Computer-Aided Drug Design of β-Secretase, γ-Secretase and Anti-Tau Inhibitors for the Discovery of Novel Alzheimer’s Therapeutics


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Amyloid-β Pathway and its Druggable Targets
2.1. Function and Structure of β-Secretase
2.2. CADD for the Development of BACE1 Inhibitors
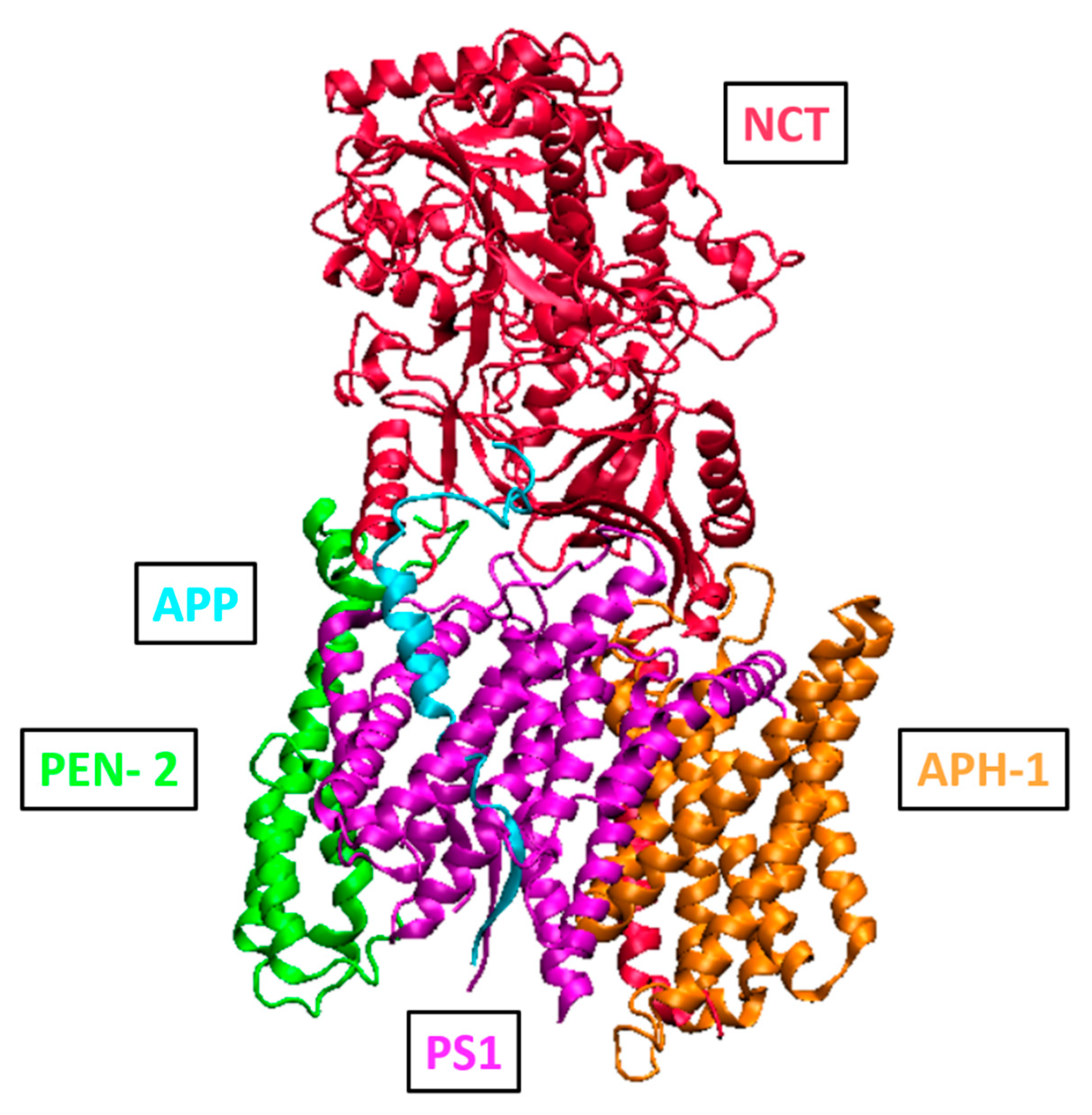
2.3. Function and Structure of γ-Secretase
2.4. CADD for the Development of γ-Secretase Inhibitors
2.5. CADD for the Development of Anti-Aβ Aggregation Inhibitors
3. Therapeutic Strategies Targeting Tau in Tauopathies
3.1. Function and Structure of Tau
3.2. CADD for the Development of Anti-Tau Inhibitors
4. Future Perspectives
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease; |
| CNS | Central nervous system |
| Aβ | Amyloid-β; |
| Tau | Tubulin-associated unit |
| NFTs | Neurofibrillary tangles |
| CADD | Computer-aided drug design |
| QSAR | Quantitative structure-activity relationship |
| APP | Amyloid precursor protein |
| Aβ40 | Amyloid-β peptide consisting of 40 residues |
| Aβ42 | Amyloid-β peptide consisting of 42 residues |
| BACE1 | APP cleaving enzyme I |
| BACE2 | APP cleaving enzyme II |
| TGN | Trans-Golgi network |
| MM-GBSA | Molecular mechanics generalized Born surface area |
| MD | Molecular dynamics |
| SOMFA | Self-organizing molecular field analysis |
| QM/MM | Quantum mechanics/molecular mechanics |
| i-CLiP | Intramembrane cleaving proteases |
| PS | Presenilin |
| NCT | Nicastrin |
| APH-1 | Anterior pharynx defective 1 |
| PEN- 2 | Presenilin enhancer 2 |
| NTF | N-terminal fragment |
| CTF | C-terminal fragment |
| GSIs | γ-secretase inhibitors |
| GSMs | γ-secretase modulators |
| Aβ49 | Amyloid-β peptide consisting of 49 residues |
| Aβ46 | Amyloid-β peptide consisting of 46 residues |
| Aβ43 | Amyloid-β peptide consisting of 43 residues |
| HQSAR | Hologram quantitative structure-activity relationship |
| PHFs | Paired helical filaments |
| SFs | Straight filaments; |
| GSK-3β | Glycogen synthase kinase-3β |
| Cdk5 | Cyclin-dependent kinase 5 |
| CK1 | Casein kinase 1 |
| PKA | Protein kinase A |
| PP2A | Protein phosphatase 2 |
| microED | Micro-electron diffraction |
| Cryo-EM | Cryo-electron microscopy |
| PET | Interactions of positron emission tomography |
| RMSD | Root mean square deviation |
| HDAC6 | Histone deacetylase 6 |
| MIF | Molecular interaction fields. |
References
- Alzheimer’s Association. 2019 Alzheimer’s disease facts and figures. Alzheimers Dement. 2019, 15, 321–387. [Google Scholar] [CrossRef]
- Nichols, E.; Szoeke, C.E.I.; Vollset, S.E.; Abbasi, N.; Abd-Allah, F.; Abdela, J.; Aichour, M.T.E.; Akinyemi, R.O.; Alahdab, F.; Asgedom, S.W.; et al. Global, regional, and national burden of Alzheimer’s disease and other dementias, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 88–106. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Gu, B.J.; Masters, C.L.; Wang, Y.J. A systemic view of Alzheimer disease—Insights from amyloid-beta metabolism beyond the brain. Nat. Rev. Neurol. 2017, 13, 612–623. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Xu, S.; Wu, T.; Shao, Y.; Luo, L.; Zhou, L.; Ou, S.; Tang, H.; Huang, W.; Guo, K.; et al. Abnormal platelet amyloid-β precursor protein metabolism in SAMP8 mice: Evidence for peripheral marker in Alzheimer’s disease. J. Cell. Physiol. 2019, 234, 23528–23536. [Google Scholar] [CrossRef] [PubMed]
- Šimić, G.; Babić Leko, M.; Wray, S.; Harrington, C.; Delalle, I.; Jovanov-Milošević, N.; Bažadona, D.; Buée, L.; De Silva, R.; Di Giovanni, G.; et al. Tau Protein Hyperphosphorylation and Aggregation in Alzheimer’s Disease and Other Tauopathies, and Possible Neuroprotective Strategies. Biomolecules 2016, 6, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alonso, A.D.; Cohen, L.S.; Corbo, C.; Morozova, V.; ElIdrissi, A.; Phillips, G.; Kleiman, F.E. Hyperphosphorylation of Tau Associates with Changes in Its Function Beyond Microtubule Stability. Front. Cell. Neurosci. 2018, 12, 338. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.-J.; Zhang, X.; Chen, W.-W. Role of oxidative stress in Alzheimer’s disease. Biomed. Rep. 2016, 4, 519–522. [Google Scholar] [CrossRef] [Green Version]
- Uttara, B.; Singh, A.V.; Zamboni, P.; Mahajan, R.T. Oxidative stress and neurodegenerative diseases: A review of upstream and downstream antioxidant therapeutic options. Curr. Neuropharmacol. 2009, 7, 65–74. [Google Scholar] [CrossRef] [Green Version]
- Bouvier, D.S.; Jones, E.V.; Quesseveur, G.; Davoli, M.A.; Ferreira, T.A.; Quirion, R.; Mechawar, N.; Murai, K.K. High Resolution Dissection of Reactive Glial Nets in Alzheimer’s Disease. Sci. Rep. 2016, 6, 24544. [Google Scholar] [CrossRef] [Green Version]
- Dario, T.; Michael, T.H. Microglia in Alzheimer’s Disease: The Good, the Bad and the Ugly. Curr. Alzheimer Res. 2016, 13, 370–380. [Google Scholar]
- Solas, M.; Puerta, E.; Ramirez, M.J. Treatment Options in Alzheimer s Disease: The GABA Story. Curr. Pharm. Des. 2015, 21, 4960–4971. [Google Scholar] [CrossRef] [PubMed]
- Graham, W.V.; Bonito-Oliva, A.; Sakmar, T.P. Update on Alzheimer’s Disease Therapy and Prevention Strategies. Annu. Rev. Med. 2017, 68, 413–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cummings, J.; Lee, G.; Ritter, A.; Sabbagh, M.; Zhong, K. Alzheimer’s disease drug development pipeline: 2019. Alzheimers Dement. 2019, 5, 272–293. [Google Scholar] [CrossRef] [PubMed]
- Indrani, B. Current Therapy and Computational Drug Designing Approaches for Neurodegenerative Diseases -with Focus on Alzheimer’s and Parkinson’s. Curr. Signal Transduct. Ther. 2019, 14, 122–128. [Google Scholar]
- Kametani, F.; Hasegawa, M. Reconsideration of Amyloid Hypothesis and Tau Hypothesis in Alzheimer’s Disease. Front. Neurosci. 2018, 12, 25. [Google Scholar] [CrossRef] [Green Version]
- Vassar, R.; Bennett, B.D.; Babu-Khan, S.; Kahn, S.; Mendiaz, E.A.; Denis, P.; Teplow, D.B.; Ross, S.; Amarante, P.; Loeloff, R.; et al. β-Secretase Cleavage of Alzheimer’s Amyloid Precursor Protein by the Transmembrane Aspartic Protease BACE. Science 1999, 286, 735–741. [Google Scholar] [CrossRef] [Green Version]
- Dislich, B.; Lichtenthaler, S. The Membrane-Bound Aspartyl Protease BACE1: Molecular and Functional Properties in Alzheimer’s Disease and Beyond. Front. Physiol. 2012, 3, 8. [Google Scholar] [CrossRef] [Green Version]
- Hussain, I.; Powell, D.; Howlett, D.R.; Tew, D.G.; Meek, T.D.; Chapman, C.; Gloger, I.S.; Murphy, K.E.; Southan, C.D.; Ryan, D.M.; et al. Identification of a novel aspartic protease (Asp 2) as beta-secretase. Mol. Cell. Neurosci. 1999, 14, 419–427. [Google Scholar] [CrossRef]
- Sinha, S.; Anderson, J.P.; Barbour, R.; Basi, G.S.; Caccavello, R.; Davis, D.; Doan, M.; Dovey, H.F.; Frigon, N.; Hong, J.; et al. Purification and cloning of amyloid precursor protein beta-secretase from human brain. Nature 1999, 402, 537–540. [Google Scholar] [CrossRef]
- Kandalepas, P.C.; Sadleir, K.R.; Eimer, W.A.; Zhao, J.; Nicholson, D.A.; Vassar, R. The Alzheimer’s β-secretase BACE1 localizes to normal presynaptic terminals and to dystrophic presynaptic terminals surrounding amyloid plaques. Acta Neuropathol. 2013, 126, 329–352. [Google Scholar] [CrossRef] [Green Version]
- Saunders, A.J.; Kim, T.-W.; Tanzi, R.E. BACE Maps to Chromosome 11 and a BACE Homolog, BACE2, Reside in the Obligate Down Syndrome Region of Chromosome 21. Science 1999, 286, 1255. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Wang, Y.; Qing, H.; Christensen, M.A.; Liu, Y.; Zhou, W.; Tong, Y.; Xiao, C.; Huang, Y.; Zhang, S.; et al. Distinct transcriptional regulation and function of the human BACE2 and BACE1 genes. FASEB J. 2005, 19, 739–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, L.; Koelsch, G.; Lin, X.; Wu, S.; Terzyan, S.; Ghosh, A.K.; Zhang, X.C.; Tang, J. Structure of the protease domain of memapsin 2 (beta-secretase) complexed with inhibitor. Science 2000, 290, 150–153. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Vuillard, L.; Cleasby, A.; Murray, C.W.; Yon, J. Apo and Inhibitor Complex Structures of BACE (β-secretase). J. Mol. Biol. 2004, 343, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Lu, W.; Lu, Y.; Zhong, M.; Sun, J.; Thomas, A.E.; Wilkinson, J.M.; Fucini, R.V.; Lam, M.; Randal, M.; et al. Aminoethylenes: A Tetrahedral Intermediate Isostere Yielding Potent Inhibitors of the Aspartyl Protease BACE-1. J. Med. Chem. 2006, 49, 839–842. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Kumaragurubaran, N.; Hong, L.; Lei, H.; Hussain, K.A.; Liu, C.-F.; Devasamudram, T.; Weerasena, V.; Turner, R.; Koelsch, G.; et al. Design, Synthesis and X-ray Structure of Protein−Ligand Complexes: Important Insight into Selectivity of Memapsin 2 (β-Secretase) Inhibitors. J. Am. Chem. Soc. 2006, 128, 5310–5311. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Judd, T.C.; Bartberger, M.D.; Brown, J.; Chen, K.; Fremeau, R.T.; Hickman, D.; Hitchcock, S.A.; Jordan, B.; Li, V.; et al. From Fragment Screening to in Vivo Efficacy: Optimization of a Series of 2-Aminoquinolines as Potent Inhibitors of Beta-Site Amyloid Precursor Protein Cleaving Enzyme 1 (BACE1). J. Med. Chem. 2011, 54, 5836–5857. [Google Scholar] [CrossRef]
- Xu, Y.; Li, M.J.; Greenblatt, H.; Chen, W.; Paz, A.; Dym, O.; Peleg, Y.; Chen, T.; Shen, X.; He, J.; et al. Flexibility of the flap in the active site of BACE1 as revealed by crystal structures and molecular dynamics simulations. Acta Crystallogr. D Biol. Crystallogr. 2012, 68, 13–25. [Google Scholar] [CrossRef]
- Cheng, Y.; Brown, J.; Judd, T.C.; Lopez, P.; Qian, W.; Powers, T.S.; Chen, J.J.; Bartberger, M.D.; Chen, K.; Dunn, R.T.; et al. An Orally Available BACE1 Inhibitor That Affords Robust CNS Aβ Reduction without Cardiovascular Liabilities. ACS Med. Chem. Lett. 2015, 6, 210–215. [Google Scholar] [CrossRef] [Green Version]
- Ruderisch, N.; Schlatter, D.; Kuglstatter, A.; Guba, W.; Huber, S.; Cusulin, C.; Benz, J.; Rufer, A.C.; Hoernschemeyer, J.; Schweitzer, C.; et al. Potent and Selective BACE-1 Peptide Inhibitors Lower Brain Aβ Levels Mediated by Brain Shuttle Transport. EBioMedicine 2017, 24, 76–92. [Google Scholar] [CrossRef] [Green Version]
- Fuchino, K.; Mitsuoka, Y.; Masui, M.; Kurose, N.; Yoshida, S.; Komano, K.; Yamamoto, T.; Ogawa, M.; Unemura, C.; Hosono, M.; et al. Rational Design of Novel 1,3-Oxazine Based β-Secretase (BACE1) Inhibitors: Incorporation of a Double Bond to Reduce P-gp Efflux Leading to Robust Aβ Reduction in the Brain. J. Med. Chem. 2018, 61, 5122–5137. [Google Scholar] [CrossRef]
- Fujimoto, K.; Matsuoka, E.; Asada, N.; Tadano, G.; Yamamoto, T.; Nakahara, K.; Fuchino, K.; Ito, H.; Kanegawa, N.; Moechars, D.; et al. Structure-Based Design of Selective β-Site Amyloid Precursor Protein Cleaving Enzyme 1 (BACE1) Inhibitors: Targeting the Flap to Gain Selectivity over BACE2. J. Med. Chem. 2019, 62, 5080–5095. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Shin, D.; Koelsch, G.; Lin, X.; Ermolieff, J.; Tang, J. Design of Potent Inhibitors for Human Brain Memapsin 2 (β-Secretase). J. Am. Chem. Soc. 2000, 122, 3522–3523. [Google Scholar] [CrossRef] [PubMed]
- Turner, R.T.; Koelsch, G.; Hong, L.; Castenheira, P.; Ghosh, A.; Tang, J. Subsite Specificity of Memapsin 2 (β-Secretase): Implications for Inhibitor Design. Biochemistry 2001, 40, 10001–10006. [Google Scholar] [CrossRef] [PubMed]
- Hong, L.; Turner, R.T.; Koelsch, G.; Shin, D.; Ghosh, A.K.; Tang, J. Crystal Structure of Memapsin 2 (β-Secretase) in Complex with an Inhibitor OM00-3. Biochemistry 2002, 41, 10963–10967. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Osswald, H.L. BACE1 (β-secretase) inhibitors for the treatment of Alzheimer’s disease. Chem. Soc. Rev. 2014, 43, 6765–6813. [Google Scholar] [CrossRef] [Green Version]
- Hamada, Y.; Ishiura, S.; Kiso, Y. BACE1 Inhibitor Peptides: Can an Infinitely Small kcat Value Turn the Substrate of an Enzyme into Its Inhibitor? ACS Med. Chem. Lett. 2012, 3, 193–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malamas, M.S.; Erdei, J.; Gunawan, I.; Turner, J.; Hu, Y.; Wagner, E.; Fan, K.; Chopra, R.; Olland, A.; Bard, J.; et al. Design and Synthesis of 5,5′-Disubstituted Aminohydantoins as Potent and Selective Human β-Secretase (BACE1) Inhibitors. J. Med. Chem. 2010, 53, 1146–1158. [Google Scholar] [CrossRef] [PubMed]
- Mandal, M.; Zhu, Z.; Cumming, J.N.; Liu, X.; Strickland, C.; Mazzola, R.D.; Caldwell, J.P.; Leach, P.; Grzelak, M.; Hyde, L.; et al. Design and Validation of Bicyclic Iminopyrimidinones as Beta Amyloid Cleaving Enzyme-1 (BACE1) Inhibitors: Conformational Constraint to Favor a Bioactive Conformation. J. Med. Chem. 2012, 55, 9331–9345. [Google Scholar] [CrossRef]
- Stamford, A.W.; Scott, J.D.; Li, S.W.; Babu, S.; Tadesse, D.; Hunter, R.; Wu, Y.; Misiaszek, J.; Cumming, J.N.; Gilbert, E.J.; et al. Discovery of an Orally Available, Brain Penetrant BACE1 Inhibitor That Affords Robust CNS Aβ Reduction. ACS Med. Chem. Lett. 2012, 3, 897–902. [Google Scholar] [CrossRef]
- Rueeger, H.; Lueoend, R.; Rogel, O.; Rondeau, J.M.; Mobitz, H.; Machauer, R.; Jacobson, L.; Staufenbiel, M.; Desrayaud, S.; Neumann, U. Discovery of cyclic sulfone hydroxyethylamines as potent and selective beta-site APP-cleaving enzyme 1 (BACE1) inhibitors: Structure-based design and in vivo reduction of amyloid beta-peptides. J. Med. Chem. 2012, 55, 3364–3386. [Google Scholar] [CrossRef] [PubMed]
- Azimi, S.; Zonouzi, A.; Firuzi, O.; Iraji, A.; Saeedi, M.; Mahdavi, M.; Edraki, N. Discovery of imidazopyridines containing isoindoline-1,3-dione framework as a new class of BACE1 inhibitors: Design, synthesis and SAR analysis. Eur. J. Med. Chem. 2017, 138, 729–737. [Google Scholar] [CrossRef] [PubMed]
- Iraji, A.; Firuzi, O.; Khoshneviszadeh, M.; Tavakkoli, M.; Mahdavi, M.; Nadri, H.; Edraki, N.; Miri, R. Multifunctional iminochromene-2H-carboxamide derivatives containing different aminomethylene triazole with BACE1 inhibitory, neuroprotective and metal chelating properties targeting Alzheimer’s disease. Eur. J. Med. Chem. 2017, 141, 690–702. [Google Scholar] [CrossRef] [PubMed]
- Yan, G.; Hao, L.; Niu, Y.; Huang, W.; Wang, W.; Xu, F.; Liang, L.; Wang, C.; Jin, H.; Xu, P. 2-Substituted-thio-N-(4-substituted-thiazol/1H-imidazol-2-yl) acetamides as BACE1 inhibitors: Synthesis, biological evaluation and docking studies. Eur. J. Med. Chem. 2017, 137, 462–475. [Google Scholar] [CrossRef] [PubMed]
- Yonezawa, S.; Yamamoto, T.; Yamakawa, H.; Muto, C.; Hosono, M.; Hattori, K.; Higashino, K.; Yutsudo, T.; Iwamoto, H.; Kondo, Y.; et al. Conformational Restriction Approach to β-Secretase (BACE1) Inhibitors: Effect of a Cyclopropane Ring to Induce an Alternative Binding Mode. J. Med. Chem. 2012, 55, 8838–8858. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.-J.; Guernon, J.; Yang, F.; Snyder, L.; Shi, J.; McClure, A.; Rajamani, R.; Park, H.; Ng, A.; Lewis, H.; et al. Targeting the BACE1 Active Site Flap Leads to a Potent Inhibitor That Elicits Robust Brain Aβ Reduction in Rodents. ACS Med. Chem. Lett. 2016, 7, 271–276. [Google Scholar] [CrossRef] [Green Version]
- Speranta, A.; Adina, L.M.; Dan, F.M.; Aurelia, D.; Maria, L.F. Computer-Aided Drug Design Applied to Beta and Gamma Secretase Inhibitors-Perspectives for New Alzheimer Disease Therapy. Curr. Enzyme Inhib. 2006, 2, 311–328. [Google Scholar]
- Mok, N.Y.; Chadwick, J.; Kellett, K.A.B.; Casas-Arce, E.; Hooper, N.M.; Johnson, A.P.; Fishwick, C.W.G. Discovery of Biphenylacetamide-Derived Inhibitors of BACE1 Using de Novo Structure-Based Molecular Design. J. Med. Chem. 2013, 56, 1843–1852. [Google Scholar] [CrossRef]
- Polgar, T.; Magyar, C.; Simon, I.; Keseru, G.M. Impact of ligand protonation on virtual screening against beta-secretase (BACE1). J. Chem. Inf. Model. 2007, 47, 2366–2373. [Google Scholar] [CrossRef]
- Polgar, T.; Keseru, G.M. Virtual screening for beta-secretase (BACE1) inhibitors reveals the importance of protonation states at Asp32 and Asp228. J. Med. Chem. 2005, 48, 3749–3755. [Google Scholar] [CrossRef]
- Vijayan, R.S.; Prabu, M.; Mascarenhas, N.M.; Ghoshal, N. Hybrid structure-based virtual screening protocol for the identification of novel BACE1 inhibitors. J. Chem. Inf. Model. 2009, 49, 647–657. [Google Scholar] [CrossRef] [PubMed]
- Nastase, A.F.; Boyd, D.B. Simple Structure-Based Approach for Predicting the Activity of Inhibitors of Beta-Secretase (BACE1) Associated with Alzheimer’s Disease. J. Chem. Inf. Model. 2012, 52, 3302–3307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakraborty, S.; Ramachandran, B.; Basu, S. Encompassing receptor flexibility in virtual screening using ensemble docking-based hybrid QSAR: Discovery of novel phytochemicals for BACE1 inhibition. Mol. Biosyst. 2014, 10, 2684–2692. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Chakraborty, S.; Basu, S. Hybrid approach to sieve out natural compounds against dual targets in Alzheimer’s Disease. Sci. Rep. 2019, 9, 3714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.; Roy, S.; Tripathi, S.; Sharma, A. Molecular docking based virtual screening of natural compounds as potential BACE1 inhibitors: 3D QSAR pharmacophore mapping and molecular dynamics analysis. J. Biomol. Struct. Dyn. 2016, 34, 239–249. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, S.; Bandyopadhyay, J.; Chakraborty, S.; Basu, S. Multi-target screening mines hesperidin as a multi-potent inhibitor: Implication in Alzheimer’s disease therapeutics. Eur. J. Med. Chem. 2016, 121, 810–822. [Google Scholar] [CrossRef]
- Ponzoni, I.; Sebastián-Pérez, V.; Martínez, M.J.; Roca, C.; De la Cruz Pérez, C.; Cravero, F.; Vazquez, G.E.; Páez, J.A.; Díaz, M.F.; Campillo, N.E. QSAR Classification Models for Predicting the Activity of Inhibitors of Beta-Secretase (BACE1) Associated with Alzheimer’s Disease. Sci. Rep. 2019, 9. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Zhou, M.; Wu, F.; Li, R.; Ding, Z. Self-organizing molecular field analysis on human β-secretase nonpeptide inhibitors: 5,5-disubstituted aminohydantoins. Eur. J. Med. Chem. 2011, 46, 58–64. [Google Scholar] [CrossRef]
- Helena, N.; Isela, G.-P.; Jose, E.R.-B.; Manolo, E.-C.; Xerardo, G.-M.; Francisco, P.-P. Review of Synthesis, Biological Assay and QSAR Studies of β-Secretase Inhibitors. Curr. Comput. Aided Drug Des. 2011, 7, 263–275. [Google Scholar]
- Park, H.; Lee, S. Determination of the Active Site Protonation State of β-Secretase from Molecular Dynamics Simulation and Docking Experiment: Implications for Structure-Based Inhibitor Design. J. Am. Chem. Soc. 2003, 125, 16416–16422. [Google Scholar] [CrossRef]
- Hernández-Rodríguez, M.; Correa-Basurto, J.; Gutiérrez, A.; Vitorica, J.; Rosales-Hernández, M.C. Asp32 and Asp228 determine the selective inhibition of BACE1 as shown by docking and molecular dynamics simulations. Eur. J. Med. Chem. 2016, 124, 1142–1154. [Google Scholar] [CrossRef]
- Frush, E.H.; Sekharan, S.; Keinan, S. In Silico Prediction of Ligand Binding Energies in Multiple Therapeutic Targets and Diverse Ligand Sets—A Case Study on BACE1, TYK2, HSP90, and PERK Proteins. J. Phys. Chem. B 2017, 121, 8142–8148. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wang, J.; Yin, B.; Pang, L.; Wang, W.; Zhu, W. Molecular Mechanism of Binding Selectivity of Inhibitors toward BACE1 and BACE2 Revealed by Multiple Short Molecular Dynamics Simulations and Free-Energy Predictions. ACS Chem. Neurosci. 2019, 10, 4303–4318. [Google Scholar] [CrossRef] [PubMed]
- Ciordia, M.; Pérez-Benito, L.; Delgado, F.; Trabanco, A.A.; Tresadern, G. Application of Free Energy Perturbation for the Design of BACE1 Inhibitors. J. Chem. Inf. Model. 2016, 56, 1856–1871. [Google Scholar] [CrossRef] [PubMed]
- Krishnaswamy, S.; Betz, A. Exosites Determine Macromolecular Substrate Recognition by Prothrombinase. Biochemistry 1997, 36, 12080–12086. [Google Scholar] [CrossRef] [PubMed]
- Das, C.; Berezovska, O.; Diehl, T.S.; Genet, C.; Buldyrev, I.; Tsai, J.-Y.; Hyman, B.T.; Wolfe, M.S. Designed Helical Peptides Inhibit an Intramembrane Protease. J. Am. Chem. Soc. 2003, 125, 11794–11795. [Google Scholar] [CrossRef] [PubMed]
- Kornacker, M.G.; Lai, Z.; Witmer, M.; Ma, J.; Hendrick, J.; Lee, V.G.; Riexinger, D.J.; Mapelli, C.; Metzler, W.; Copeland, R.A. An inhibitor binding pocket distinct from the catalytic active site on human beta-APP cleaving enzyme. Biochemistry 2005, 44, 11567–11573. [Google Scholar] [CrossRef]
- Drag, M.; Salvesen, G.S. Emerging principles in protease-based drug discovery. Nat. Rev. Drug Discov. 2010, 9, 690–701. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Liu, Y.; Lazarus, R. Allosteric inhibition of BACE1 by an exosite-binding antibody. Curr. Opin. Struct. Biol. 2013, 23, 797–805. [Google Scholar] [CrossRef]
- Gutierrez, L.J.; Angelina, E.; Gyebrovszki, A.; Fulop, L.; Peruchena, N.; Baldoni, H.A.; Penke, B.; Enriz, R.D. New small-size peptides modulators of the exosite of BACE1 obtained from a structure-based design. J. Biomol. Struct. Dyn. 2017, 35, 413–426. [Google Scholar] [CrossRef]
- Atwal, J.K.; Chen, Y.; Chiu, C.; Mortensen, D.L.; Meilandt, W.J.; Liu, Y.; Heise, C.E.; Hoyte, K.; Luk, W.; Lu, Y.; et al. A Therapeutic Antibody Targeting BACE1 Inhibits Amyloid-β Production in Vivo. Sci. Transl. Med. 2011, 3, 84ra43. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, L.J.; Enriz, R.D.; Baldoni, H.A. Structural and Thermodynamic Characteristics of the Exosite Binding Pocket on the Human BACE1: A Molecular Modeling Approach. J. Phys. Chem. A 2010, 114, 10261–10269. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez, L.J.; Andujar, S.A.; Enriz, R.D.; Baldoni, H.A. Structural and functional insights into the anti-BACE1 Fab fragment that recognizes the BACE1 exosite. J. Biomol. Struct. Dyn. 2014, 32, 1421–1433. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, M.S. Structure and Function of the γ-Secretase Complex. Biochemistry 2019, 58, 2953–2966. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, Y.; Xu, H.; Zhang, Y.-W. The γ-secretase complex: From structure to function. Front. Cell. Neurosci. 2014, 8, 427. [Google Scholar] [CrossRef] [Green Version]
- Frånberg, J.; Welander, H.; Aoki, M.; Winblad, B.; Tjernberg, L.O.; Frykman, S. Rat Brain γ-Secretase Activity Is Highly Influenced by Detergents. Biochemistry 2007, 46, 7647–7654. [Google Scholar] [CrossRef]
- Frykman, S.; Hur, J.-Y.; Frånberg, J.; Aoki, M.; Winblad, B.; Nahalkova, J.; Behbahani, H.; Tjernberg, L.O. Synaptic and Endosomal Localization of Active γ-Secretase in Rat Brain. PLoS ONE 2010, 5, e8948. [Google Scholar] [CrossRef] [Green Version]
- De Strooper, B. Aph-1, Pen-2, and Nicastrin with Presenilin Generate an Active γ-Secretase Complex. Neuron 2003, 38, 9–12. [Google Scholar] [CrossRef] [Green Version]
- Iwatsubo, T. The γ-secretase complex: Machinery for intramembrane proteolysis. Curr. Opin. Neurobiol. 2004, 14, 379–383. [Google Scholar] [CrossRef]
- Kimberly, W.T.; LaVoie, M.J.; Ostaszewski, B.L.; Ye, W.; Wolfe, M.S.; Selkoe, D.J. γ-Secretase is a membrane protein complex comprised of presenilin, nicastrin, aph-1, and pen-2. Proc. Natl. Acad. Sci. USA 2003, 100, 6382–6387. [Google Scholar] [CrossRef] [Green Version]
- Sato, T.; Diehl, T.S.; Narayanan, S.; Funamoto, S.; Ihara, Y.; De Strooper, B.; Steiner, H.; Haass, C.; Wolfe, M.S. Active γ-Secretase Complexes Contain Only One of Each Component. J. Biol. Chem. 2007, 282, 33985–33993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, X.-C.; Rajendra, E.; Yang, G.; Shi, Y.; Scheres, S.H.W. Sampling the conformational space of the catalytic subunit of human γ-secretase. eLife 2015, 4, e11182. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.-C.; Yan, C.; Yang, G.; Lu, P.; Ma, D.; Sun, L.; Zhou, R.; Scheres, S.H.W.; Shi, Y. An atomic structure of human γ-secretase. Nature 2015, 525, 212–217. [Google Scholar] [CrossRef] [Green Version]
- Yang, G.; Zhou, R.; Zhou, Q.; Guo, X.; Yan, C.; Ke, M.; Lei, J.; Shi, Y. Structural basis of Notch recognition by human γ-secretase. Nature 2019, 565, 192–197. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Yang, G.; Guo, X.; Zhou, Q.; Lei, J.; Shi, Y. Recognition of the amyloid precursor protein by human γ-secretase. Science 2019, 363, eaaw0930. [Google Scholar] [CrossRef]
- Wolfe, M.S.; Xia, W.; Ostaszewski, B.L.; Diehl, T.S.; Kimberly, W.T.; Selkoe, D.J. Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and γ-secretase activity. Nature 1999, 398, 513–517. [Google Scholar] [CrossRef]
- Thinakaran, G.; Borchelt, D.R.; Lee, M.K.; Slunt, H.H.; Spitzer, L.; Kim, G.; Ratovitsky, T.; Davenport, F.; Nordstedt, C.; Seeger, M.; et al. Endoproteolysis of Presenilin 1 and Accumulation of Processed Derivatives in Vivo. Neuron 1996, 17, 181–190. [Google Scholar] [CrossRef] [Green Version]
- Takasugi, N.; Tomita, T.; Hayashi, I.; Tsuruoka, M.; Niimura, M.; Takahashi, Y.; Thinakaran, G.; Iwatsubo, T. The role of presenilin cofactors in the γ-secretase complex. Nature 2003, 422, 438–441. [Google Scholar] [CrossRef]
- Shah, S.; Lee, S.-F.; Tabuchi, K.; Hao, Y.-H.; Yu, C.; LaPlant, Q.; Ball, H.; Dann, C.E.; Südhof, T.; Yu, G. Nicastrin Functions as a γ-Secretase-Substrate Receptor. Cell 2005, 122, 435–447. [Google Scholar] [CrossRef] [Green Version]
- Golde, T.E.; Petrucelli, L.; Lewis, J. Targeting Aβ and tau in Alzheimer’s disease, an early interim report. Exp. Neurol. 2010, 223, 252–266. [Google Scholar] [CrossRef] [Green Version]
- Wolfe, M.S. Inhibition and Modulation of γ-Secretase for Alzheimer’s Disease. Neurotherapeutics 2008, 5, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Golde, T.E.; Koo, E.H.; Felsenstein, K.M.; Osborne, B.A.; Miele, L. γ-Secretase inhibitors and modulators. Biochim. Biophys. Acta 2013, 1828, 2898–2907. [Google Scholar] [CrossRef] [Green Version]
- Wong, G.T.; Manfra, D.; Poulet, F.M.; Zhang, Q.; Josien, H.; Bara, T.; Engstrom, L.; Pinzon-Ortiz, M.; Fine, J.S.; Lee, H.J.; et al. Chronic treatment with the gamma-secretase inhibitor LY-411,575 inhibits beta-amyloid peptide production and alters lymphopoiesis and intestinal cell differentiation. J. Biol. Chem. 2004, 279, 12876–12882. [Google Scholar] [CrossRef] [Green Version]
- Ling, I.F.; Golde, T.E.; Galasko, D.R.; Koo, E.H. Modulation of Aβ42 in vivo by γ-secretase modulator in primates and humans. Alzheimers Res. Ther. 2015, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arun Ekiri Vaidyanathan, R.; Karthic, K.; Arun, M.; Nandini, S. In Silico Screening of Drugs to Find Potential Gamma-Secretase Inhibitors Using Pharmacophore Modeling, QSAR and Molecular Docking Studies. Comb. Chem. High Throughput Screen. 2014, 17, 770–780. [Google Scholar]
- Lee, J.Y.; Feng, Z.; Xie, X.-Q.; Bahar, I. Allosteric Modulation of Intact γ-Secretase Structural Dynamics. Biophys. J. 2017, 113, 2634–2649. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Zhang, W.; Han, W. Initial Substrate Binding of γ-Secretase: The Role of Substrate Flexibility. ACS Chem. Neurosci. 2017, 8, 1279–1290. [Google Scholar] [CrossRef]
- Gertsik, N.; am Ende, C.W.; Geoghegan, K.F.; Nguyen, C.; Mukherjee, P.; Mente, S.; Seneviratne, U.; Johnson, D.S.; Li, Y.-M. Mapping the Binding Site of BMS-708163 on γ-Secretase with Cleavable Photoprobes. Cell Chem. Biol. 2017, 24, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Somavarapu, A.K.; Kepp, K.P. Membrane Dynamics of γ-Secretase Provides a Molecular Basis for β-Amyloid Binding and Processing. ACS Chem. Neurosci. 2017, 8, 2424–2436. [Google Scholar] [CrossRef] [Green Version]
- Hitzenberger, M.; Zacharias, M. Structural Modeling of γ-Secretase Aβn Complex Formation and Substrate Processing. ACS Chem. Neurosci. 2019, 10, 1826–1840. [Google Scholar] [CrossRef]
- Pester, O.; Barrett, P.J.; Hornburg, D.; Hornburg, P.; Pröbstle, R.; Widmaier, S.; Kutzner, C.; Dürrbaum, M.; Kapurniotu, A.; Sanders, C.R.; et al. The Backbone Dynamics of the Amyloid Precursor Protein Transmembrane Helix Provides a Rationale for the Sequential Cleavage Mechanism of γ-Secretase. J. Am. Chem. Soc. 2013, 135, 1317–1329. [Google Scholar] [CrossRef] [Green Version]
- Hitzenberger, M.; Zacharias, M. Uncovering the Binding Mode of γ -Secretase Inhibitors. ACS Chem. Neurosci. 2019, 10, 3398–3403. [Google Scholar] [CrossRef]
- Tang, N.; Somavarapu, A.K.; Kepp, K.P. Molecular Recipe for γ-Secretase Modulation from Computational Analysis of 60 Active Compounds. ACS Omega 2018, 3, 18078–18088. [Google Scholar] [CrossRef] [Green Version]
- Aguayo-Ortiz, R.; Dominguez, L. Simulating the γ-secretase enzyme: Recent advances and future directions. Biochimie 2018, 147, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Afantitis, A.; Melagraki, G.; Koutentis, P.A.; Sarimveis, H.; Kollias, G. Ligand-based virtual screening procedure for the prediction and the identification of novel β-amyloid aggregation inhibitors using Kohonen maps and Counterpropagation Artificial Neural Networks. Eur. J. Med. Chem. 2011, 46, 497–508. [Google Scholar] [CrossRef] [PubMed]
- Eskici, G.; Gur, M. Computational design of new Peptide inhibitors for amyloid Beta (Aβ) aggregation in Alzheimer’s disease: Application of a novel methodology. PLoS ONE 2013, 8, e66178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.; Yoon, J.; Shin, S. Computational Study on Structure and Aggregation Pathway of Aβ42 Amyloid Protofibril. J. Phys. Chem. B 2019, 123, 7859–7868. [Google Scholar] [CrossRef]
- Zhou, Z.-L.; Ho, Y.; Liu, H.-L.; Elumalai, P.; Chen, W.-H. Computer-aided discovery of novel non-peptide inhibitors against amyloid-beta (Aβ) peptide aggregation for treating Alzheimer’s disease. Mol. Simul. 2015, 41, 622–632. [Google Scholar] [CrossRef]
- Mehrazma, B.; Opare, S.; Petoyan, A.; Rauk, A. D-Amino Acid Pseudopeptides as Potential Amyloid-Beta Aggregation Inhibitors. Molecules 2018, 23, 2387. [Google Scholar] [CrossRef] [Green Version]
- Novick, P.A.; Lopes, D.H.; Branson, K.M.; Esteras-Chopo, A.; Graef, I.A.; Bitan, G.; Pande, V.S. Design of β-Amyloid Aggregation Inhibitors from a Predicted Structural Motif. J. Med. Chem. 2012, 55, 3002–3010. [Google Scholar] [CrossRef] [Green Version]
- Aswathy, L.; Jisha, R.S.; Masand, V.H.; Gajbhiye, J.M.; Shibi, I.G. Design of novel amyloid β aggregation inhibitors using QSAR, pharmacophore modeling, molecular docking and ADME prediction. In Silico Pharmacol. 2018, 6. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Simone, A.D.; Schenk, D.; Toth, G.; Dobson, C.M.; Vendruscolo, M. Identification of small-molecule binding pockets in the soluble monomeric form of the Aβ42 peptide. J. Chem. Phys. 2013, 139, 035101. [Google Scholar] [CrossRef] [Green Version]
- Shinzato, T.; Sato, R.; Suzuki, K.; Tomioka, S.; Sogawa, H.; Shulga, S.; Blume, Y.; Kurita, N. Proposal of therapeutic curcumin derivatives for Alzheimer’s disease based on ab initio molecular simulations. Chem. Phys. Lett. 2019, 738, 136883. [Google Scholar] [CrossRef]
- Lu, J.; Cao, Q.; Wang, C.; Zheng, J.; Luo, F.; Xie, J.; Li, Y.; Ma, X.; He, L.; Eisenberg, D.; et al. Structure-Based Peptide Inhibitor Design of Amyloid-β Aggregation. Front. Mol. Neurosci. 2019, 12, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medina, M. An Overview on the Clinical Development of Tau-Based Therapeutics. Int. J. Mol. Sci. 2018, 19, 1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Congdon, E.E.; Sigurdsson, E.M. Tau-targeting therapies for Alzheimer disease. Nat. Rev. Neurol. 2018, 14, 399–415. [Google Scholar] [CrossRef]
- Fichou, Y.; Al-Hilaly, Y.K.; Devred, F.; Smet-Nocca, C.; Tsvetkov, P.O.; Verelst, J.; Winderickx, J.; Geukens, N.; Vanmechelen, E.; Perrotin, A.; et al. The elusive tau molecular structures: Can we translate the recent breakthroughs into new targets for intervention? Acta Neuropathol. Commun. 2019, 7, 31. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, H.; Goedert, M. Phosphorylation of microtubule-associated protein tau by AMPK-related kinases. J. Neurochem. 2012, 120, 165–176. [Google Scholar] [CrossRef]
- Schedin-Weiss, S.; Winblad, B.; Tjernberg, L.O. The role of protein glycosylation in Alzheimer disease. FEBS J. 2014, 281, 46–62. [Google Scholar] [CrossRef]
- Cohen, T.J.; Guo, J.L.; Hurtado, D.E.; Kwong, L.K.; Mills, I.P.; Trojanowski, J.Q.; Lee, V.M.Y. The acetylation of tau inhibits its function and promotes pathological tau aggregation. Nat. Commun. 2011, 2, 252. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Sierra, F.; Mondragon-Rodriguez, S.; Basurto-Islas, G. Truncation of tau protein and its pathological significance in Alzheimer’s disease. J. Alzheimer’s Dis. 2008, 14, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Morishima-Kawashima, M.; Hasegawa, M.; Takio, K.; Suzuki, M.; Yoshida, H.; Watanabe, A.; Titani, K.; Ihara, Y. Hyperphosphorylation of Tau in PHF. Neurobiol. Aging 1995, 16, 365–371. [Google Scholar] [CrossRef]
- Bretteville, A.; Ando, K.; Ghestem, A.; Loyens, A.; Begard, S.; Beauvillain, J.C.; Sergeant, N.; Hamdane, M.; Buee, L. Two-dimensional electrophoresis of tau mutants reveals specific phosphorylation pattern likely linked to early tau conformational changes. PLoS ONE 2009, 4, e4843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buée, L.; Bussière, T.; Buée-Scherrer, V.; Delacourte, A.; Hof, P.R. Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res. Rev. 2000, 33, 95–130. [Google Scholar] [CrossRef]
- Chen, X.; Reichert, M.C.; Gan, L. Chapter 5—Molecular Pathways in Alzheimer’s Disease and Cognitive Function: New Insights into Pathobiology of Tau. In Genes, Environment and Alzheimer’s Disease; Lazarov, O., Tesco, G., Eds.; Academic Press: San Diego, CA, USA, 2016; pp. 135–167. [Google Scholar]
- Liu, F.; Grundke-Iqbal, I.; Iqbal, K.; Gong, C.-X. Contributions of protein phosphatases PP1, PP2A, PP2B and PP5 to the regulation of tau phosphorylation. Eur. J. Neurosci. 2005, 22, 1942–1950. [Google Scholar] [CrossRef]
- Sato, Y.; Naito, Y.; Grundke-Iqbal, I.; Iqbal, K.; Endo, T. Analysis of N-glycans of pathological tau: Possible occurrence of aberrant processing of tau in Alzheimer’s disease. FEBS Lett. 2001, 496, 152–160. [Google Scholar] [CrossRef] [Green Version]
- Arnold, C.S.; Johnson, G.V.; Cole, R.N.; Dong, D.L.; Lee, M.; Hart, G.W. The microtubule-associated protein tau is extensively modified with O-linked N-acetylglucosamine. J. Biol. Chem. 1996, 271, 28741–28744. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Shan, X.; Yuzwa, S.A.; Vocadlo, D.J. The emerging link between O-GlcNAc and Alzheimer disease. J. Biol. Chem. 2014, 289, 34472–34481. [Google Scholar] [CrossRef] [Green Version]
- Grinberg, L.T.; Wang, X.; Wang, C.; Sohn, P.D.; Theofilas, P.; Sidhu, M.; Arevalo, J.B.; Heinsen, H.; Huang, E.J.; Rosen, H.; et al. Argyrophilic grain disease differs from other tauopathies by lacking tau acetylation. Acta Neuropathol. 2013, 125, 581–593. [Google Scholar] [CrossRef] [Green Version]
- Cook, C.; Carlomagno, Y.; Gendron, T.F.; Dunmore, J.; Scheffel, K.; Stetler, C.; Davis, M.; Dickson, D.; Jarpe, M.; DeTure, M.; et al. Acetylation of the KXGS motifs in tau is a critical determinant in modulation of tau aggregation and clearance. Hum. Mol. Genet. 2014, 23, 104–116. [Google Scholar] [CrossRef]
- Weingarten, M.D.; Lockwood, A.H.; Hwo, S.Y.; Kirschner, M.W. A protein factor essential for microtubule assembly. Proc. Natl. Acad. Sci. USA 1975, 72, 1858–1862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cleveland, D.W.; Hwo, S.-Y.; Kirschner, M.W. Purification of tau, a microtubule-associated protein that induces assembly of microtubules from purified tubulin. J. Mol. Biol. 1977, 116, 207–225. [Google Scholar] [CrossRef]
- Cleveland, D.W.; Hwo, S.-Y.; Kirschner, M.W. Physical and chemical properties of purified tau factor and the role of tau in microtubule assembly. J. Mol. Biol. 1977, 116, 227–247. [Google Scholar] [CrossRef]
- Jameson, L.; Frey, T.; Zeeberg, B.; Dalldorf, F.; Caplow, M. Inhibition of microtubule assembly by phosphorylation of microtubule-associated proteins. Biochemistry 1980, 19, 2472–2479. [Google Scholar] [CrossRef]
- Gu, Y.; Oyama, F.; Ihara, Y. Tau is widely expressed in rat tissues. J. Neurochem. 1996, 67, 1235–1244. [Google Scholar] [CrossRef]
- Goedert, M.; Wischik, C.M.; Crowther, R.A.; Walker, J.E.; Klug, A. Cloning and sequencing of the cDNA encoding a core protein of the paired helical filament of Alzheimer disease: Identification as the microtubule-associated protein tau. Proc. Natl. Acad. Sci. USA 1988, 85, 4051–4055. [Google Scholar] [CrossRef] [Green Version]
- Goedert, M.; Spillantini, M.G.; Jakes, R.; Rutherford, D.; Crowther, R.A. Multiple isoforms of human microtubule-associated protein tau: Sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron 1989, 3, 519–526. [Google Scholar] [CrossRef]
- Steiner, B.; Mandelkow, E.M.; Biernat, J.; Gustke, N.; Meyer, H.E.; Schmidt, B.; Mieskes, G.; Söling, H.D.; Drechsel, D.; Kirschner, M.W.; et al. Phosphorylation of microtubule-associated protein tau: Identification of the site for Ca2(+)-calmodulin dependent kinase and relationship with tau phosphorylation in Alzheimer tangles. EMBO J. 1990, 9, 3539–3544. [Google Scholar] [CrossRef]
- Mandelkow, E.-M.; Mandelkow, E. Biochemistry and cell biology of tau protein in neurofibrillary degeneration. Cold Spring Harb. Perspect. Med. 2012, 2, a006247. [Google Scholar] [CrossRef]
- von Bergen, M.; Friedhoff, P.; Biernat, J.; Heberle, J.; Mandelkow, E.-M.; Mandelkow, E. Assembly of τ protein into Alzheimer paired helical filaments depends on a local sequence motif (306VQIVYK311) forming β structure. Proc. Natl. Acad. Sci. USA 2000, 97, 5129–5134. [Google Scholar] [CrossRef] [Green Version]
- Seidler, P.M.; Boyer, D.R.; Rodriguez, J.A.; Sawaya, M.R.; Cascio, D.; Murray, K.; Gonen, T.; Eisenberg, D.S. Structure-based inhibitors of tau aggregation. Nat. Chem. 2017, 10, 170–176. [Google Scholar] [CrossRef] [PubMed]
- de la Cruz, M.J.; Hattne, J.; Shi, D.; Seidler, P.; Rodriguez, J.; Reyes, F.E.; Sawaya, M.R.; Cascio, D.; Weiss, S.C.; Kim, S.K.; et al. Atomic-resolution structures from fragmented protein crystals with the cryoEM method MicroED. Nat. Methods 2017, 14, 399–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabrales Fontela, Y.; Kadavath, H.; Biernat, J.; Riedel, D.; Mandelkow, E.; Zweckstetter, M. Multivalent cross-linking of actin filaments and microtubules through the microtubule-associated protein Tau. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Kadavath, H.; Jaremko, M.; Jaremko, L.; Biernat, J.; Mandelkow, E.; Zweckstetter, M. Folding of the Tau Protein on Microtubules. Angew. Chem. Int. Ed. Engl. 2015, 54, 10347–10351. [Google Scholar] [CrossRef]
- Fitzpatrick, A.W.P.; Falcon, B.; He, S.; Murzin, A.G.; Murshudov, G.; Garringer, H.J.; Crowther, R.A.; Ghetti, B.; Goedert, M.; Scheres, S.H.W. Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature 2017, 547, 185–190. [Google Scholar] [CrossRef] [Green Version]
- Falcon, B.; Zhang, W.; Schweighauser, M.; Murzin, A.G.; Vidal, R.; Garringer, H.J.; Ghetti, B.; Scheres, S.H.W.; Goedert, M. Tau filaments from multiple cases of sporadic and inherited Alzheimer’s disease adopt a common fold. Acta Neuropathol. 2018, 136, 699–708. [Google Scholar] [CrossRef] [Green Version]
- Liang, Z.; Zhang, B.; Su, W.W.; Williams, P.G.; Li, Q.X. C-Glycosylflavones Alleviate Tau Phosphorylation and Amyloid Neurotoxicity through GSK3β Inhibition. ACS Chem. Neurosci. 2016, 7, 912–923. [Google Scholar] [CrossRef]
- Llorach-Pares, L.; Nonell-Canals, A.; Sanchez-Martinez, M.; Avila, C. Computer-Aided Drug Design Applied to Marine Drug Discovery: Meridianins as Alzheimer’s Disease Therapeutic Agents. Mar. Drugs 2017, 15, 366. [Google Scholar] [CrossRef] [Green Version]
- Zeb, A.; Son, M.; Yoon, S.; Kim, J.H.; Park, S.J.; Lee, K.W. Computational Simulations Identified Two Candidate Inhibitors of Cdk5/p25 to Abrogate Tau-associated Neurological Disorders. Comput. Struct. Biotechnol. J. 2019, 17, 579–590. [Google Scholar] [CrossRef]
- Dessalew, N.; Patel, D.S.; Bharatam, P.V. 3D-QSAR and molecular docking studies on pyrazolopyrimidine derivatives as glycogen synthase kinase-3β inhibitors. J. Mol. Graph. Model. 2007, 25, 885–895. [Google Scholar] [CrossRef]
- Rejc, L.; Šmid, L.; Kepe, V.; Podlipnik, Č.; Golobič, A.; Bresjanac, M.; Barrio, J.R.; Petrič, A.; Košmrlj, J. Design, Syntheses, and in Vitro Evaluation of New Fluorine-18 Radiolabeled Tau-Labeling Molecular Probes. J. Med. Chem. 2017, 60, 8741–8757. [Google Scholar] [CrossRef] [PubMed]
- Murugan, N.A.; Nordberg, A.; Ågren, H. Different Positron Emission Tomography Tau Tracers Bind to Multiple Binding Sites on the Tau Fibril: Insight from Computational Modeling. ACS Chem. Neurosci. 2018, 9, 1757–1767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bijari, N.; Balalaie, S.; Akbari, V.; Golmohammadi, F.; Moradi, S.; Adibi, H.; Khodarahmi, R. Effective suppression of the modified PHF6 peptide/1N4R Tau amyloid aggregation by intact curcumin, not its degradation products: Another evidence for the pigment as preventive/therapeutic “functional food”. Int. J. Biol. Macromol. 2018, 120, 1009–1022. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Gao, X.; Sun, W.; Yao, T.; Shi, S.; Ji, L. Molecular Hairpin: A Possible Model for Inhibition of Tau Aggregation by Tannic Acid. Biochemistry 2013, 52, 1893–1902. [Google Scholar] [CrossRef]
- Gadakar, P.K.; Phukan, S.; Dattatreya, P.; Balaji, V.N. Pose prediction accuracy in docking studies and enrichment of actives in the active site of GSK-3beta. J. Chem. Inf. Model. 2007, 47, 1446–1459. [Google Scholar] [CrossRef]
- Kiss, R.; Csizmadia, G.; Solti, K.; Keresztes, A.; Zhu, M.; Pickhardt, M.; Mandelkow, E.; Tóth, G. Structural Basis of Small Molecule Targetability of Monomeric Tau Protein. ACS Chem. Neurosci. 2018, 9, 2997–3006. [Google Scholar] [CrossRef]
- Pradeepkiran, J.A.; Reddy, P.H. Structure Based Design and Molecular Docking Studies for Phosphorylated Tau Inhibitors in Alzheimer’s Disease. Cells 2019, 8, 260. [Google Scholar] [CrossRef] [Green Version]
- Zeb, A.; Park, C.; Rampogu, S.; Son, M.; Lee, G.; Lee, K.W. Structure-Based Drug Designing Recommends HDAC6 Inhibitors to Attenuate Microtubule-Associated Tau-Pathogenesis. ACS Chem. Neurosci. 2019, 10, 1326–1335. [Google Scholar] [CrossRef]
- Mohamed, T.; Hoang, T.; Jelokhani-Niaraki, M.; Rao, P.P. Tau-derived-hexapeptide 306VQIVYK311 aggregation inhibitors: Nitrocatechol moiety as a pharmacophore in drug design. ACS Chem. Neurosci. 2013, 4, 1559–1570. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Zhong, H.; Liu, X.; Zhou, S.; Tan, S.; Liu, H.; Yao, X. Disclosing the Mechanism of Spontaneous Aggregation and Template-Induced Misfolding of the Key Hexapeptide (PHF6) of Tau Protein Based on Molecular Dynamics Simulation. ACS Chem. Neurosci. 2019, 10, 4810–4823. [Google Scholar] [CrossRef]
- Liu, H.; Liu, X.; Zhou, S.; An, X.; Liu, H.; Yao, X. Disclosing the Template-Induced Misfolding Mechanism of Tau Protein by Studying the Dissociation of the Boundary Chain from the Formed Tau Fibril Based on a Steered Molecular Dynamics Simulation. ACS Chem. Neurosci. 2019, 10, 1854–1865. [Google Scholar] [CrossRef]
- Susimaire, P.-M.; Carlos Henrique Tomich de Paula da, S.; Vinicius Barreto da, S. In Silico Binding Mode Proposed for Flavonoid Ligands of Tau Protein with Interest in Alzheimer’s Disease. Curr. Bioact. Compd. 2013, 9, 21–26. [Google Scholar]
- Susimaire, P.-M.; Vinicius, B.D.S.; Carlton, A.T.; Carlos, H.T.P.D.S. Pharmacophore-based Drug Design of Novel Potential Tau Ligands for Alzheimer’s Disease Treatment. Curr. Phys. Chem. 2014, 4, 35–44. [Google Scholar]
- Luo, Y.; Ma, B.; Nussinov, R.; Wei, G. Structural Insight into Tau Protein’s Paradox of Intrinsically Disordered Behavior, Self-Acetylation Activity, and Aggregation. J. Phys. Chem. Lett. 2014, 5, 3026–3031. [Google Scholar] [CrossRef]
- Baggett, D.W.; Nath, A. The Rational Discovery of a Tau Aggregation Inhibitor. Biochemistry 2018, 57, 6099–6107. [Google Scholar] [CrossRef]
- Kim, H.; Han, H. Computer-Aided Multi-Target Management of Emergent Alzheimer’s Disease. Bioinformation 2018, 14, 167–180. [Google Scholar] [CrossRef] [PubMed]
- Agatonovic-Kustrin, S.; Kettle, C.; Morton, D.W. A molecular approach in drug development for Alzheimer’s disease. Biomed. Pharmacother. 2018, 106, 553–565. [Google Scholar] [CrossRef] [PubMed]
- Calcoen, D.; Elias, L.; Yu, X. What does it take to produce a breakthrough drug? Nat. Rev. Drug Discov. 2015, 14, 161–162. [Google Scholar] [CrossRef] [PubMed]
- Doody, R.S.; Raman, R.; Farlow, M.; Iwatsubo, T.; Vellas, B.; Joffe, S.; Kieburtz, K.; He, F.; Sun, X.; Thomas, R.G.; et al. A phase 3 trial of semagacestat for treatment of Alzheimer’s disease. N. Engl. J. Med. 2013, 369, 341–350. [Google Scholar] [CrossRef]
- Vandenberghe, R.; Rinne, J.O.; Boada, M.; Katayama, S.; Scheltens, P.; Vellas, B.; Tuchman, M.; Gass, A.; Fiebach, J.B.; Hill, D.; et al. Bapineuzumab for mild to moderate Alzheimer’s disease in two global, randomized, phase 3 trials. Alzheimers Res. Ther. 2016, 8, 18. [Google Scholar] [CrossRef] [Green Version]
- The Lancet, N. Solanezumab: Too late in mild Alzheimer’s disease? Lancet Neurol. 2017, 16, 97. [Google Scholar] [CrossRef] [Green Version]
- Gauthier, S.; Albert, M.; Fox, N.; Goedert, M.; Kivipelto, M.; Mestre-Ferrandiz, J.; Middleton, L.T. Why has therapy development for dementia failed in the last two decades? Alzheimers Dement. 2016, 12, 60–64. [Google Scholar] [CrossRef] [PubMed]
- Mehta, D.; Jackson, R.; Paul, G.; Shi, J.; Sabbagh, M. Why do trials for Alzheimer’s disease drugs keep failing? A discontinued drug perspective for 2010-2015. Expert Opin. Investig. Drugs 2017, 26, 735–739. [Google Scholar] [CrossRef] [PubMed]
- Anderson, R.M.; Hadjichrysanthou, C.; Evans, S.; Wong, M.M. Why do so many clinical trials of therapies for Alzheimer’s disease fail? Lancet 2017, 390, 2327–2329. [Google Scholar] [CrossRef]
- Schneider, L. A resurrection of aducanumab for Alzheimer’s disease. Lancet Neurol. 2019. [Google Scholar] [CrossRef] [Green Version]
- Ossenkoppele, R.; van der Flier, W.M.; Verfaillie, S.C.; Vrenken, H.; Versteeg, A.; van Schijndel, R.A.; Sikkes, S.A.; Twisk, J.; Adriaanse, S.M.; Zwan, M.D.; et al. Long-term effects of amyloid, hypometabolism, and atrophy on neuropsychological functions. Neurology 2014, 82, 1768–1775. [Google Scholar] [CrossRef]
- Duyckaerts, C. Tau pathology in children and young adults: Can you still be unconditionally baptist? Acta Neuropathol. 2011, 121, 145–147. [Google Scholar] [CrossRef] [Green Version]
- Pascoal, T.A.; Mathotaarachchi, S.; Shin, M.; Benedet, A.L.; Mohades, S.; Wang, S.; Beaudry, T.; Kang, M.S.; Soucy, J.P.; Labbe, A.; et al. Synergistic interaction between amyloid and tau predicts the progression to dementia. Alzheimers Dement. 2017, 13, 644–653. [Google Scholar] [CrossRef]
- Cummings, J.; Morstorf, T.; Lee, G. Alzheimer’s drug-development pipeline: 2016. Alzheimers Dement. (N. Y.) 2016, 2, 222–232. [Google Scholar] [CrossRef] [Green Version]
- Dias, K.S.; Viegas, C., Jr. Multi-Target Directed Drugs: A Modern Approach for Design of New Drugs for the treatment of Alzheimer’s Disease. Curr. Neuropharmacol. 2014, 12, 239–255. [Google Scholar] [CrossRef] [Green Version]
- Sato, C.; Barthelemy, N.R.; Mawuenyega, K.G.; Patterson, B.W.; Gordon, B.A.; Jockel-Balsarotti, J.; Sullivan, M.; Crisp, M.J.; Kasten, T.; Kirmess, K.M.; et al. Tau Kinetics in Neurons and the Human Central Nervous System. Neuron 2018, 97, 1284–1298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartolini, M.; Bertucci, C.; Cavrini, V.; Andrisano, V. beta-Amyloid aggregation induced by human acetylcholinesterase: Inhibition studies. Biochem. Pharmacol. 2003, 65, 407–416. [Google Scholar] [CrossRef]
- Dong, H.; Yuede, C.M.; Coughlan, C.A.; Murphy, K.M.; Csernansky, J.G. Effects of donepezil on amyloid-beta and synapse density in the Tg2576 mouse model of Alzheimer’s disease. Brain Res. 2009, 1303, 169–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okuda, M.; Hijikuro, I.; Fujita, Y.; Teruya, T.; Kawakami, H.; Takahashi, T.; Sugimoto, H. Design and synthesis of curcumin derivatives as tau and amyloid beta dual aggregation inhibitors. Bioorg. Med. Chem. Lett. 2016, 26, 5024–5028. [Google Scholar] [CrossRef]
- Mariano, M.; Schmitt, C.; Miralinaghi, P.; Catto, M.; Hartmann, R.W.; Carotti, A.; Engel, M. First selective dual inhibitors of tau phosphorylation and Beta-amyloid aggregation, two major pathogenic mechanisms in Alzheimer’s disease. ACS Chem. Neurosci. 2014, 5, 1198–1202. [Google Scholar] [CrossRef]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mouchlis, V.D.; Melagraki, G.; Zacharia, L.C.; Afantitis, A. Computer-Aided Drug Design of β-Secretase, γ-Secretase and Anti-Tau Inhibitors for the Discovery of Novel Alzheimer’s Therapeutics. Int. J. Mol. Sci. 2020, 21, 703. https://doi.org/10.3390/ijms21030703
Mouchlis VD, Melagraki G, Zacharia LC, Afantitis A. Computer-Aided Drug Design of β-Secretase, γ-Secretase and Anti-Tau Inhibitors for the Discovery of Novel Alzheimer’s Therapeutics. International Journal of Molecular Sciences. 2020; 21(3):703. https://doi.org/10.3390/ijms21030703
Chicago/Turabian StyleMouchlis, Varnavas D., Georgia Melagraki, Lefteris C. Zacharia, and Antreas Afantitis. 2020. "Computer-Aided Drug Design of β-Secretase, γ-Secretase and Anti-Tau Inhibitors for the Discovery of Novel Alzheimer’s Therapeutics" International Journal of Molecular Sciences 21, no. 3: 703. https://doi.org/10.3390/ijms21030703