatin toxicity is not HIF-dependent (Table 2). The role of hypoxia in cisplatin nephrotoxicity remains unresolved.
Therapy of TM nephrotoxicity, controversies, outlook, and conclusions
Chelation of TMs is an obvious therapeutic strategy to prevent further damage, once they have entered the body (reviewed in Flora and Pachauri 2010; Smith 2013). Another important aspect of acute and chronic TM nephrotoxicity is unregulated and overwhelming formation of free radicals (see “Transition metals: environmental presence, exposure, general modes of toxicity in the kidney”), which disrupt physiological ROS signaling, including mitochondrial ROS-dependent activation of the HPHE pathway (see “Contribution of mitochondrial ROS to HPHE signaling” and Fig. 1). In addition, ROS cause detrimental effects to cells through oxidation of proteins, lipids and nucleic acids, which results in cell damage and death. Consequently, application of natural or synthetic antioxidants may be beneficial in the prevention and attenuation of TM-induced renal damage (Flora 2009; Forman and Zhang 2021; Halliwell 2011).
Modulation of the different components of HPHE signaling may also be advocated using pharmacological PHD inhibitors (PHIs) to induce endogenous defense mechanisms, as described with “hypoxic preconditioning” for various causes of AKI and CKD, including TMs (reviewed in Shu et al. 2019; Tanaka 2016; Tiwari and Kapitsinou 2021); see also “General considerations” in “Impact of toxic metal ions on the renal HPHE signaling axis”). PHIs are specific drugs that activate HPHE signaling and were developed to stimulate the production of endogenous EPO in anemia of CKD (Haase 2021). Three oral PHIs (daprodustat, vadadustat, and roxadustat) have advanced to global phase III clinical development (see Table 3 for an overview). On the plus side, PHIs could be useful in the prophylaxis or treatment of TM nephrotoxicity by affecting different mechanisms regulated by HPHE signaling (provided the HPHE axis is intact; see above). They may reduce cell death by inducing HIF activation (Bernhardt et al. 2006a; Conde et al. 2012; Tanaka et al. 2005a; Weidemann et al. 2008), increase HIF-dependent EPO production (for review see Moore and Bellomo 2011), and promote tissue regeneration by HIF activation (Ceradini et al. 2004).
However, regarding therapy of acute versus chronic TM-induced nephrotoxicity a more differentiated approach and a note of caution are necessary. In TM-induced AKI, the literature suggests that PHIs could be useful to increase survival and nephroprotection (reviewed in Li et al. 2021; Shu et al. 2019; Tiwari and Kapitsinou 2021). In contrast, therapeutic application of PHIs for CKD induced by TM ions may be a double-edged sword, first because of the putative proapoptotic (reviewed in Mazure and Pouyssegur 2010; Piret et al. 2002) and pro-fibrotic potential of HPHE activation, due to the interdependence between inflammatory (e.g. nuclear factor kappa B) and HPHE signaling pathways (reviewed in Faivre et al. 2021; Imtiyaz and Simon 2010; Taylor et al. 2016). Second, HIFs induce the expression of proteins involved in Fe/TM transport and homeostasis (e.g. DCYTB, SLC11A2, TF, TFRC, FTH1, HAMP1, MT1A) (reviewed in Shah and Xie 2014; Simpson and McKie 2015). Hence, activation of these two processes could exacerbate chronic renal damage induced by TMs (see also “Transition metals: environmental presence, exposure, general modes of toxicity in the kidney” and “Impact of toxic metal ions on the renal HPHE signaling axis”). Moreover, the carcinogenic potential of metal ions, such as Cd, in the kidney (Hartwig 2013), may be accelerated by activation of HPHE signaling, as exemplified by the proposed role of HIF2A in promoting clear cell renal cell carcinoma (Choueiri et al. 2021; Dufies et al. 2021). On the other hand, PHIs could be beneficial in advanced kidney cancer (reviewed in Burrows and Maxwell 2021). When carefully balancing pros and cons, at the present stage the use of PHIs in humans to slow down or revert CKD induced by nephrotoxic TM ions does not seem recommendable and awaits further developments in the field.
To conclude, acute exposure to relatively high concentrations of TM ions leads to cell death primarily associated with either apoptosis or necrosis, with the latter triggering inflammation and fibrosis. Consequently, these endpoints of acute renal damage may promote disruption of the HPHE pathway, preventing a protective response to damage. Indeed, acute (and chronic) exposure to TMs, such as Fe, Ni, Cd, Cr6+ or Pt has been shown to damage the kidney and disrupt HPHE signaling (see “Impact of toxic metal ions on the renal HPHE signaling axis” and Table 2). On the other hand, exposure to low TM ion concentrations may trigger activation of renal HPHE signaling, e.g. by mimicking hypoxia, but also depends on the exposure time. Indeed, Co as well as Fe and Ni (at doses that are not nephrotoxic) do induce HPHE signaling and protect against kidney damage (“preconditioning”; see “General considerations” in “Impact of toxic metal ions on the renal HPHE signaling axis”). Hence, activation of the renal HPHE signaling axis (e.g., by application of pharmacological PHIs under specific circumstances) may delay the onset of acute or chronic renal dysfunction induced by TMs. Strikingly, current knowledge of the field is still very sketchy. For instance, for Ni and Cr6+, so far no animal studies on renal HPHE signaling have been performed. Further, for Fe, Ni, Cd and Cr6+ no studies investigating HPHE signaling are available in renal cell lines. Clearly, mechanistic elucidation of the role of renal hypoxia–HIF–PHD–EPO signaling in TM nephrotoxicity is necessary before contemplating a rational approach to prevention or therapy.
References
Adams L, Franco MC, Estevez AG (2015) Reactive nitrogen species in cellular signaling. Exp Biol Med (maywood) 240(6):711–717. https://doi.org/10.1177/1535370215581314
Adedoyin O, Boddu R, Traylor A et al (2018) Heme oxygenase-1 mitigates ferroptosis in renal proximal tubule cells. Am J Physiol Renal Physiol 314(5):F702–F714. https://doi.org/10.1152/ajprenal.00044.2017
Agarwal A, Nick HS (2000) Renal response to tissue injury: lessons from heme oxygenase-1 GeneAblation and expression. J Am Soc Nephrol 11(5):965–973. https://doi.org/10.1681/ASN.V115965
Ahn JM, You SJ, Lee YM et al (2012) Hypoxia-inducible factor activation protects the kidney from gentamicin-induced acute injury. PLoS ONE 7(11):e48952. https://doi.org/10.1371/journal.pone.0048952
Akizawa T, Tsubakihara Y, Nangaku M et al (2017) Effects of daprodustat, a novel hypoxia-inducible factor prolyl hydroxylase inhibitor on anemia management in Japanese hemodialysis subjects. Am J Nephrol 45(2):127–135. https://doi.org/10.1159/000454818
Alam J, Killeen E, Gong P et al (2003) Heme activates the heme oxygenase-1 gene in renal epithelial cells by stabilizing Nrf2. Am J Physiol Renal Physiol 284(4):F743–F752. https://doi.org/10.1152/ajprenal.00376.2002
Al-Kahtani MA, Abdel-Moneim AM, Elmenshawy OM, El-Kersh MA (2014) Hemin attenuates cisplatin-induced acute renal injury in male rats. Oxid Med Cell Longev 2014:476430. https://doi.org/10.1155/2014/476430
Andrew AS, Klei LR, Barchowsky A (2001) Nickel requires hypoxia-inducible factor-1 alpha, not redox signaling, to induce plasminogen activator inhibitor-1. Am J Physiol Lung Cell Mol Physiol 281(3):L607–L615. https://doi.org/10.1152/ajplung.2001.281.3.L607
Appelhoff RJ, Tian YM, Raval RR et al (2004) Differential function of the prolyl hydroxylases PHD1, PHD2, and PHD3 in the regulation of hypoxia-inducible factor. J Biol Chem 279(37):38458–38465. https://doi.org/10.1074/jbc.M406026200
Aquino-Galvez A, Gonzalez-Avila G, Jimenez-Sanchez LL et al (2019) Dysregulated expression of hypoxia-inducible factors augments myofibroblasts differentiation in idiopathic pulmonary fibrosis. Respir Res 20(1):130. https://doi.org/10.1186/s12931-019-1100-4
Ariazi JL, Duffy KJ, Adams DF et al (2017) Discovery and preclinical characterization of GSK1278863 (daprodustat), a small molecule hypoxia inducible factor-prolyl hydroxylase inhibitor for anemia. J Pharmacol Exp Ther 363(3):336–347. https://doi.org/10.1124/jpet.117.242503
Arreola-Mendoza L, Reyes JL, Melendez E et al (2006) Alpha-tocopherol protects against the renal damage caused by potassium dichromate. Toxicology 218(2–3):237–246. https://doi.org/10.1016/j.tox.2005.11.010
Asada N, Takase M, Nakamura J et al (2011) Dysfunction of fibroblasts of extrarenal origin underlies renal fibrosis and renal anemia in mice. J Clin Invest 121(10):3981–3990. https://doi.org/10.1172/JCI57301
Ashley-Martin J, Iannotti L, Lesorogol C et al (2021) Heavy metal blood concentrations in association with sociocultural characteristics, anthropometry and anemia among Kenyan adolescents. Int J Environ Health Res 2:1–15. https://doi.org/10.1080/09603123.2021.1929871
ATSDR (2004) toxicological profile for cobalt. In: Agency for Toxic Substance and Disease Registry US (ed). Department of Health and Humans Services, Public Health Service, Centers for Disease Control, Atlanta, GA, U.S.A.
ATSDR (2005) toxicological profile for nickel (update). In: Agency for Toxic Substance and Disease Registry US (ed). Department of Health and Humans Services, Public Health Service, Centers for Disease Control, Atlanta, GA, U.S.A.
ATSDR (2012) Toxicological profile for chromium. In: Agency for Toxic Substance and Disease Registry US (ed). Agency for Toxic Substances and Disease Registry (ATSDR) Toxicological Profiles. Department of Health and Humans Services, Public Health Service, Centers for Disease Control, Atlanta, GA, U.S.A.
ATSDR (2019) The ATSDR 2019 substance priority list (SPL). In: Agency for Toxic Substance and Disease Registry US (ed). Department of Health and Humans Services, Public Health Service, Centers for Disease Control, Atlanta, GA, U.S.A., p https://www.atsdr.cdc.gov/spl/index.html
ATSDR (2021) Toxicological profiles. In: Agency for Toxic Substance and Disease Registry US (ed). Department of Health and Humans Services, Public Health Service, Centers for Disease Control, Atlanta, GA, U.S.A., p https://www.atsdr.cdc.gov/toxprofiledocs/index.html
Bachmann S, Le Hir M, Eckardt KU (1993) Co-localization of erythropoietin mRNA and ecto-5’-nucleotidase immunoreactivity in peritubular cells of rat renal cortex indicates that fibroblasts produce erythropoietin. J Histochem Cytochem 41(3):335–341. https://doi.org/10.1177/41.3.8429197
Barabas K, Milner R, Lurie D, Adin C (2008) Cisplatin: a review of toxicities and therapeutic applications. Vet Comp Oncol 6(1):1–18. https://doi.org/10.1111/j.1476-5829.2007.00142.x
Basnakian AG, Apostolov EO, Yin X, Napirei M, Mannherz HG, Shah SV (2005) Cisplatin nephrotoxicity is mediated by deoxyribonuclease I. J Am Soc Nephrol 16(3):697–702. https://doi.org/10.1681/ASN.2004060494
Batie CJ, LaHaie E, Ballou DP (1987) Purification and characterization of phthalate oxygenase and phthalate oxygenase reductase from Pseudomonas cepacia. J Biol Chem 262(4):1510–1518
Bayhan T, Unal S, Cirak E et al (2017) Heavy metal levels in patients with ineffective erythropoiesis. Transfus Apher Sci 56(4):539–543. https://doi.org/10.1016/j.transci.2017.07.021
Belisario DC, Kopecka J, Pasino M et al (2020) hypoxia dictates metabolic rewiring of tumors: implications for chemoresistance. Cells. https://doi.org/10.3390/cells9122598
Bell EL, Emerling BM, Chandel NS (2005) Mitochondrial regulation of oxygen sensing. Mitochondrion 5(5):322–332. https://doi.org/10.1016/j.mito.2005.06.005
Bell EL, Klimova TA, Eisenbart J et al (2007) The Qo site of the mitochondrial complex III is required for the transduction of hypoxic signaling via reactive oxygen species production. J Cell Biol 177(6):1029–1036. https://doi.org/10.1083/jcb.200609074
Bernhardt WM, Campean V, Kany S et al (2006a) Preconditional activation of hypoxia-inducible factors ameliorates ischemic acute renal failure. J Am Soc Nephrol 17(7):1970–1978. https://doi.org/10.1681/ASN.2005121302
Bernhardt WM, Schmitt R, Rosenberger C et al (2006b) Expression of hypoxia-inducible transcription factors in developing human and rat kidneys. Kidney Int 69(1):114–122. https://doi.org/10.1038/sj.ki.5000062
Bernhardt WM, Warnecke C, Willam C, Tanaka T, Wiesener MS, Eckardt KU (2007) Organ protection by hypoxia and hypoxia-inducible factors. Methods Enzymol 435:221–245. https://doi.org/10.1016/S0076-6879(07)35012-X
Berra E, Benizri E, Ginouves A, Volmat V, Roux D, Pouyssegur J (2003) HIF prolyl-hydroxylase 2 is the key oxygen sensor setting low steady-state levels of HIF-1alpha in normoxia. EMBO J 22(16):4082–4090. https://doi.org/10.1093/emboj/cdg392
Besarab A, Provenzano R, Hertel J et al (2015) Randomized placebo-controlled dose-ranging and pharmacodynamics study of roxadustat (FG-4592) to treat anemia in nondialysis-dependent chronic kidney disease (NDD-CKD) patients. Nephrol Dial Transplant 30(10):1665–1673. https://doi.org/10.1093/ndt/gfv302
Beyer S, Kristensen MM, Jensen KS, Johansen JV, Staller P (2008) The histone demethylases JMJD1A and JMJD2B are transcriptional targets of hypoxia-inducible factor HIF. J Biol Chem 283(52):36542–36552. https://doi.org/10.1074/jbc.M804578200
Bleackley MR, Macgillivray RT (2011) Transition metal homeostasis: from yeast to human disease. Biometals 24(5):785–809. https://doi.org/10.1007/s10534-011-9451-4
Bolisetty S, Traylor A, Joseph R, Zarjou A, Agarwal A (2016) Proximal tubule-targeted heme oxygenase-1 in cisplatin-induced acute kidney injury. Am J Physiol Renal Physiol 310(5):F385–F394. https://doi.org/10.1152/ajprenal.00335.2015
Bracken CP, Fedele AO, Linke S et al (2006) Cell-specific regulation of hypoxia-inducible factor (HIF)-1alpha and HIF-2alpha stabilization and transactivation in a graded oxygen environment. J Biol Chem 281(32):22575–22585. https://doi.org/10.1074/jbc.M600288200
Brahimi-Horn C, Mazure N, Pouyssegur J (2005) Signalling via the hypoxia-inducible factor-1alpha requires multiple posttranslational modifications. Cell Signal 17(1):1–9. https://doi.org/10.1016/j.cellsig.2004.04.010
Brauch H, Weirich G, Klein B, Rabstein S, Bolt HM, Bruning T (2004) VHL mutations in renal cell cancer: does occupational exposure to trichloroethylene make a difference? Toxicol Lett 151(1):301–310. https://doi.org/10.1016/j.toxlet.2003.12.074
Bridges CC, Zalups RK (2005) Molecular and ionic mimicry and the transport of toxic metals. Toxicol Appl Pharmacol 204(3):274–308
Bridges CC, Zalups RK (2017) The aging kidney and the nephrotoxic effects of mercury. J Toxicol Environ Health B Crit Rev 20(2):55–80. https://doi.org/10.1080/10937404.2016.1243501
Brigandi RA, Johnson B, Oei C et al (2016) A novel hypoxia-inducible factor-prolyl hydroxylase inhibitor (GSK1278863) for anemia in CKD: a 28-day, phase 2A randomized trial. Am J Kidney Dis 67(6):861–871. https://doi.org/10.1053/j.ajkd.2015.11.021
Briggs KJ, Koivunen P, Cao S et al (2016) Paracrine induction of HIF by glutamate in breast cancer: EglN1 senses cysteine. Cell 166(1):126–139. https://doi.org/10.1016/j.cell.2016.05.042
Brooks C, Wang J, Yang T, Dong Z (2007) Characterization of cell clones isolated from hypoxia-selected renal proximal tubular cells. Am J Physiol Renal Physiol 292(1):F243–F252. https://doi.org/10.1152/ajprenal.00236.2006
Brunelle JK, Bell EL, Quesada NM et al (2005) Oxygen sensing requires mitochondrial ROS but not oxidative phosphorylation. Cell Metab 1(6):409–414. https://doi.org/10.1016/j.cmet.2005.05.002
Bugert P, Wilhelm M, Kovacs G (1997) FHIT gene and the FRA3B region are not involved in the genetics of renal cell carcinomas. Genes Chromosomes Cancer 20(1):9–15
Burrows N, Maxwell PH (2021) Hypoxia-inducible factor 2 inhibitors show promise in advanced kidney cancer. Nat Rev Urol 18(9):516–517. https://doi.org/10.1038/s41585-021-00487-9
Cancer Genome Atlas Research N, Weinstein JN, Collisson EA et al (2013) The Cancer Genome Atlas Pan-Cancer analysis project. Nat Genet 45(10):1113–1120. https://doi.org/10.1038/ng.2764
Carreau A, El Hafny-Rahbi B, Matejuk A, Grillon C, Kieda C (2011) Why is the partial oxygen pressure of human tissues a crucial parameter? Small molecules and hypoxia. J Cell Mol Med 15(6):1239–1253. https://doi.org/10.1111/j.1582-4934.2011.01258.x
Ceradini DJ, Kulkarni AR, Callaghan MJ et al (2004) Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nat Med 10(8):858–864. https://doi.org/10.1038/nm1075
Chandel NS, Maltepe E, Goldwasser E, Mathieu CE, Simon MC, Schumacker PT (1998) Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc Natl Acad Sci USA 95(20):11715–11720. https://doi.org/10.1073/pnas.95.20.11715
Chandel NS, McClintock DS, Feliciano CE et al (2000) Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: a mechanism of O2 sensing. J Biol Chem 275(33):25130–25138. https://doi.org/10.1074/jbc.M001914200
Chemistry IUoPaA (1997) Compendium of chemical terminology, 2nd edn. Blackwell Scientific Publications, Oxford
Chen C, Pore N, Behrooz A, Ismail-Beigi F, Maity A (2001) Regulation of glut1 mRNA by hypoxia-inducible factor-1 interaction between H-ras and hypoxia. J Biol Chem 276(12):9519–9525. https://doi.org/10.1074/jbc.M010144200
Chen H, Davidson T, Singleton S, Garrick MD, Costa M (2005) Nickel decreases cellular iron level and converts cytosolic aconitase to iron-regulatory protein 1 in A549 cells. Toxicol Appl Pharmacol 206(3):275–287. https://doi.org/10.1016/j.taap.2004.11.011
Chen CY, Lin TK, Chang YC et al (2010) Nickel(II)-induced oxidative stress, apoptosis, G2/M arrest, and genotoxicity in normal rat kidney cells. J Toxicol Environ Health A 73(8):529–539. https://doi.org/10.1080/15287390903421250
Chen N, Qian J, Chen J et al (2017) Phase 2 studies of oral hypoxia-inducible factor prolyl hydroxylase inhibitor FG-4592 for treatment of anemia in China. Nephrol Dial Transplant 32(8):1373–1386. https://doi.org/10.1093/ndt/gfx011
Chen QY, DesMarais T, Costa M (2019a) Metals and mechanisms of carcinogenesis. Annu Rev Pharmacol Toxicol 59:537–554. https://doi.org/10.1146/annurev-pharmtox-010818-021031
Chen QY, Murphy A, Sun H, Costa M (2019b) Molecular and epigenetic mechanisms of Cr(VI)-induced carcinogenesis. Toxicol Appl Pharmacol 377:114636. https://doi.org/10.1016/j.taap.2019.114636
Chervona Y, Arita A, Costa M (2012) Carcinogenic metals and the epigenome: understanding the effect of nickel, arsenic, and chromium. Metallomics Integr Biomet Sci 4(7):619–627. https://doi.org/10.1039/c2mt20033c
Choi J, Chang JY, Hong J, Shin S, Park JS, Oh S (2017) Low-level toxic metal exposure in healthy weaning-age infants: association with growth, dietary intake, and iron deficiency. Int J Environ Res Public Health 14(4):388. https://doi.org/10.3390/ijerph14040388
Choueiri TK, Bauer TM, Papadopoulos KP et al (2021) Inhibition of hypoxia-inducible factor-2alpha in renal cell carcinoma with belzutifan: a phase 1 trial and biomarker analysis. Nat Med 27(5):802–805. https://doi.org/10.1038/s41591-021-01324-7
Chowdhury R, Leung IK, Tian YM et al (2016) Structural basis for oxygen degradation domain selectivity of the HIF prolyl hydroxylases. Nat Commun 7:12673. https://doi.org/10.1038/ncomms12673
Christensen EI, Birn H (2002) Megalin and cubilin: multifunctional endocytic receptors. Nat Rev Mol Cell Biol 3:256–266
Chun YS, Choi E, Kim GT et al (2000) Cadmium blocks hypoxia-inducible factor (HIF)-1-mediated response to hypoxia by stimulating the proteasome-dependent degradation of HIF-1alpha. Eur J Biochem 267(13):4198–4204. https://doi.org/10.1046/j.1432-1327.2000.01453.x
Ciarimboli G (2014) Membrane transporters as mediators of cisplatin side-effects. Anticancer Res 34(1):547–550
Ciarimboli G, Ludwig T, Lang D et al (2005) Cisplatin nephrotoxicity is critically mediated via the human organic cation transporter 2. Am J Pathol 167(6):1477–1484
Clanton TL (2007) Hypoxia-induced reactive oxygen species formation in skeletal muscle. J Appl Physiol (1985) 102(6):2379–2388. https://doi.org/10.1152/japplphysiol.01298.2006
Cochran KW, Maassab HF (1970) Inhibition of a cold variant of influenza virus by selected chemicals. Arch Environ Health 21(3):312–315. https://doi.org/10.1080/00039896.1970.10667244
Conde E, Alegre L, Blanco-Sanchez I et al (2012) Hypoxia inducible factor 1-alpha (HIF-1 alpha) is induced during reperfusion after renal ischemia and is critical for proximal tubule cell survival. PLoS ONE 7(3):e33258. https://doi.org/10.1371/journal.pone.0033258
Courjault F, Leroy D, Coquery L, Toutain H (1993) Platinum complex-induced dysfunction of cultured renal proximal tubule cells. A comparative study of carboplatin and transplatin with cisplatin. Arch Toxicol 67(5):338–346. https://doi.org/10.1007/BF01973705
Crichton RR (2017) Metal toxicity—an introduction. In: Crichton R, Ward RJ, Hider RC (eds) Metal chelation in medicine. RSC metallobiology. The Royal Society of Chemistry, London, pp 1–23
Dame C, Sola MC, Lim KC et al (2004) Hepatic erythropoietin gene regulation by GATA-4. J Biol Chem 279(4):2955–2961. https://doi.org/10.1074/jbc.M310404200
Dang CV, Semenza GL (1999) Oncogenic alterations of metabolism. Trends Biochem Sci 24(2):68–72. https://doi.org/10.1016/s0968-0004(98)01344-9
Dartsch PC, Hildenbrand S, Kimmel R, Schmahl FW (1998) Investigations on the nephrotoxicity and hepatotoxicity of trivalent and hexavalent chromium compounds. Int Arch Occup Environ Health 71(Suppl):S40–S45
Das KK, Das SN, Dhundasi SA (2008) Nickel, its adverse health effects & oxidative stress. Indian J Med Res 128(4):412–425
Dasari S, Tchounwou PB (2014) Cisplatin in cancer therapy: molecular mechanisms of action. Eur J Pharmacol 740:364–378. https://doi.org/10.1016/j.ejphar.2014.07.025
Davidson T, Chen H, Garrick MD, D’Angelo G, Costa M (2005) Soluble nickel interferes with cellular iron homeostasis. Mol Cell Biochem 279(1–2):157–162. https://doi.org/10.1007/s11010-005-8288-y
Davidson TL, Chen H, Di Toro DM, D’Angelo G, Costa M (2006) Soluble nickel inhibits HIF-prolyl-hydroxylases creating persistent hypoxic signaling in A549 cells. Mol Carcinog 45(7):479–489. https://doi.org/10.1002/mc.20176
Dawson TL, Gores GJ, Nieminen AL, Herman B, Lemasters JJ (1993) Mitochondria as a source of reactive oxygen species during reductive stress in rat hepatocytes. Am J Physiol 264(4 Pt 1):C961–C967. https://doi.org/10.1152/ajpcell.1993.264.4.C961
Dengler VL, Galbraith M, Espinosa JM (2014) Transcriptional regulation by hypoxia inducible factors. Crit Rev Biochem Mol Biol 49(1):1–15. https://doi.org/10.3109/10409238.2013.838205
Denkhaus E, Salnikow K (2002) Nickel essentiality, toxicity, and carcinogenicity. Crit Rev Oncol Hematol 42(1):35–56. https://doi.org/10.1016/s1040-8428(01)00214-1
Depping R, Jelkmann W, Kosyna FK (2015) Nuclear-cytoplasmatic shuttling of proteins in control of cellular oxygen sensing. J Mol Med 93(6):599–608. https://doi.org/10.1007/s00109-015-1276-0
Dhillon S (2019) Roxadustat: first global approval. Drugs 79(5):563–572. https://doi.org/10.1007/s40265-019-01077-1
Dhillon S (2020) Daprodustat: first approval. Drugs 80(14):1491–1497. https://doi.org/10.1007/s40265-020-01384-y
Diaz-Vivancos P, de Simone A, Kiddle G, Foyer CH (2015) Glutathione—linking cell proliferation to oxidative stress. Free Radic Biol Med 89:1154–1164. https://doi.org/10.1016/j.freeradbiomed.2015.09.023
Dikalov SI, Harrison DG (2014) Methods for detection of mitochondrial and cellular reactive oxygen species. Antioxid Redox Signal 20(2):372–382. https://doi.org/10.1089/ars.2012.4886
Dobritzsch D, Grancharov K, Hermsen C, Krauss GJ, Schaumloffel D (2020) Inhibitory effect of metals on animal and plant glutathione transferases. J Trace Elem Med Biol 57:48–56. https://doi.org/10.1016/j.jtemb.2019.09.007
Drose S, Brandt U (2012) Molecular mechanisms of superoxide production by the mitochondrial respiratory chain. Adv Exp Med Biol 748:145–169. https://doi.org/10.1007/978-1-4614-3573-0_6
Duan C (2016) Hypoxia-inducible factor 3 biology: complexities and emerging themes. Am J Physiol Cell Physiol 310(4):C260–C269. https://doi.org/10.1152/ajpcell.00315.2015
Dufies M, Verbiest A, Cooley LS et al (2021) Plk1, upregulated by HIF-2, mediates metastasis and drug resistance of clear cell renal cell carcinoma. Commun Biol 4(1):166. https://doi.org/10.1038/s42003-021-01653-w
Eckardt KU, Koury ST, Tan CC et al (1993) Distribution of erythropoietin producing cells in rat kidneys during hypoxic hypoxia. Kidney Int 43(4):815–823. https://doi.org/10.1038/ki.1993.115
Eckardt KU, Bernhardt WM, Weidemann A et al (2005) Role of hypoxia in the pathogenesis of renal disease. Kidney Int 68 (Suppl. 99):S46–S51. https://doi.org/10.1111/j.1523-1755.2005.09909.x
Eliopoulos N, Zhao J, Forner K, Birman E, Young YK, Bouchentouf M (2011) Erythropoietin gene-enhanced marrow mesenchymal stromal cells decrease cisplatin-induced kidney injury and improve survival of allogeneic mice. Mol Ther 19(11):2072–2083. https://doi.org/10.1038/mt.2011.162
Ema M, Taya S, Yokotani N, Sogawa K, Matsuda Y, Fujii-Kuriyama Y (1997) A novel bHLH-PAS factor with close sequence similarity to hypoxia-inducible factor 1alpha regulates the VEGF expression and is potentially involved in lung and vascular development. Proc Natl Acad Sci USA 94(9):4273–4278. https://doi.org/10.1073/pnas.94.9.4273
Epstein AC, Gleadle JM, McNeill LA et al (2001) C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell 107(1):43–54. https://doi.org/10.1016/s0092-8674(01)00507-4
Eremina V, Sood M, Haigh J et al (2003) Glomerular-specific alterations of VEGF-A expression lead to distinct congenital and acquired renal diseases. J Clin Investig 111(5):707–716. https://doi.org/10.1172/JCI17423
Eremina V, Jefferson JA, Kowalewska J et al (2008) VEGF inhibition and renal thrombotic microangiopathy. N Engl J Med 358(11):1129–1136. https://doi.org/10.1056/NEJMoa0707330
Erslev AJ (1991) Erythropoietin. N Engl J Med 324(19):1339–1344. https://doi.org/10.1056/NEJM199105093241907
Evans RG, Gardiner BS, Smith DW, O’Connor PM (2008) Intrarenal oxygenation: unique challenges and the biophysical basis of homeostasis. Am J Physiol Renal Physiol 295(5):F1259–F1270. https://doi.org/10.1152/ajprenal.90230.2008
Faivre A, Scholz CC, de Seigneux S (2021) Hypoxia in chronic kidney disease: towards a paradigm shift? Nephrol Dial Transplant 36(10):1782–1790. https://doi.org/10.1093/ndt/gfaa091
Ferguson CJ, Wareing M, Ward DT, Green R, Smith CP, Riccardi D (2001) Cellular localization of divalent metal transporter DMT-1 in rat kidney. Am J Physiol Renal Physiol 280(5):F803–F814. https://doi.org/10.1152/ajprenal.2001.280.5.F803
Fisher JW, Langston JW (1968) Effects of testosterone, cobalt and hypoxia on erythropoietin production in the isolated perfused dog kidney. Ann N Y Acad Sci 149(1):75–87. https://doi.org/10.1111/j.1749-6632.1968.tb15139.x
Flashman E, Davies SL, Yeoh KK, Schofield CJ (2010) Investigating the dependence of the hypoxia-inducible factor hydroxylases (factor inhibiting HIF and prolyl hydroxylase domain 2) on ascorbate and other reducing agents. Biochem J 427(1):135–142. https://doi.org/10.1042/BJ20091609
Flora SJ (2009) Structural, chemical and biological aspects of antioxidants for strategies against metal and metalloid exposure. Oxid Med Cell Longev 2(4):191–206. https://doi.org/10.4161/oxim.2.4.9112
Flora SJ, Pachauri V (2010) Chelation in metal intoxication. Int J Environ Res Public Health 7(7):2745–2788. https://doi.org/10.3390/ijerph7072745
Fong GH, Takeda K (2008) Role and regulation of prolyl hydroxylase domain proteins. Cell Death Differ 15(4):635–641. https://doi.org/10.1038/cdd.2008.10
Forman HJ, Zhang H (2021) Targeting oxidative stress in disease: promise and limitations of antioxidant therapy. Nat Rev Drug Discov 20(9):689–709. https://doi.org/10.1038/s41573-021-00233-1
Forsythe JA, Jiang BH, Iyer NV et al (1996) Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol Cell Biol 16(9):4604–4613. https://doi.org/10.1128/MCB.16.9.4604
Fraga CG (2005) Relevance, essentiality and toxicity of trace elements in human health. Mol Aspects Med 26(4–5):235–244. https://doi.org/10.1016/j.mam.2005.07.013
Franchini I, Mutti A (1988) Selected toxicological aspects of chromium(VI) compounds. Sci Total Environ 71(3):379–387. https://doi.org/10.1016/0048-9697(88)90210-0
Fukai T, Ushio-Fukai M (2011) Superoxide dismutases: role in redox signaling, vascular function, and diseases. Antioxid Redox Signal 15(6):1583–1606. https://doi.org/10.1089/ars.2011.3999
Galluzzi L, Bravo-San Pedro JM, Kepp O, Kroemer G (2016) Regulated cell death and adaptive stress responses. Cell Mol Life Sci 73(11–12):2405–2410. https://doi.org/10.1007/s00018-016-2209-y
Ganz T (2013) Systemic iron homeostasis. Physiol Rev 93(4):1721–1741. https://doi.org/10.1152/physrev.00008.2013
Ganz T (2018) Erythropoietin and iron—a conflicted alliance? Kidney Int 94(5):851–853. https://doi.org/10.1016/j.kint.2018.07.027
Gao S, Zhou J, Zhao Y, Toselli P, Li W (2013) Hypoxia-response element (HRE)-directed transcriptional regulation of the rat lysyl oxidase gene in response to cobalt and cadmium. Toxicol Sci 132(2):379–389. https://doi.org/10.1093/toxsci/kfs327
Genchi G, Carocci A, Lauria G, Sinicropi MS, Catalano A (2020) Nickel: human health and environmental toxicology. Int J Environ Res Public Health. 17(3):679. https://doi.org/10.3390/ijerph17030679
Glover TW, Wilson TE, Arlt MF (2017) Fragile sites in cancer: more than meets the eye. Nat Rev Cancer 17(8):489–501. https://doi.org/10.1038/nrc.2017.52
Goldberg MA, Dunning SP, Bunn HF (1988) Regulation of the erythropoietin gene: evidence that the oxygen sensor is a heme protein. Science 242(4884):1412–1415. https://doi.org/10.1126/science.2849206
Gozzelino R, Jeney V, Soares MP (2010) Mechanisms of cell protection by heme oxygenase-1. Annu Rev Pharmacol Toxicol 50:323–354. https://doi.org/10.1146/annurev.pharmtox.010909.105600
Gruber M, Hu CJ, Johnson RS, Brown EJ, Keith B, Simon MC (2007) Acute postnatal ablation of Hif-2alpha results in anemia. Proc Natl Acad Sci USA 104(7):2301–2306. https://doi.org/10.1073/pnas.0608382104
Gu YZ, Moran SM, Hogenesch JB, Wartman L, Bradfield CA (1998) Molecular characterization and chromosomal localization of a third alpha-class hypoxia inducible factor subunit, HIF3alpha. Gene Expr 7(3):205–213
Gumbleton M, Nicholls PJ (1988) Dose-response and time-response biochemical and histological study of potassium dichromate-induced nephrotoxicity in the rat. Food Chem Toxicol 26(1):37–44. https://doi.org/10.1016/0278-6915(88)90039-7
Gunshin H, Mackenzie B, Berger UV et al (1997) Cloning and characterization of a mammalian proton-coupled metal-ion transporter. Nature 388(6641):482–488. https://doi.org/10.1038/41343
Gupta AK, Jadhav SH, Tripathy NK, Nityanand S (2015) Fetal kidney stem cells ameliorate cisplatin induced acute renal failure and promote renal angiogenesis. World J Stem Cells 7(4):776–788. https://doi.org/10.4252/wjsc.v7.i4.776
Guzy RD, Hoyos B, Robin E et al (2005) Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab 1(6):401–408. https://doi.org/10.1016/j.cmet.2005.05.001
Haase VH (2017) HIF-prolyl hydroxylases as therapeutic targets in erythropoiesis and iron metabolism. Hemodial Int 21(Suppl 1):S110–S124. https://doi.org/10.1111/hdi.12567
Haase VH (2021) Hypoxia-inducible factor-prolyl hydroxylase inhibitors in the treatment of anemia of chronic kidney disease. Kidney Int Suppl 11(1):8–25. https://doi.org/10.1016/j.kisu.2020.12.002
Haase VH, Chertow GM, Block GA et al (2019) Effects of vadadustat on hemoglobin concentrations in patients receiving hemodialysis previously treated with erythropoiesis-stimulating agents. Nephrol Dial Transplant 34(1):90–99. https://doi.org/10.1093/ndt/gfy055
Hadaczek P, Siprashvili Z, Markiewski M et al (1998) Absence or reduction of Fhit expression in most clear cell renal carcinomas. Cancer Res 58(14):2946–2951
Hagen T, Taylor CT, Lam F, Moncada S (2003) Redistribution of intracellular oxygen in hypoxia by nitric oxide: effect on HIF1alpha. Science 302(5652):1975–1978. https://doi.org/10.1126/science.1088805
Halliwell B (1991) Reactive oxygen species in living systems: source, biochemistry, and role in human disease. Am J Med 91(3C):14S-22S. https://doi.org/10.1016/0002-9343(91)90279-7
Halliwell B (2011) Free radicals and antioxidants—quo vadis? Trends Pharmacol Sci 32(3):125–130. https://doi.org/10.1016/j.tips.2010.12.002
Hancock RL, Dunne K, Walport LJ, Flashman E, Kawamura A (2015) Epigenetic regulation by histone demethylases in hypoxia. Epigenomics 7(5):791–811. https://doi.org/10.2217/epi.15.24
Hara S, Hamada J, Kobayashi C, Kondo Y, Imura N (2001) Expression and characterization of hypoxia-inducible factor (HIF)-3alpha in human kidney: suppression of HIF-mediated gene expression by HIF-3alpha. Biochem Biophys Res Commun 287(4):808–813. https://doi.org/10.1006/bbrc.2001.5659
Hartwig A (2013) Cadmium and cancer. Met Ions Life Sci 11:491–507. https://doi.org/10.1007/978-94-007-5179-8_15
Haynes WM, Lide DR, Bruno TJ (2016) Abundance of elements in the Earth’s crust and in the sea. CRC handbook of chemistry and physics, 97th edn. CRC Press, Boca Raton, pp 14–17
Hayyan M, Hashim MA, AlNashef IM (2016) Superoxide Ion: Generation and Chemical Implications. Chem Rev 116(5):3029–3085. https://doi.org/10.1021/acs.chemrev.5b00407
Hentze MW, Muckenthaler MU, Galy B, Camaschella C (2010) Two to tango: regulation of Mammalian iron metabolism. Cell 142(1):24–38. https://doi.org/10.1016/j.cell.2010.06.028
Heyman SN, Rosen S, Rosenberger C (2011) Hypoxia-inducible factors and the prevention of acute organ injury. Crit Care 15(2):209. https://doi.org/10.1186/cc9991
Hiratsuka H, Katsuta O, Toyota N, Tsuchitani M, Umemura T, Marumo F (1996) Chronic cadmium exposure-induced renal anemia in ovariectomized rats. Toxicol Appl Pharmacol 137(2):228–236. https://doi.org/10.1006/taap.1996.0076
Holditch SJ, Brown CN, Lombardi AM, Nguyen KN, Edelstein CL (2019) Recent advances in models, mechanisms, biomarkers, and interventions in cisplatin-induced acute kidney injury. Int J Mol Sci 20(12):3011. https://doi.org/10.3390/ijms20123011
Holdstock L, Meadowcroft AM, Maier R et al (2016) Four-week studies of oral hypoxia-inducible factor-prolyl hydroxylase inhibitor GSK1278863 for treatment of anemia. J Am Soc Nephrol 27(4):1234–1244. https://doi.org/10.1681/ASN.2014111139
Holmquist-Mengelbier L, Fredlund E, Lofstedt T et al (2006) Recruitment of HIF-1alpha and HIF-2alpha to common target genes is differentially regulated in neuroblastoma: HIF-2alpha promotes an aggressive phenotype. Cancer Cell 10(5):413–423. https://doi.org/10.1016/j.ccr.2006.08.026
Hopfer SM, Sunderman FW Jr, Reid MC, Goldwasser E (1984) Increased immunoreactive erythropoietin in serum and kidney extracts. Res Commun Chem Pathol Pharmacol 43(2):299–305
Horak E, Sunderman FW Jr (1980) Nephrotoxicity of nickel carbonyl in rats. Ann Clin Lab Sci 10(5):425–431
Horiguchi H, Sato M, Konno N, Fukushima M (1996) Long-term cadmium exposure induces anemia in rats through hypoinduction of erythropoietin in the kidneys. Arch Toxicol 71(1–2):11–19. https://doi.org/10.1007/s002040050352
Horiguchi H, Kayama F, Oguma E, Willmore WG, Hradecky P, Bunn HF (2000) Cadmium and platinum suppression of erythropoietin production in cell culture: clinical implications. Blood 96(12):3743–3747
Horiguchi H, Oguma E, Kayama F (2006) Cadmium and cisplatin damage erythropoietin-producing proximal renal tubular cells. Arch Toxicol 80(10):680–686. https://doi.org/10.1007/s00204-006-0093-1
Horiguchi H, Oguma E, Kayama F (2011) Cadmium induces anemia through interdependent progress of hemolysis, body iron accumulation, and insufficient erythropoietin production in rats. Toxicol Sci 122(1):198–210. https://doi.org/10.1093/toxsci/kfr100
Hosseini SA, Horton S, Saldivar JC et al (2013) Common chromosome fragile sites in human and murine epithelial cells and FHIT/FRA3B loss-induced global genome instability. Genes Chromosomes Cancer 52(11):1017–1029. https://doi.org/10.1002/gcc.22097
Hosseini MJ, Shaki F, Ghazi-Khansari M, Pourahmad J (2014) Toxicity of copper on isolated liver mitochondria: impairment at complexes I, II, and IV leads to increased ROS production. Cell Biochem Biophys 70(1):367–381. https://doi.org/10.1007/s12013-014-9922-7
Hou J, Cai S, Kitajima Y et al (2013) 5-Aminolevulinic acid combined with ferrous iron induces carbon monoxide generation in mouse kidneys and protects from renal ischemia-reperfusion injury. Am J Physiol Renal Physiol 305(8):F1149–F1157. https://doi.org/10.1152/ajprenal.00275.2013
Hu CJ, Iyer S, Sataur A, Covello KL, Chodosh LA, Simon MC (2006) Differential regulation of the transcriptional activities of hypoxia-inducible factor 1 alpha (HIF-1alpha) and HIF-2alpha in stem cells. Mol Cell Biol 26(9):3514–3526. https://doi.org/10.1128/MCB.26.9.3514-3526.2006
Huang X (2003) Iron overload and its association with cancer risk in humans: evidence for iron as a carcinogenic metal. Mutat Res 533(1–2):153–171
Huang LE, Arany Z, Livingston DM, Bunn HF (1996) Activation of hypoxia-inducible transcription factor depends primarily upon redox-sensitive stabilization of its alpha subunit. J Biol Chem 271(50):32253–32259. https://doi.org/10.1074/jbc.271.50.32253
Huang LE, Ho V, Arany Z et al (1997) Erythropoietin gene regulation depends on heme-dependent oxygen sensing and assembly of interacting transcription factors. Kidney Int 51(2):548–552. https://doi.org/10.1038/ki.1997.76
Huebner K, Croce CM (2001) FRA3B and other common fragile sites: the weakest links. Nat Rev Cancer 1(3):214–221. https://doi.org/10.1038/35106058
Illing AC, Shawki A, Cunningham CL, Mackenzie B (2012) Substrate profile and metal-ion selectivity of human divalent metal-ion transporter-1. J Biol Chem 287(36):30485–30496. https://doi.org/10.1074/jbc.M112.364208
Imagawa S, Suzuki N, Ohmine K et al (2002) GATA suppresses erythropoietin gene expression through GATA site in mouse erythropoietin gene promoter. Int J Hematol 75(4):376–381. https://doi.org/10.1007/BF02982127
Imtiyaz HZ, Simon MC (2010) Hypoxia-inducible factors as essential regulators of inflammation. Curr Top Microbiol Immunol 345:105–120. https://doi.org/10.1007/82_2010_74
Ivan M, Kondo K, Yang H et al (2001) HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science 292(5516):464–468. https://doi.org/10.1126/science.1059817
Jaakkola P, Mole DR, Tian YM et al (2001) Targeting of HIF-alpha to the von Hippel–Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 292(5516):468–472. https://doi.org/10.1126/science.1059796
Jacobo-Estrada T, Cardenas-Gonzalez M, Santoyo-Sanchez MP, Thévenod F, Barbier O (2018) Intrauterine exposure to cadmium reduces HIF-1 DNA-binding ability in rat fetal kidneys. Toxics 6(3):53. https://doi.org/10.3390/toxics6030053
Jamieson D, Chance B, Cadenas E, Boveris A (1986) The relation of free radical production to hyperoxia. Annu Rev Physiol 48:703–719. https://doi.org/10.1146/annurev.ph.48.030186.003415
Jing Y, Liu LZ, Jiang Y et al (2012) Cadmium increases HIF-1 and VEGF expression through ROS, ERK, and AKT signaling pathways and induces malignant transformation of human bronchial epithelial cells. Toxicol Sci 125(1):10–19. https://doi.org/10.1093/toxsci/kfr256
Kaczmarek M, Timofeeva OA, Karaczyn A, Malyguine A, Kasprzak KS, Salnikow K (2007) The role of ascorbate in the modulation of HIF-1alpha protein and HIF-dependent transcription by chromium(VI) and nickel(II). Free Radic Biol Med 42(8):1246–1257. https://doi.org/10.1016/j.freeradbiomed.2007.01.026
Kaczmarek M, Cachau RE, Topol IA, Kasprzak KS, Ghio A, Salnikow K (2009) Metal ions-stimulated iron oxidation in hydroxylases facilitates stabilization of HIF-1 alpha protein. Toxicol Sci 107(2):394–403. https://doi.org/10.1093/toxsci/kfn251
Kaelin WG (2005) Proline hydroxylation and gene expression. Annu Rev Biochem 74:115–128. https://doi.org/10.1146/annurev.biochem.74.082803.133142
Kalkavan H, Green DR (2018) MOMP, cell suicide as a BCL-2 family business. Cell Death Differ 25(1):46–55. https://doi.org/10.1038/cdd.2017.179
Kaneko H, Katoh T, Hirano I et al (2017) Induction of erythropoietin gene expression in epithelial cells by chemicals identified in GATA inhibitor screenings. Genes Cells 22(11):939–952. https://doi.org/10.1111/gtc.12537
Kapitsinou PP, Liu Q, Unger TL et al (2010) Hepatic HIF-2 regulates erythropoietic responses to hypoxia in renal anemia. Blood 116(16):3039–3048. https://doi.org/10.1182/blood-2010-02-270322
Kapitsinou PP, Sano H, Michael M et al (2014) Endothelial HIF-2 mediates protection and recovery from ischemic kidney injury. J Clin Invest 124(6):2396–2409. https://doi.org/10.1172/JCI69073
Karaczyn A, Ivanov S, Reynolds M, Zhitkovich A, Kasprzak KS, Salnikow K (2006) Ascorbate depletion mediates up-regulation of hypoxia-associated proteins by cell density and nickel. J Cell Biochem 97(5):1025–1035. https://doi.org/10.1002/jcb.20705
Karasawa T, Steyger PS (2015) An integrated view of cisplatin-induced nephrotoxicity and ototoxicity. Toxicol Lett 237(3):219–227. https://doi.org/10.1016/j.toxlet.2015.06.012
Keir LS, Firth R, Aponik L et al (2017) VEGF regulates local inhibitory complement proteins in the eye and kidney. J Clin Investig 127(1):199–214. https://doi.org/10.1172/JCI86418
Khadangi F, Azzi A (2019) Vitamin E—the next 100 years. IUBMB Life 71(4):411–415. https://doi.org/10.1002/iub.1990
Kim JY, Lee JY (2017) Targeting tumor adaption to chronic hypoxia: implications for drug resistance, and how it can be overcome. Int J Mol Sci 18(9):1854. https://doi.org/10.3390/ijms18091854
Klaassen CD (2019) Casarett and Doull’s toxicology: the basic science of poisons, 9th edn. McGraw-Hill, New York
Klimova T, Chandel NS (2008) Mitochondrial complex III regulates hypoxic activation of HIF. Cell Death Differ 15(4):660–666. https://doi.org/10.1038/sj.cdd.4402307
Kobayashi H, Liu Q, Binns TC et al (2016) Distinct subpopulations of FOXD1 stroma-derived cells regulate renal erythropoietin. J Clin Investig 126(5):1926–1938. https://doi.org/10.1172/JCI83551
Kong D, Zhuo L, Gao C et al (2013) Erythropoietin protects against cisplatin-induced nephrotoxicity by attenuating endoplasmic reticulum stress-induced apoptosis. J Nephrol 26(1):219–227. https://doi.org/10.5301/jn.5000177
Koury ST, Koury MJ, Bondurant MC, Caro J, Graber SE (1989) Quantitation of erythropoietin-producing cells in kidneys of mice by in situ hybridization: correlation with hematocrit, renal erythropoietin mRNA, and serum erythropoietin concentration. Blood 74(2):645–651
Krieg AJ, Rankin EB, Chan D, Razorenova O, Fernandez S, Giaccia AJ (2010) Regulation of the histone demethylase JMJD1A by hypoxia-inducible factor 1 alpha enhances hypoxic gene expression and tumor growth. Mol Cell Biol 30(1):344–353. https://doi.org/10.1128/MCB.00444-09
Kudo Y, Kakinuma Y, Mori Y et al (2005) Hypoxia-inducible factor-1alpha is involved in the attenuation of experimentally induced rat glomerulonephritis. Nephron Exp Nephrol 100(2):e95-103. https://doi.org/10.1159/000084575
Kurt B, Kurtz A (2015) Plasticity of renal endocrine function. Am J Physiol Regul Integr Comp Physiol 308(6):R455–R466. https://doi.org/10.1152/ajpregu.00568.2013
Lando D, Peet DJ, Gorman JJ, Whelan DA, Whitelaw ML, Bruick RK (2002a) FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes Dev 16(12):1466–1471. https://doi.org/10.1101/gad.991402
Lando D, Peet DJ, Whelan DA, Gorman JJ, Whitelaw ML (2002b) Asparagine hydroxylation of the HIF transactivation domain a hypoxic switch. Science 295(5556):858–861. https://doi.org/10.1126/science.1068592
Leaf DE, Swinkels DW (2016) Catalytic iron and acute kidney injury. Am J Physiol Renal Physiol 311(5):F871–F876. https://doi.org/10.1152/ajprenal.00388.2016
Lee WK, Thévenod F (2020) Cell organelles as targets of mammalian cadmium toxicity. Arch Toxicol 94:1017–1049. https://doi.org/10.1007/s00204-020-02692-8
Lee PJ, Jiang BH, Chin BY et al (1997) Hypoxia-inducible factor-1 mediates transcriptional activation of the heme oxygenase-1 gene in response to hypoxia. J Biol Chem 272(9):5375–5381
Lee WK, Wolff NA, Thévenod F (2009) Organic cation transporters: physiology, toxicology and special focus on ethidium as a novel substrate. Curr Drug Metab 10(6):617–631
Lee G, Won HS, Lee YM et al (2016) Oxidative dimerization of PHD2 is responsible for its inactivation and contributes to metabolic reprogramming via HIF-1alpha activation. Sci Rep 6:18928. https://doi.org/10.1038/srep18928
Lee P, Chandel NS, Simon MC (2020) Cellular adaptation to hypoxia through hypoxia inducible factors and beyond. Nat Rev Mol Cell Biol 21(5):268–283. https://doi.org/10.1038/s41580-020-0227-y
Lejding T, Mowitz M, Isaksson M et al (2018) A retrospective investigation of hexavalent chromium allergy in southern Sweden. Contact Dermatitis 78(6):386–392. https://doi.org/10.1111/cod.12969
LeRoy AF (1975) Interactions of platinum metals and their complexes in biological systems. Environ Health Perspect 10:73–83. https://doi.org/10.1289/ehp.751073
Lever JM, Boddu R, George JF, Agarwal A (2016) Heme oxygenase-1 in kidney health and disease. Antioxid Redox Signal 25(3):165–183. https://doi.org/10.1089/ars.2016.6659
Leyssens L, Vinck B, Van Der Straeten C, Wuyts F, Maes L (2017) Cobalt toxicity in humans-A review of the potential sources and systemic health effects. Toxicology 387:43–56. https://doi.org/10.1016/j.tox.2017.05.015
Li Q, Chen H, Huang X, Costa M (2006) Effects of 12 metal ions on iron regulatory protein 1 (IRP-1) and hypoxia-inducible factor-1 alpha (HIF-1alpha) and HIF-regulated genes. Toxicol Appl Pharmacol 213(3):245–255. https://doi.org/10.1016/j.taap.2005.11.006
Li Q, Liang X, Yang Y, Zeng X, Zhong X, Huang C (2020) Panax notoginseng saponins ameliorate cisplatin-induced mitochondrial injury via the HIF-1alpha/mitochondria/ROS pathway. FEBS Open Bio 10(1):118–126. https://doi.org/10.1002/2211-5463.12760
Li S, Wen L, Hu X, Wei Q, Dong Z (2021) HIF in nephrotoxicity during cisplatin chemotherapy: regulation, function and therapeutic potential. Cancers (basel) 13(2):180. https://doi.org/10.3390/cancers13020180
Lieb ME, Menzies K, Moschella MC, Ni R, Taubman MB (2002) Mammalian EGLN genes have distinct patterns of mRNA expression and regulation. Biochem Cell Biol 80(4):421–426. https://doi.org/10.1139/o02-115
Limon-Pacheco J, Gonsebatt ME (2009) The role of antioxidants and antioxidant-related enzymes in protective responses to environmentally induced oxidative stress. Mutat Res 674(1–2):137–147. https://doi.org/10.1016/j.mrgentox.2008.09.015
Lin X, David CA, Donnelly JB et al (2008) A chemical genomics screen highlights the essential role of mitochondria in HIF-1 regulation. Proc Natl Acad Sci USA 105(1):174–179. https://doi.org/10.1073/pnas.0706585104
Lin TJ, Huang YL, Chang JS et al (2018) Optimal dosage and early intervention of l-ascorbic acid inhibiting K2Cr2O7-induced renal tubular cell damage. J Trace Elem Med Biol 48:1–7. https://doi.org/10.1016/j.jtemb.2018.02.022
Linehan WM, Ricketts CJ (2013) The metabolic basis of kidney cancer. Semin Cancer Biol 23(1):46–55. https://doi.org/10.1016/j.semcancer.2012.06.002
Linehan WM, Rouault TA (2013) Molecular pathways: fumarate hydratase-deficient kidney cancer—targeting the Warburg effect in cancer. Clin Cancer Res 19(13):3345–3352. https://doi.org/10.1158/1078-0432.CCR-13-0304
Linehan WM, Srinivasan R, Schmidt LS (2010) The genetic basis of kidney cancer: a metabolic disease. Nat Rev Urol 7(5):277–285. https://doi.org/10.1038/nrurol.2010.47
Linehan WM, Schmidt LS, Crooks DR et al (2019) The metabolic basis of kidney cancer. Cancer Discov 9(8):1006–1021. https://doi.org/10.1158/2159-8290.CD-18-1354
Linkermann A, Skouta R, Himmerkus N et al (2014) Synchronized renal tubular cell death involves ferroptosis. Proc Natl Acad Sci USA 111(47):16836–16841. https://doi.org/10.1073/pnas.1415518111
Liu W, Chen H, Wong N, Haynes W, Baker CM, Wang X (2017) Pseudohypoxia induced by miR-126 deactivation promotes migration and therapeutic resistance in renal cell carcinoma. Cancer Lett 394:65–75. https://doi.org/10.1016/j.canlet.2017.02.025
Loenarz C, Schofield CJ (2011) Physiological and biochemical aspects of hydroxylations and demethylations catalyzed by human 2-oxoglutarate oxygenases. Trends Biochem Sci 36(1):7–18. https://doi.org/10.1016/j.tibs.2010.07.002
Lopez-Rodriguez G, Galvan M, Gonzalez-Unzaga M, Hernandez Avila J, Perez-Labra M (2017) Blood toxic metals and hemoglobin levels in Mexican children. Environ Monit Assess 189(4):179. https://doi.org/10.1007/s10661-017-5886-6
Lynes MA, Kang YJ, Sensi SL, Perdrizet GA, Hightower LE (2007) Heavy metal ions in normal physiology, toxic stress, and cytoprotection. Ann N Y Acad Sci 1113:159–172. https://doi.org/10.1196/annals.1391.010
Mansfield KD, Guzy RD, Pan Y et al (2005) Mitochondrial dysfunction resulting from loss of cytochrome c impairs cellular oxygen sensing and hypoxic HIF-alpha activation. Cell Metab 1(6):393–399. https://doi.org/10.1016/j.cmet.2005.05.003
Markham A (2020) Vadadustat: first approval. Drugs 80(13):1365–1371. https://doi.org/10.1007/s40265-020-01383-z
Martin ER, Smith MT, Maroni BJ, Zuraw QC, deGoma EM (2017) Clinical trial of vadadustat in patients with anemia secondary to stage 3 or 4 chronic kidney disease. Am J Nephrol 45(5):380–388. https://doi.org/10.1159/000464476
Martinou JC, Youle RJ (2011) Mitochondria in apoptosis: Bcl-2 family members and mitochondrial dynamics. Dev Cell 21(1):92–101. https://doi.org/10.1016/j.devcel.2011.06.017
Matsumoto M, Makino Y, Tanaka T et al (2003) Induction of renoprotective gene expression by cobalt ameliorates ischemic injury of the kidney in rats. J Am Soc Nephrol 14(7):1825–1832. https://doi.org/10.1097/01.asn.0000074239.22357.06
Maxwell P, Salnikow K (2004) HIF-1: an oxygen and metal responsive transcription factor. Cancer Biol Ther 3(1):29–35. https://doi.org/10.4161/cbt.3.1.547
Maxwell PH, Osmond MK, Pugh CW et al (1993) Identification of the renal erythropoietin-producing cells using transgenic mice. Kidney Int 44(5):1149–1162. https://doi.org/10.1038/ki.1993.362
Maxwell PH, Wiesener MS, Chang GW et al (1999) The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 399(6733):271–275. https://doi.org/10.1038/20459
Mazure NM, Pouyssegur J (2010) Hypoxia-induced autophagy: cell death or cell survival? Curr Opin Cell Biol 22(2):177–180. https://doi.org/10.1016/j.ceb.2009.11.015
McNeill LA, Flashman E, Buck MR et al (2005) Hypoxia-inducible factor prolyl hydroxylase 2 has a high affinity for ferrous iron and 2-oxoglutarate. Mol Biosyst 1(4):321–324. https://doi.org/10.1039/b511249b
McSweeney KR, Gadanec LK, Qaradakhi T, Ali BA, Zulli A, Apostolopoulos V (2021) Mechanisms of cisplatin-induced acute kidney injury: pathological mechanisms, pharmacological interventions, and genetic mitigations. Cancers (basel) 13(7):1572. https://doi.org/10.3390/cancers13071572
Mecinovic J, Chowdhury R, Lienard BM et al (2008) ESI-MS studies on prolyl hydroxylase domain 2 reveal a new metal binding site. ChemMedChem 3(4):569–572. https://doi.org/10.1002/cmdc.200700233
Metzen E, Berchner-Pfannschmidt U, Stengel P et al (2003) Intracellular localisation of human HIF-1 alpha hydroxylases: implications for oxygen sensing. J Cell Sci 116(Pt 7):1319–1326. https://doi.org/10.1242/jcs.00318
Mohamed HE, El-Swefy SE, Mohamed RH, Ghanim AM (2013) Effect of erythropoietin therapy on the progression of cisplatin induced renal injury in rats. Exp Toxicol Pathol 65(1–2):197–203. https://doi.org/10.1016/j.etp.2011.08.006
Moore E, Bellomo R (2011) Erythropoietin (EPO) in acute kidney injury. Ann Intensive Care 1(1):3. https://doi.org/10.1186/2110-5820-1-3
Morais C, Johnson DW, Vesey DA, Gobe GC (2013) Functional significance of erythropoietin in renal cell carcinoma. BMC Cancer 13:14. https://doi.org/10.1186/1471-2407-13-14
Morita M, Ohneda O, Yamashita T et al (2003) HLF/HIF-2alpha is a key factor in retinopathy of prematurity in association with erythropoietin. EMBO J 22(5):1134–1146. https://doi.org/10.1093/emboj/cdg117
Moulis JM, Thévenod F (2010) New perspectives in cadmium toxicity: an introduction. Biometals 23(5):763–768. https://doi.org/10.1007/s10534-010-9365-6
Moussavian MR, Slotta JE, Kollmar O, Menger MD, Schilling MK, Gronow G (2007) Hemoglobin induces cytotoxic damage of glycine-preserved renal tubules. Transpl Int 20(10):884–894. https://doi.org/10.1111/j.1432-2277.2007.00538.x
Munro D, Treberg JR (2017) A radical shift in perspective: mitochondria as regulators of reactive oxygen species. J Exp Biol 220(Pt 7):1170–1180. https://doi.org/10.1242/jeb.132142
Nagai T, Yasuoka Y, Izumi Y et al (2014) Reevaluation of erythropoietin production by the nephron. Biochem Biophys Res Commun 449(2):222–228. https://doi.org/10.1016/j.bbrc.2014.05.014
Nangaku M, Eckardt KU (2007) Hypoxia and the HIF system in kidney disease. J Mol Med 85(12):1325–1330. https://doi.org/10.1007/s00109-007-0278-y
Nath M, Agarwal A (2020) New insights into the role of heme oxygenase-1 in acute kidney injury. Kidney Res Clin Pract 39(4):387–401. https://doi.org/10.23876/j.krcp.20.091
Nath KA, Balla G, Vercellotti GM et al (1992) Induction of heme oxygenase is a rapid, protective response in rhabdomyolysis in the rat. J Clin Investig 90(1):267–270. https://doi.org/10.1172/JCI115847
Nezu M, Souma T, Yu L et al (2017) Transcription factor Nrf2 hyperactivation in early-phase renal ischemia-reperfusion injury prevents tubular damage progression. Kidney Int 91(2):387–401. https://doi.org/10.1016/j.kint.2016.08.023
Ngaha EO (1981) Renal effects of potassium dichromate in the rat: comparison of urinary enzyme excretion with corresponding tissue patterns. Gen Pharmacol 12(6):497–500. https://doi.org/10.1016/0306-3623(81)90078-1
Nishimura K, Iitaka S, Nakagawa H (2021) Effect of trivalent chromium on erythropoietin production and the prevention of insulin resistance in HepG2 cells. Arch Biochem Biophys 708:108960. https://doi.org/10.1016/j.abb.2021.108960
Nuyts GD, Van Vlem E, Thys J et al (1995) New occupational risk factors for chronic renal failure. Lancet 346(8966):7–11. https://doi.org/10.1016/s0140-6736(95)92648-8
Obara N, Imagawa S, Nakano Y, Suzuki N, Yamamoto M, Nagasawa T (2003) Suppression of erythropoietin gene expression by cadmium depends on inhibition of HIF-1, not stimulation of GATA-2. Arch Toxicol 77(5):267–273. https://doi.org/10.1007/s00204-003-0444-0
Obara N, Suzuki N, Kim K, Nagasawa T, Imagawa S, Yamamoto M (2008) Repression via the GATA box is essential for tissue-specific erythropoietin gene expression. Blood 111(10):5223–5232. https://doi.org/10.1182/blood-2007-10-115857
Obone E, Chakrabarti SK, Bai C, Malick MA, Lamontagne L, Subramanian KS (1999) Toxicity and bioaccumulation of nickel sulfate in Sprague-Dawley rats following 13 weeks of subchronic exposure. J Toxicol Environ Health A 57(6):379–401. https://doi.org/10.1080/009841099157593
Oshima K, Ikeda Y, Horinouchi Y et al (2017) Iron suppresses erythropoietin expression via oxidative stress-dependent hypoxia-inducible factor-2 alpha inactivation. Lab Investig 97(5):555–566. https://doi.org/10.1038/labinvest.2017.11
Paliege A, Rosenberger C, Bondke A et al (2010) Hypoxia-inducible factor-2alpha-expressing interstitial fibroblasts are the only renal cells that express erythropoietin under hypoxia-inducible factor stabilization. Kidney Int 77(4):312–318. https://doi.org/10.1038/ki.2009.460
Patlolla AK, Barnes C, Yedjou C, Velma VR, Tchounwou PB (2009) Oxidative stress, DNA damage, and antioxidant enzyme activity induced by hexavalent chromium in Sprague-Dawley rats. Environ Toxicol 24(1):66–73. https://doi.org/10.1002/tox.20395
Pedraza-Chaverri J, Barrera D, Medina-Campos ON et al (2005) Time course study of oxidative and nitrosative stress and antioxidant enzymes in K2Cr2O7-induced nephrotoxicity. BMC Nephrol 6:4. https://doi.org/10.1186/1471-2369-6-4
Pergola PE, Spinowitz BS, Hartman CS, Maroni BJ, Haase VH (2016) Vadadustat, a novel oral HIF stabilizer, provides effective anemia treatment in nondialysis-dependent chronic kidney disease. Kidney Int 90(5):1115–1122. https://doi.org/10.1016/j.kint.2016.07.019
Piret JP, Mottet D, Raes M, Michiels C (2002) Is HIF-1alpha a pro- or an anti-apoptotic protein? Biochem Pharmacol 64(5–6):889–892. https://doi.org/10.1016/s0006-2952(02)01155-3
Pollard PJ, Loenarz C, Mole DR et al (2008) Regulation of Jumonji-domain-containing histone demethylases by hypoxia-inducible factor (HIF)-1alpha. Biochem J 416(3):387–394. https://doi.org/10.1042/BJ20081238
Provenzano R, Besarab A, Wright S et al (2016) Roxadustat (FG-4592) versus epoetin alfa for anemia in patients receiving maintenance hemodialysis: a phase 2, randomized, 6- to 19-week, open-label, active-comparator, dose-ranging, safety and exploratory efficacy study. Am J Kidney Dis 67(6):912–924. https://doi.org/10.1053/j.ajkd.2015.12.020
Qi L, Luo Q, Zhang Y, Jia F, Zhao Y, Wang F (2019) Advances in toxicological research of the anticancer drug cisplatin. Chem Res Toxicol 32(8):1469–1486. https://doi.org/10.1021/acs.chemrestox.9b00204
Rankin EB, Biju MP, Liu Q et al (2007) Hypoxia-inducible factor-2 (HIF-2) regulates hepatic erythropoietin in vivo. J Clin Invest 117(4):1068–1077. https://doi.org/10.1172/JCI30117
Rauch S, Morrison GM (2008) Environmental relevance of the platinum-group elements. Elements 4(4):259–263. https://doi.org/10.2113/GSELEMENTS.4.4.259%JElements
Rjiba-Touati K, Boussema IA, Belarbia A, Achour A, Bacha H (2011) Protective effect of recombinant human erythropoietin against cisplatin-induced oxidative stress and nephrotoxicity in rat kidney. Int J Toxicol 30(5):510–517. https://doi.org/10.1177/1091581810411931
Rosenberg B, Van Camp L, Grimley EB, Thomson AJ (1967) The inhibition of growth or cell division in Escherichia coli by different ionic species of platinum(IV) complexes. J Biol Chem 242(6):1347–1352
Rosenberg B, VanCamp L, Trosko JE, Mansour VH (1969) Platinum compounds: a new class of potent antitumour agents. Nature 222(5191):385–386. https://doi.org/10.1038/222385a0
Rosenberger C, Mandriota S, Jurgensen JS et al (2002) Expression of hypoxia-inducible factor-1alpha and -2alpha in hypoxic and ischemic rat kidneys. J Am Soc Nephrol 13(7):1721–1732. https://doi.org/10.1097/01.asn.0000017223.49823.2a
Rossignol F, Vache C, Clottes E (2002) Natural antisense transcripts of hypoxia-inducible factor 1alpha are detected in different normal and tumour human tissues. Gene 299(1–2):135–140. https://doi.org/10.1016/s0378-1119(02)01049-1
Rubio-Navarro A, Vazquez-Carballo C, Guerrero-Hue M et al (2019) Nrf2 plays a protective role against intravascular hemolysis-mediated acute kidney injury. Front Pharmacol 10:740. https://doi.org/10.3389/fphar.2019.00740
Sabolic I (2006) Common mechanisms in nephropathy induced by toxic metals. Nephron Physiol 104(3):p107–p114. https://doi.org/10.1159/000095539
Salnikow K, An WG, Melillo G, Blagosklonny MV, Costa M (1999) Nickel-induced transformation shifts the balance between HIF-1 and p53 transcription factors. Carcinogenesis 20(9):1819–1823. https://doi.org/10.1093/carcin/20.9.1819
Salnikow K, Blagosklonny MV, Ryan H, Johnson R, Costa M (2000a) Carcinogenic nickel induces genes involved with hypoxic stress. Cancer Res 60(1):38–41
Salnikow K, Su W, Blagosklonny MV, Costa M (2000b) Carcinogenic metals induce hypoxia-inducible factor-stimulated transcription by reactive oxygen species-independent mechanism. Cancer Res 60(13):3375–3378
Salnikow K, Donald SP, Bruick RK, Zhitkovich A, Phang JM, Kasprzak KS (2004) Depletion of intracellular ascorbate by the carcinogenic metals nickel and cobalt results in the induction of hypoxic stress. J Biol Chem 279(39):40337–40344. https://doi.org/10.1074/jbc.M403057200
Sano R, Reed JC (2013) ER stress-induced cell death mechanisms. Biochim Biophys Acta 1833(12):3460–3470. https://doi.org/10.1016/j.bbamcr.2013.06.028
Satarug S, G CG, D AV, Phelps KR (2020) Cadmium and Lead Exposure, Nephrotoxicity, and Mortality. Toxics 8(4):86. doi:https://doi.org/10.3390/toxics8040086
Sato Y, Yoshizato T, Shiraishi Y et al (2013) Integrated molecular analysis of clear-cell renal cell carcinoma. Nat Genet 45(8):860–867. https://doi.org/10.1038/ng.2699
Schaaf GJ, Maas RF, de Groene EM, Fink-Gremmels J (2002) Management of oxidative stress by heme oxygenase-1 in cisplatin-induced toxicity in renal tubular cells. Free Radical Res 36(8):835–843. https://doi.org/10.1080/1071576021000005267
Schley G, Klanke B, Schodel J et al (2011) Hypoxia-inducible transcription factors stabilization in the thick ascending limb protects against ischemic acute kidney injury. J Am Soc Nephrol 22(11):2004–2015. https://doi.org/10.1681/ASN.2010121249
Schley G, Klanke B, Kalucka J et al (2019) Mononuclear phagocytes orchestrate prolyl hydroxylase inhibition-mediated renoprotection in chronic tubulointerstitial nephritis. Kidney Int 96(2):378–396. https://doi.org/10.1016/j.kint.2019.02.016
Schodel J, Ratcliffe PJ (2019) Mechanisms of hypoxia signalling: new implications for nephrology. Nat Rev Nephrol 15(10):641–659. https://doi.org/10.1038/s41581-019-0182-z
Schodel J, Klanke B, Weidemann A et al (2009) HIF-prolyl hydroxylases in the rat kidney: physiologic expression patterns and regulation in acute kidney injury. Am J Pathol 174(5):1663–1674. https://doi.org/10.2353/ajpath.2009.080687
Schodel J, Bohr D, Klanke B et al (2010) Factor inhibiting HIF limits the expression of hypoxia-inducible genes in podocytes and distal tubular cells. Kidney Int 78(9):857–867. https://doi.org/10.1038/ki.2010.284
Schwerdt G, Freudinger R, Schuster C, Weber F, Thews O, Gekle M (2005) Cisplatin-induced apoptosis is enhanced by hypoxia and by inhibition of mitochondria in renal collecting duct cells. Toxicol Sci 85(1):735–742. https://doi.org/10.1093/toxsci/kfi117
Scindia Ph DY, Leeds MdJ, Swaminathan MdS (2019) Iron Homeostasis in Healthy Kidney and its Role in Acute Kidney Injury. Semin Nephrol 39(1):76–84. https://doi.org/10.1016/j.semnephrol.2018.10.006
Scire A, Cianfruglia L, Minnelli C et al (2019) Glutathione compartmentalization and its role in glutathionylation and other regulatory processes of cellular pathways. BioFactors 45(2):152–168. https://doi.org/10.1002/biof.1476
Scortegagna M, Ding K, Zhang Q et al (2005) HIF-2alpha regulates murine hematopoietic development in an erythropoietin-dependent manner. Blood 105(8):3133–3140. https://doi.org/10.1182/blood-2004-05-1695
Seagroves TN, Ryan HE, Lu H et al (2001) Transcription factor HIF-1 is a necessary mediator of the pasteur effect in mammalian cells. Mol Cell Biol 21(10):3436–3444. https://doi.org/10.1128/MCB.21.10.3436-3444.2001
Seiken G, Grillo FG, Schaudies RP, Johnson JP (1994) Modulation of renal EGF in dichromate-induced acute renal failure treated with thyroid hormone. Kidney Int 45(6):1622–1627. https://doi.org/10.1038/ki.1994.213
Semenza GL, Wang GL (1992) A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol Cell Biol 12(12):5447–5454. https://doi.org/10.1128/mcb.12.12.5447-5454.1992
Shah YM, Xie L (2014) Hypoxia-inducible factors link iron homeostasis and erythropoiesis. Gastroenterology 146(3):630–642. https://doi.org/10.1053/j.gastro.2013.12.031
Sharma BK, Singhal PC, Chugh KS (1978) Intravascular haemolysis and acute renal failure following potassium dichromate poisoning. Postgrad Med J 54(632):414–415. https://doi.org/10.1136/pgmj.54.632.414
Sharma R, Singh VJ, Chawla PA (2022) Advancements in the Use of Platinum Complexes as Anticancer Agents. Anticancer Agents Med Chem. 22(5):821-835. https://doi.org/10.2174/1871520621666210805150705
Shen X, Chi Y, Xiong K (2019) The effect of heavy metal contamination on humans and animals in the vicinity of a zinc smelting facility. PLoS ONE 14(10):e0207423. https://doi.org/10.1371/journal.pone.0207423
Shenoy N, Dronca R, Quevedo F et al (2017) Low hypoxia inducible factor-1alpha (HIF-1alpha) expression in testicular germ cell tumors - a major reason for enhanced chemosensitivity? Chin J Cancer Res 29(4):374–378. https://doi.org/10.21147/j.issn.1000-9604.2017.04.11
Shiao YH, Rice JM, Anderson LM, Diwan BA, Hard GC (1998) von Hippel-Lindau gene mutations in N-nitrosodimethylamine-induced rat renal epithelial tumors. J Natl Cancer Inst 90(22):1720–1723. https://doi.org/10.1093/jnci/90.22.1720
Shiao YH, Kamata SI, Li LM et al (2002) Mutations in the VHL gene from potassium bromate-induced rat clear cell renal tumors. Cancer Lett 187(1–2):207–214. https://doi.org/10.1016/s0304-3835(02)00407-x
Shibuya M (2013) VEGFR and type-V RTK activation and signaling. Cold Spring Harb Perspect Biol 5(10):a009092. https://doi.org/10.1101/cshperspect.a009092
Shimizu H, Takahashi T, Suzuki T et al (2000) Protective effect of heme oxygenase induction in ischemic acute renal failure. Crit Care Med 28(3):809–817
Shiraishi F, Curtis LM, Truong L et al (2000) Heme oxygenase-1 gene ablation or expression modulates cisplatin-induced renal tubular apoptosis. Am J Physiol Renal Physiol 278(5):F726–F736. https://doi.org/10.1152/ajprenal.2000.278.5.F726
Shridhar V, Wang L, Rosati R et al (1997) Frequent breakpoints in the region surrounding FRA3B in sporadic renal cell carcinomas. Oncogene 14(11):1269–1277. https://doi.org/10.1038/sj.onc.1201100
Shu S, Wang Y, Zheng M et al (2019) Hypoxia and hypoxia-inducible factors in kidney injury and repair. Cells 8(3):207. https://doi.org/10.3390/cells8030207
Shulman A, Dwyer FP (1964) Metal chelates in biological systems. In: Dwyer FP, Mellor DP (eds) chelating agents and metal chelates. Academic Press, New York, pp 383–439
Siddique A, Kowdley KV (2012) Review article: the iron overload syndromes. Aliment Pharmacol Ther 35(8):876–893. https://doi.org/10.1111/j.1365-2036.2012.05051.x
Simonsen LO, Harbak H, Bennekou P (2012) Cobalt metabolism and toxicology–a brief update. The Science of the Total Environment 432:210–215. https://doi.org/10.1016/j.scitotenv.2012.06.009
Simpson RJ, McKie AT (2015) Iron and oxygen sensing: a tale of 2 interacting elements? Metallomics Integr Biomet Sci 7(2):223–231. https://doi.org/10.1039/c4mt00225c
Slotki I, Cabantchik ZI (2015) The Labile Side of Iron Supplementation in CKD. J Am Soc Nephrol 26(11):2612–2619. https://doi.org/10.1681/ASN.2015010052
Smith SW (2013) The role of chelation in the treatment of other metal poisonings. J Med Toxicol 9(4):355–369. https://doi.org/10.1007/s13181-013-0343-6
Smith CP, Thévenod F (2009) Iron transport and the kidney. Biochim Biophys Acta 1790(7):724–730. https://doi.org/10.1016/j.bbagen.2008.10.010
Smith KS, Huyck HLO (1999) An overview of the abundance, relative mobility, bioavailability, and human toxicity of metals. In: Plumlee GS, Logdson MJ (eds) The environmental geochemistry of mineral deposits part A: processes, techniques, and health issues. Reviews in economic geology, vol 6. Society of Economic Geologists Inc, Littleton, p 571
Soilleux EJ, Turley H, Tian YM, Pugh CW, Gatter KC, Harris AL (2005) Use of novel monoclonal antibodies to determine the expression and distribution of the hypoxia regulatory factors PHD-1, PHD-2, PHD-3 and FIH in normal and neoplastic human tissues. Histopathology 47(6):602–610. https://doi.org/10.1111/j.1365-2559.2005.02280.x
Soudani N, Sefi M, Ben Amara I, Boudawara T, Zeghal N (2010) Protective effects of Selenium (Se) on Chromium (VI) induced nephrotoxicity in adult rats. Ecotoxicol Environ Saf 73(4):671–678. https://doi.org/10.1016/j.ecoenv.2009.10.002
Soudani N, Sefi M, Bouaziz H, Chtourou Y, Boudawara T, Zeghal N (2011) Nephrotoxicity induced by chromium (VI) in adult rats and their progeny. Hum Exp Toxicol 30(9):1233–1245. https://doi.org/10.1177/0960327110387454
Souma T, Nezu M, Nakano D et al (2016) Erythropoietin Synthesis in Renal Myofibroblasts Is Restored by Activation of Hypoxia Signaling. J Am Soc Nephrol 27(2):428–438. https://doi.org/10.1681/ASN.2014121184
Standeven AM, Wetterhahn KE (1991) Ascorbate is the principal reductant of chromium (VI) in rat liver and kidney ultrafiltrates. Carcinogenesis 12(9):1733–1737. https://doi.org/10.1093/carcin/12.9.1733
Steinhoff A, Pientka FK, Mockel S et al (2009) Cellular oxygen sensing: Importins and exportins are mediators of intracellular localisation of prolyl-4-hydroxylases PHD1 and PHD2. Biochem Biophys Res Commun 387(4):705–711. https://doi.org/10.1016/j.bbrc.2009.07.090
Stohs SJ, Bagchi D (1995) Oxidative mechanisms in the toxicity of metal ions. Free Radic Biol Med 18(2):321–336
Stolze IP, Tian YM, Appelhoff RJ et al (2004) Genetic analysis of the role of the asparaginyl hydroxylase factor inhibiting hypoxia-inducible factor (FIH) in regulating hypoxia-inducible factor (HIF) transcriptional target genes [corrected]. J Biol Chem 279(41):42719–42725. https://doi.org/10.1074/jbc.M406713200
Sukosd F, Kuroda N, Beothe T, Kaur AP, Kovacs G (2003) Deletion of chromosome 3p14.2-p25 involving the VHL and FHIT genes in conventional renal cell carcinoma. Cancer Res 63(2):455–7
Sunderman FW Jr (1987) Metal induction of heme oxygenase. Ann N Y Acad Sci 514:65–80. https://doi.org/10.1111/j.1749-6632.1987.tb48762.x
Sung H, Ferlay J, Siegel RL et al (2021) Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin 71(3):209–249. https://doi.org/10.3322/caac.21660
Suzuki N, Matsuo-Tezuka Y, Sasaki Y et al (2018) Iron attenuates erythropoietin production by decreasing hypoxia-inducible transcription factor 2alpha concentrations in renal interstitial fibroblasts. Kidney Int 94(5):900–911. https://doi.org/10.1016/j.kint.2018.06.028
Takeda K, Cowan A, Fong GH (2007) Essential role for prolyl hydroxylase domain protein 2 in oxygen homeostasis of the adult vascular system. Circulation 116(7):774–781. https://doi.org/10.1161/CIRCULATIONAHA.107.701516
Takeda K, Aguila HL, Parikh NS et al (2008) Regulation of adult erythropoiesis by prolyl hydroxylase domain proteins. Blood 111(6):3229–3235. https://doi.org/10.1182/blood-2007-09-114561
Tanaka T (2016) Expanding roles of the hypoxia-response network in chronic kidney disease. Clin Exp Nephrol 20(6):835–844. https://doi.org/10.1007/s10157-016-1241-4
Tanaka T, Kojima I, Ohse T et al (2005a) Hypoxia-inducible factor modulates tubular cell survival in cisplatin nephrotoxicity. Am J Physiol Renal Physiol 289(5):F1123–F1133. https://doi.org/10.1152/ajprenal.00081.2005
Tanaka T, Kojima I, Ohse T et al (2005b) Cobalt promotes angiogenesis via hypoxia-inducible factor and protects tubulointerstitium in the remnant kidney model. Lab Invest 85(10):1292–1307. https://doi.org/10.1038/labinvest.3700328
Tanaka T, Matsumoto M, Inagi R et al (2005c) Induction of protective genes by cobalt ameliorates tubulointerstitial injury in the progressive Thy1 nephritis. Kidney Int 68(6):2714–2725. https://doi.org/10.1111/j.1523-1755.2005.00742.x
Taylor CT, Doherty G, Fallon PG, Cummins EP (2016) Hypoxia-dependent regulation of inflammatory pathways in immune cells. J Clin Invest 126(10):3716–3724. https://doi.org/10.1172/JCI84433
Theilig F, Enke AK, Scolari B, Polzin D, Bachmann S, Koesters R (2011) Tubular deficiency of von Hippel-Lindau attenuates renal disease progression in anti-GBM glomerulonephritis. Am J Pathol 179(5):2177–2188. https://doi.org/10.1016/j.ajpath.2011.07.012
Thévenod F (2003) Nephrotoxicity and the proximal tubule. Insights from Cadmium Nephron Physiol 93:87–93. https://doi.org/10.1159/000070241
Thévenod F (2009) Cadmium and cellular signaling cascades: To be or not to be? Toxicol Appl Pharmacol 238:221–239. https://doi.org/10.1016/j.taap.2009.01.013
Thévenod F (2018) Iron and Its Role in Cancer Defense: A Double-Edged Sword. Met Ions Life Sci 18:437–467. https://doi.org/10.1515/9783110470734-021
Thévenod F, Lee WK (2013) Toxicology of cadmium and its damage to Mammalian organs. Met Ions Life Sci 11:415–490. https://doi.org/10.1007/978-94-007-5179-8_14
Thévenod F, Wolff NA (2016) Iron transport in the kidney: implications for physiology and cadmium nephrotoxicity. Metallomics Integr Biomet Sci 8:17–42. https://doi.org/10.1039/c5mt00215j
Thévenod F, Lee WK, Garrick MD (2020) Iron and Cadmium Entry Into Renal Mitochondria: Physiological and Toxicological Implications. Front Cell Dev Biol 8:848. https://doi.org/10.3389/fcell.2020.00848
Tiwari R, Kapitsinou PP (2021) Role of endothelial prolyl-4-hydroxylase domain protein/hypoxia-inducible factor axis in acute kidney injury. Nephron. https://doi.org/10.1159/000518632
Tokar EJ, Benbrahim-Tallaa L, Waalkes MP (2011) Metal ions in human cancer development. Met Ions Life Sci 8:375–401
Torti SV, Manz DH, Paul BT, Blanchette-Farra N, Torti FM (2018) Iron and Cancer. Annu Rev Nutr 38:97–125. https://doi.org/10.1146/annurev-nutr-082117-051732
Toyokuni S (2009) Role of iron in carcinogenesis: cancer as a ferrotoxic disease. Cancer Sci 100(1):9–16. https://doi.org/10.1111/j.1349-7006.2008.01001.x
Tracz MJ, Alam J, Nath KA (2007) Physiology and pathophysiology of heme: implications for kidney disease. J Am Soc Nephrol 18(2):414–420. https://doi.org/10.1681/ASN.2006080894
Tsai TL, Kuo CC, Pan WH et al (2017) The decline in kidney function with chromium exposure is exacerbated with co-exposure to lead and cadmium. Kidney Int 92(3):710–720. https://doi.org/10.1016/j.kint.2017.03.013
Tsave O, Yavropoulou MP, Kafantari M, Gabriel C, Yovos JG, Salifoglou A (2016) The adipogenic potential of Cr(III). A molecular approach exemplifying metal-induced enhancement of insulin mimesis in diabetes mellitus II. J Inorg Biochem 163:323–331. https://doi.org/10.1016/j.jinorgbio.2016.07.015
Ufelle AC, Barchowsky A (2019) Toxic effects of metals. In: Klaassen CD (ed) Casarett and Doull’s toxicology : the basic science of poisons, 9th edn. McGraw-Hill, New York, pp 1107–1162
Urakami-Takebayashi Y, Kuroda Y, Murata T, Miyazaki M, Nagai J (2018) Pioglitazone induces hypoxia-inducible factor 1 activation in human renal proximal tubular epithelial cell line HK-2. Biochem Biophys Res Commun 503(3):1682–1688. https://doi.org/10.1016/j.bbrc.2018.07.099
Valko M, Morris H, Cronin MT (2005) Metals, toxicity and oxidative stress. Curr Med Chem 12(10):1161–1208
Van Avondt K, Nur E, Zeerleder S (2019) Mechanisms of haemolysis-induced kidney injury. Nat Rev Nephrol 15(11):671–692. https://doi.org/10.1038/s41581-019-0181-0
van den Berg A, Draaijers TG, Kok K et al (1997) Normal FHIT transcripts in renal cell cancer- and lung cancer-derived cell lines, including a cell line with a homozygous deletion in the FRA3B region. Genes Chromosomes Cancer 19(4):220–227. https://doi.org/10.1002/(sici)1098-2264(199708)19:4%3c220::aid-gcc3%3e3.0.co;2-z
van Raaij S, van Swelm R, Bouman K et al (2018) Tubular iron deposition and iron handling proteins in human healthy kidney and chronic kidney disease. Sci Rep 8(1):9353. https://doi.org/10.1038/s41598-018-27107-8
van Swelm RP, Wetzels JF, Verweij VG et al (2016) Renal handling of circulating and renal-synthesized hepcidin and its protective effects against hemoglobin-mediated kidney injury. J Am Soc Nephrol 27(9):2720–2732. https://doi.org/10.1681/ASN.2015040461
van Swelm RPL, Vos M, Verhoeven F, Thévenod F, Swinkels DW (2018) Endogenous hepcidin synthesis protects the distal nephron against hemin and hemoglobin mediated necroptosis. Cell Death Dis 9(5):550. https://doi.org/10.1038/s41419-018-0568-z
van Swelm RPL, Wetzels JFM, Swinkels DW (2020) The multifaceted role of iron in renal health and disease. Nat Rev Nephrol 16(2):77–98. https://doi.org/10.1038/s41581-019-0197-5
Villalpando-Rodriguez GE, Gibson SB (2021) Reactive oxygen species (ROS) regulates different types of cell death by acting as a rheostat. Oxid Med Cell Longev 2021:9912436. https://doi.org/10.1155/2021/9912436
Villar D, Vara-Vega A, Landazuri MO, Del Peso L (2007) Identification of a region on hypoxia-inducible-factor prolyl 4-hydroxylases that determines their specificity for the oxygen degradation domains. Biochem J 408(2):231–240. https://doi.org/10.1042/BJ20071052
Volarevic V, Djokovic B, Jankovic MG et al (2019) Molecular mechanisms of cisplatin-induced nephrotoxicity: a balance on the knife edge between renoprotection and tumor toxicity. J Biomed Sci 26(1):25. https://doi.org/10.1186/s12929-019-0518-9
Vyskocil A, Viau C, Cizkova M (1994) Chronic nephrotoxicity of soluble nickel in rats. Hum Exp Toxicol 13(10):689–693. https://doi.org/10.1177/096032719401301007
Wang GL, Semenza GL (1995) Purification and characterization of hypoxia-inducible factor 1. J Biol Chem 270(3):1230–1237. https://doi.org/10.1074/jbc.270.3.1230
Wang GL, Jiang BH, Rue EA, Semenza GL (1995) Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci USA 92(12):5510–5514. https://doi.org/10.1073/pnas.92.12.5510
Wang Y, Fang J, Leonard SS, Rao KM (2004) Cadmium inhibits the electron transfer chain and induces reactive oxygen species. Free Radic Biol Med 36(11):1434–1443. https://doi.org/10.1016/j.freeradbiomed.2004.03.010
Wang J, Biju MP, Wang MH, Haase VH, Dong Z (2006) Cytoprotective effects of hypoxia against cisplatin-induced tubular cell apoptosis: involvement of mitochondrial inhibition and p53 suppression. J Am Soc Nephrol 17(7):1875–1885. https://doi.org/10.1681/ASN.2005121371
Wang T, Jia G, Zhang J et al (2011) Renal impairment caused by chronic occupational chromate exposure. Int Arch Occup Environ Health 84(4):393–401. https://doi.org/10.1007/s00420-010-0569-4
Wang YF, Shyu HW, Chang YC et al (2012a) Nickel (II)-induced cytotoxicity and apoptosis in human proximal tubule cells through a ROS- and mitochondria-mediated pathway. Toxicol Appl Pharmacol 259(2):177–186. https://doi.org/10.1016/j.taap.2011.12.022
Wang Z, Schley G, Turkoglu G et al (2012b) The protective effect of prolyl-hydroxylase inhibition against renal ischaemia requires application prior to ischaemia but is superior to EPO treatment. Nephrol Dial Transplant 27(3):929–936. https://doi.org/10.1093/ndt/gfr379
Wang W, Wang W, Jiang Y et al (2014) Human adipose-derived stem cells modified by HIF-1alpha accelerate the recovery of cisplatin-induced acute renal injury in vitro. Biotechnol Lett 36(3):667–676. https://doi.org/10.1007/s10529-013-1389-x
Wang WW, Li ZZ, Wang W et al (2015) Enhanced renoprotective effect of HIF-1alpha modified human adipose-derived stem cells on cisplatin-induced acute kidney injury in vivo. Sci Rep 5:10851. https://doi.org/10.1038/srep10851
Wardman P, Candeias LP (1996) Fenton chemistry: an introduction. Radiat Res 145(5):523–531
Warnecke C, Zaborowska Z, Kurreck J et al (2004) Differentiating the functional role of hypoxia-inducible factor (HIF)-1alpha and HIF-2alpha (EPAS-1) by the use of RNA interference: erythropoietin is a HIF-2alpha target gene in Hep3B and Kelly cells. FASEB J 18(12):1462–1464. https://doi.org/10.1096/fj.04-1640fje
Watson JA, Watson CJ, McCann A, Baugh J (2010) Epigenetics, the epicenter of the hypoxic response. Epigenetics 5(4):293–296. https://doi.org/10.4161/epi.5.4.11684
Weber H (1983) Long-term study of the distribution of soluble chromate-51 in the rat after a single intratracheal administration. J Toxicol Environ Health 11(4–6):749–764. https://doi.org/10.1080/15287398309530382
Wedeen RP, Qian LF (1991) Chromium-induced kidney disease. Environ Health Perspect 92:71–74. https://doi.org/10.1289/ehp.92-1519395
Weidemann A, Bernhardt WM, Klanke B et al (2008) HIF activation protects from acute kidney injury. J Am Soc Nephrol 19(3):486–494. https://doi.org/10.1681/ASN.2007040419
Weinhouse C (2021) The roles of inducible chromatin and transcriptional memory in cellular defense system responses to redox-active pollutants. Free Radic Biol Med 170:85–108. https://doi.org/10.1016/j.freeradbiomed.2021.03.018
Wellmann S, Bettkober M, Zelmer A et al (2008) Hypoxia upregulates the histone demethylase JMJD1A via HIF-1. Biochem Biophys Res Commun 372(4):892–897. https://doi.org/10.1016/j.bbrc.2008.05.150
Wenger RH (2002) Cellular adaptation to hypoxia: O2-sensing protein hydroxylases, hypoxia-inducible transcription factors, and O2-regulated gene expression. FASEB J 16(10):1151–1162. https://doi.org/10.1096/fj.01-0944rev
Wenger RH, Stiehl DP (2005) Camenisch G (2005) Integration of oxygen signaling at the consensus HRE. Science’s STKE: Signal Transduct Knowl Environ 2005(306):re12. https://doi.org/10.1126/stke.3062005re12
WHO (1992) Cadmium. World Health Organization
WHO (2010) Exposure to cadmium: a major public health concern International Programme on Chemical Safety. World Health Organization. https://www.who.int/ipcs/assessment/public_health/cadmium/en/
Wiesener MS, Turley H, Allen WE et al (1998) Induction of endothelial PAS domain protein-1 by hypoxia: characterization and comparison with hypoxia-inducible factor-1alpha. Blood 92(7):2260–2268
Williams M, Bozhilov K, Ghai S, Talbot P (2017) Elements including metals in the atomizer and aerosol of disposable electronic cigarettes and electronic hookahs. PLoS ONE 12(4):e0175430. https://doi.org/10.1371/journal.pone.0175430
Xia X, Lemieux ME, Li W et al (2009) Integrative analysis of HIF binding and transactivation reveals its role in maintaining histone methylation homeostasis. Proc Natl Acad Sci USA 106(11):4260–4265. https://doi.org/10.1073/pnas.0810067106
Xiao F, Li Y, Dai L et al (2012) Hexavalent chromium targets mitochondrial respiratory chain complex I to induce reactive oxygen species-dependent caspase-3 activation in L-02 hepatocytes. Int J Mol Med 30(3):629–635. https://doi.org/10.3892/ijmm.2012.1031
Yadav G, Chambial S, Agrawal N et al (2020) Blood lead levels in antenatal women and its association with iron deficiency anemia and adverse pregnancy outcomes. J Fam Med Prim Care 9(6):3106–3111. https://doi.org/10.4103/jfmpc.jfmpc_78_20
Yamakawa K, Takahashi E, Murata M, Okui K, Yokoyama S, Nakamura Y (1992) Detailed mapping around the breakpoint of (3;8) translocation in familial renal cell carcinoma and FRA3B. Genomics 14(2):412–416. https://doi.org/10.1016/s0888-7543(05)80234-4
Yamazaki S, Souma T, Hirano I et al (2013) A mouse model of adult-onset anaemia due to erythropoietin deficiency. Nat Commun 4:1950. https://doi.org/10.1038/ncomms2950
Yasumoto K, Kowata Y, Yoshida A, Torii S, Sogawa K (2009) Role of the intracellular localization of HIF-prolyl hydroxylases. Biochim Biophys Acta 1793(5):792–797. https://doi.org/10.1016/j.bbamcr.2009.01.014
Yeh TL, Leissing TM, Abboud MI et al (2017) Molecular and cellular mechanisms of HIF prolyl hydroxylase inhibitors in clinical trials. Chem Sci 8(11):7651–7668. https://doi.org/10.1039/c7sc02103h
Ying JF, Lu ZB, Fu LQ et al (2021) The role of iron homeostasis and iron-mediated ROS in cancer. Am J Cancer Res 11(5):1895–1912
Yu X, Fang Y, Liu H et al (2012) The balance of beneficial and deleterious effects of hypoxia-inducible factor activation by prolyl hydroxylase inhibitor in rat remnant kidney depends on the timing of administration. Nephrol Dial Transplant 27(8):3110–3119. https://doi.org/10.1093/ndt/gfr754
Yu ASL, Chertow GM, Luyckx V, Marsden PA, Skorecki K, Taal MW (2019) Brenner & Rector’s the kidney, 11th edn. Elsevier, Amsterdam
Yuan Y, Beitner-Johnson D, Millhorn DE (2001) Hypoxia-inducible factor 2alpha binds to cobalt in vitro. Biochem Biophys Res Commun 288(4):849–854. https://doi.org/10.1006/bbrc.2001.5835
Zabarovsky ER, Lerman MI, Minna JD (2002) Tumor suppressor genes on chromosome 3p involved in the pathogenesis of lung and other cancers. Oncogene 21(45):6915–6935. https://doi.org/10.1038/sj.onc.1205835
Zafirov D, Petrusevska G, Sikole A et al (2008) Erythropoietin reduces cumulative nephrotoxicity from cisplatin and enhances renal tubular cell proliferation. Prilozi 29(2):167–183
Zager RA, Gamelin LM (1989) Pathogenetic mechanisms in experimental hemoglobinuric acute renal failure. Am J Physiol 256(3 Pt 2):F446–F455. https://doi.org/10.1152/ajprenal.1989.256.3.F446
Zager RA, Schimpf BA, Bredl CR, Gmur DJ (1993) Inorganic iron effects on in vitro hypoxic proximal renal tubular cell injury. J Clin Invest 91(2):702–708. https://doi.org/10.1172/JCI116251
Zager RA, Johnson AC, Frostad KB (2016) Combined iron sucrose and protoporphyrin treatment protects against ischemic and toxin-mediated acute renal failure. Kidney Int 90(1):67–76. https://doi.org/10.1016/j.kint.2016.01.022
Zager RA, Johnson ACM, Therapeutics R (2021) Iron sucrose ('RBT-3’) activates the hepatic and renal HAMP1 gene, evoking renal hepcidin loading and resistance to cisplatin nephrotoxicity. Nephrol Dial Transplant 36(3):465–474. https://doi.org/10.1093/ndt/gfaa348
Zarjou A, Kim J, Traylor AM et al (2011) Paracrine effects of mesenchymal stem cells in cisplatin-induced renal injury require heme oxygenase-1. Am J Physiol Renal Physiol 300(1):F254–F262. https://doi.org/10.1152/ajprenal.00594.2010
Zbar B, Brauch H, Talmadge C, Linehan M (1987) Loss of alleles of loci on the short arm of chromosome 3 in renal cell carcinoma. Nature 327(6124):721–724. https://doi.org/10.1038/327721a0
Zhang S, Lovejoy KS, Shima JE et al (2006) Organic cation transporters are determinants of oxaliplatin cytotoxicity. Cancer Res 66(17):8847–8857. https://doi.org/10.1158/0008-5472.CAN-06-0769
Zhang P, Yao Q, Lu L, Li Y, Chen PJ, Duan C (2014) Hypoxia-inducible factor 3 is an oxygen-dependent transcription activator and regulates a distinct transcriptional response to hypoxia. Cell Rep 6(6):1110–1121. https://doi.org/10.1016/j.celrep.2014.02.011
Zhao H, Han Y, Jiang N et al (2021) Effects of HIF-1alpha on renal fibrosis in cisplatin-induced chronic kidney disease. Clin Sci (lond) 135(10):1273–1288. https://doi.org/10.1042/CS20210061
Zhitkovich A (2005) Importance of chromium-DNA adducts in mutagenicity and toxicity of chromium(VI). Chem Res Toxicol 18(1):3–11. https://doi.org/10.1021/tx049774+
Zhou Y, Vaidya VS, Brown RP et al (2008) Comparison of kidney injury molecule-1 and other nephrotoxicity biomarkers in urine and kidney following acute exposure to gentamicin, mercury, and chromium. Toxicol Sci 101(1):159–170. https://doi.org/10.1093/toxsci/kfm260
Zhou J, Zhang W, Liang B et al (2009) PPARgamma activation induces autophagy in breast cancer cells. Int J Biochem Cell Biol 41(11):2334–2342. https://doi.org/10.1016/j.biocel.2009.06.007
Zoroddu MA, Aaseth J, Crisponi G, Medici S, Peana M, Nurchi VM (2019) The essential metals for humans: a brief overview. J Inorg Biochem 195:120–129. https://doi.org/10.1016/j.jinorgbio.2019.03.013
Acknowledgements
Frank Thévenod dedicates this review to the memory of Professor Karl-Martin Koch, MD (* 20.03.1934—† 22.12.2020), a pioneer of research on the pathogenesis of renal anemia and his MD supervisor, who triggered his passion for mechanistic and molecular foundations of renal diseases. F.T. received funding from BMBF (01DN16039), DFG (TH345), and ZBAF. W.-K.L. received financial support through the Intramural Funding Program at Witten/Herdecke University (IFF 2018-52).
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Thévenod, F., Schreiber, T. & Lee, WK. Renal hypoxia–HIF–PHD–EPO signaling in transition metal nephrotoxicity: friend or foe?. Arch Toxicol 96, 1573–1607 (2022). https://doi.org/10.1007/s00204-022-03285-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00204-022-03285-3