


Abstract
Cholangiocarcinoma is a highly malignant neoplasm of the biliary tree. It has a high rate of mortality, and currently, there is no effective chemoprevention and treatment. This study was designed to investigate the potential effect of ω3 polyunsaturated fatty acids (ω3-PUFA) on human cholangiocarcinoma cell growth and to determine their mechanisms of actions. Treatment of three human cholangiocarcinoma cells (CCLP1, HuCCT1, SG231) with two ω3-PUFAs, docosahexaenoic acid (DHA) and eicosapentaenoic acid (EPA), for 12 to 72 h resulted in a dose- and time-dependent inhibition of cell growth; in contrast, arachidonic acid, a ω6-PUFA, had no significant effect. The ω3-PUFA effect is due to the induction of apoptosis, given that DHA induced the cleaved form of PARP, caspase-3, and caspase-9. DHA and EPA treatment caused dephosphorylation (and hence, the activation) of glycogen synthase kinase-3β (GSK-3β) with a decline of β-catenin protein. Accordingly, DHA treatment also decreased the β-catenin–mediated T cell factor/lymphoid enhancer factor (TCF/LEF) reporter activity, and inhibited the expression of c-Met, a β-catenin–controlled downstream gene implicated in cholangiocarcinogenesis. The GSK-3β inhibitor, SB216763, partially prevented DHA-induced reduction of β-catenin protein and TCF/LEF reporter activity, and restored cell growth, suggesting the involvement of GSK-3β dephosphorylation in ω3-PUFA–induced β-catenin degradation. In parallel, DHA treatment also induced the formation of the β-catenin/Axin/GSK-3β binding complex, further leading to β-catenin degradation. Moreover, DHA inhibited the expression of cyclooxygenase-2 (COX-2) and enhanced the expression of 15-hydroxyprostaglandin dehydrogenase, a physiologic COX-2 antagonist, in human cholangiocarcinoma cells. These findings suggest that ω3-PUFAs block cholangiocarcinoma cell growth at least in part through inhibition of Wnt/β-catenin and COX-2 signaling pathways. Thus, utilization of ω3-PUFAs may represent an effective and safe therapeutic approach for the chemoprevention and treatment of human cholangiocarcinoma. [Cancer Res 2008;68(2):553–60]
Introduction
Cholangiocarcinoma is a malignant epithelial neoplasm of the biliary tree with a high rate of mortality (1–6). Early diagnosis of cholangiocarcinoma is difficult, and currently, there is no effective chemoprevention or treatment. The tumor often arises from background conditions that cause long-standing inflammation, injury, and reparative biliary epithelial cell proliferation, such as primary sclerosing cholangitis, clonorchiasis, hepatolithiasis, or complicated fibropolycystic diseases (1–6). Consistent with the strong association between bile duct chronic inflammation and cholangiocarcinoma, recent studies have documented an important role of cyclooxygenase-2 (COX-2)–derived prostaglandin E2 (PGE2), a potent lipid inflammatory mediator, in cholangiocarcinogenesis (1, 2, 4, 6). For example, increased COX-2 expression has been documented in cholangiocarcinoma cells and precancerous bile duct lesions but not in normal bile duct epithelial cells (BEC; refs. 7–9). Overexpression of COX-2 in cultured human cholangiocarcinoma cells enhances PGE2 production and promotes tumor growth, whereas depletion of COX-2 attenuates growth (10, 11). Treatment of cholangiocarcinoma cells with exogenous PGE2 increases tumor cell growth and prevents apoptosis (10–15). Consistent with these findings, selective COX-2 inhibitors prevent cholangiocarcinoma cell growth and invasion, in vitro and in nude mice (4, 10, 11, 14–16), although their effect may be mediated through COX-2–dependent and -independent mechanisms.
Recent evidence suggests that alteration of other growth-regulatory molecules such as Wnt/β-catenin pathway is also implicated in cholangiocarcinogenesis (17–20). β-Catenin is a key mediator in Wnt regulation of multiple cellular functions in embryogenesis and tumorigenesis (21–24). In adult tissues, β-catenin is a component of stable cell adherent complexes whereas its free form functions as a coactivator for a family of transcription factors termed T cell factor/lymphoid enhancer factor (TCF/LEF). Wnt proteins comprise a family of highly conserved secreted proteins that signal through the Frizzled receptors (21–24). In the absence of a Wnt signal, β-catenin exists within a cytoplasmic complex (β-catenin destruction complex) along with glycogen synthase kinase-3β (GSK-3β), adenomatous polyposis coli, and Axin, where it is phosphorylated and targeted for degradation by the proteasome. Activation of Wnt signaling perturbs this destruction complex, leading to cytoplasmic accumulation of β-catenin and allowing its translocation into the cell nucleus. In the nucleus, β-catenin associates with TCF/LEF that stimulate the transcription of target genes important for proliferation, differentiation, and apoptosis (21–24). Recent studies have shown that accumulation of nuclear β-catenin is induced by PGE2, in addition to the canonical Wnt/Frizzled signaling, in human colon cancer cells. Castellone et al. reported that PGE2 activates its G protein–coupled receptor, EP2, resulting in direct association of the G protein α subunit with the regulator of G protein signaling domain of Axin; this results in the release of GSK-3β from its complex with Axin, thus leading to β-catenin accumulation (25). Shao et al. showed the involvement of the cyclic AMP/protein kinase A pathway in PGE2-induced β-catenin accumulation in colon cancer cells (26). However, it remains unknown whether the COX-2/PG and Wnt/β-catenin signaling pathways converge during cholangiocarcinogenesis.
Although PGE2 derived from arachidonic acid (AA; an ω3-PUFA) is known to promote tumor growth, there is convincing experimental evidence that the ω3-PUFAs rich in fish oil, such as docosahexaenoic acid (DHA) and eicosapentaenoic acid (EPA), prevent carcinogenesis (27, 28). However, the potential effect of ω3-PUFAs on cholangiocarcinoma growth has not been studied and the molecular mechanisms for their anticancer actions remain incompletely understood. This study was designed to investigate the effect and mechanism of ω3-PUFAs in human cholangiocarcinoma cells. Our data show that DHA and EPA inhibited the growth of three human cholangiocarcinoma cells (CCLP1, SG231, and HuCCT1). Moreover, our data reveal that COX-2–derived PGE2 activates β-catenin signaling pathways in human cholangiocarcinoma cells and that ω3-PUFAs inhibit cholangiocarcinoma cell growth by simultaneously blocking β-catenin and COX-2 signaling pathways. These findings provide important preclinical evidence and molecular insight for the use of ω3-PUFAs in the chemoprevention and treatment of human cholangiocarcinoma.
Materials and Methods
Materials. α-MEM, DMEM, RPMI 1640, fetal bovine serum (FBS), glutamine, antibiotics, and LipofectAMINE plus reagent were purchased from Life Technologies, Inc. PGE2 was purchased from Calbiochem. The cell proliferation assay reagent WST-1 was purchased from Roche Molecular Biochemicals. DHA, EPA, and AA were from Cayman Chemical (DHA and EPA were purified from algae; AA was synthesized; purity >98%). The antibodies for human COX-2, 15-hydroxyprostaglandin dehydrogenase (15-PGDH) were also purchased from Cayman Chemical. The antibodies against human Axin, β-catenin, PARP, caspase-3, caspase-9, and c-Met were purchased from Santa Cruz Biotechnology. The antihuman β-actin monoclonal antibody was purchased from Sigma. The horseradish peroxidase–linked streptavidin and chemiluminescence detection reagents were from Amersham Pharmacia Biotech, Inc. The rabbit antibodies for phosphorylated Akt (Thr308), Akt, phosphorylated GSK-3β (Ser9), and GSK-3β were purchased from Cell Signaling Technology. Mouse monoclonal anti–GSK-3β was purchased from Transduction Laboratories and cytochrome c was purchased from BD Bioscience. The Bio-Rad protein assay system was obtained from Bio-Rad Laboratories. The Tris-glycine gels were obtained from Invitrogen Life Technologies, Inc.
Culture of human cholangiocarcinoma cells. Three human intrahepatic cholangiocarcinoma cell lines—HuCCT1 (obtained from the Japanese Cancer Research Resources Bank), SG231, and CCLP1 were cultured according to our previously described methods (10, 12, 29). Briefly, CCLP1 cells were cultured in DMEM supplemented with 10% FBS, 2 mmol/L of l-glutamine, and 50 μg/mL of gentamicin. SG231 cells were cultured in α-MEM with 2 mmol/L of l-glutamine, 50 μg/mL of gentamicin, 10 mmol/L of HEPES, and 10% FBS. HuCCT1 cells were cultured in RPMI 1640 supplemented with 10% FBS, 2 mmol/L of l-glutamine, and 50 μg/mL of gentamicin. The cells were cultured at 37°C in a humidified CO2 incubator. The experiments were performed when cells reached ∼80% confluence and were conducted in serum-free medium with serum deprivation for 24 h before the experiments.
Culture of primary BECs. Primary mouse BECs were isolated from normal female C57BL/6 mice (8–12 weeks old) and cultured as previously described (30, 31). Briefly, BECs were cultured on collagen gels in complete serum-free medium, which consisted of DMEM/F12 medium (Sigma) supplemented with 5.4 g/L of d-glucose, 50 μg/mL of gentamicin, antibiotic–antimycotic (100 units/mL penicillin G sodium, 100 μg/mL streptomycin sulfate, 250 ng/mL amphotericin B), 10 mmol/L of HEPES, 2.5 mg/mL of bovine serum albumin (Sigma), insulin-transferrin-selenium-X (10 mg/L insulin, 5.5 mg/L transferrin, 6.7 ng/L sodium selenite), 0.1 mmol/L of minimal essential media nonessential amino acid solution, 2 mmol/L of l-glutamine, 32 ng/mL of thyroxin (Sigma), 10 ng/mL of prostaglandin E1 (Sigma), 40 ng/mL of hydrocortisone (Sigma), 10 μmol/L of forskolin (Sigma), and 50 μg/mL of trypsin inhibitor (Sigma) for 21 days. Cells (1 × 105) in each well of a 48-well plate were subcultured and allowed to reattach in complete DMEM, which is complete serum-free medium with 50 μg/mL of bovine pituitary extract and 10 ng/mL of epidermal growth factor. Then the cells were held quiescent overnight in simple and serum-free medium, which is DMEM/F12 supplemented with 5.4 g/L of d-glucose, 50 μg/mL of gentamicin, antibiotic–antimycotic (100 units/mL penicillin G sodium, 100 μg/mL streptomycin sulfate, 250 ng/mL amphotericin B), 10 mmol/L of HEPES, and 2.5 mg/mL of bovine serum albumin.
Cell viability assay. The viability of human cholangiocarcinoma cells was determined by using the cell proliferation reagent WST-1, a tetrazolium salt that is cleaved by mitochondrial dehydrogenases in viable cells. Briefly, 100 μL of cell suspension (containing 0.5–2 × 104 cells) were plated in each well of a 96-well plate. After 24 h of culture to allow reattachment, the cells then were treated with specific reagents such as DHA or Wnt3a-conditioned medium (Wnt3a-CM) for the indicated time points. At the end of each experiment, the cell proliferation reagent WST-1 (10 μL) was added to each well, and the cells were incubated at 37°C for 0.5 to 5 h. Absorbance at 450 nm was measured using an automatic ELISA plate reader. The viability of primary mouse BECs was determined by using cell counts with trypan blue exclusion.
Immunoprecipitation. Equal amounts of cellular protein from the treated cells were incubated overnight with 10 μL of rabbit anti-human Axin polyclonal antibody at 4°C, followed by the addition of 20 μL of protein A/G PLUS agarose (Santa Cruz Biotechnology). The mixture was incubated for 2 h and then washed thrice with the cell lysis buffer [50 mmol/L HEPES (pH 7.55), 1 mmol/L EDTA, 1 mmol/L DTT, and protease inhibitor cocktail tablets from Roche Diagnostics]. The final pellets were dissolved in 20 μL of 2× protein loading buffer, and the samples were subjected to SDS-PAGE and Western blot analysis using a 1:1,000 dilution of mouse anti-human GSK-3β or β-catenin monoclonal antibody and enhanced chemiluminescence Western blot detection system (Amersham Pharmacia Biotech, Inc.).
Transient transfection of COX-2 expression plasmid in human cholangiocarcinoma cells. The cells were exposed to the mixture of LipofectAMINE plus reagents and COX-2 expression plasmid (full-length human COX-2 cDNA cloned in pcDNA3 vector), pcDNA3 control vector, or TCF/LEF-Luc reporter plasmid for 4 h. Following removal of the transfection mixtures, fresh serum-free medium was added with specific reagents as indicated in the text. The cells with optimal overexpression of COX-2 were confirmed by immunoblotting and subsequently used for further experiments.
Luciferase reporter activity assay. The cultured cells were seeded at a concentration achieving 80% confluence in 12-well plates for 18 h before transfection. The cells were transiently transfected with 0.2 μg/well of translucent TCF/LEF-Luc reporter vector, which was designed to measure the transcriptional activity of TCF/LEF-responsive genes. After transfection, the cells were treated with specific reagents such as DHA or Wnt3a-CM in serum-free medium at the indicated time periods. The cell lysates were then obtained with 1× reporter lysis buffer (Promega). The luciferase activity was assayed in a Berthold AutoLumat LB953 Luminometer by using the luciferase assay system from Promega. The relative luciferase activity was calculated after normalization of cellular proteins. All values are expressed as the percentage of activity relative to basal activity.
Immunoblotting. At the end of each indicated treatment, the cells were scraped off the plates and centrifuged, washed twice with cold PBS containing 0.5 mmol/L of phenylmethylsulfonyl fluoride and 10 μg/mL of leupeptin, and resuspended in 5-fold volume of hypotonic buffer consisting of 50 mmol/L of HEPES (pH 7.55), 1 mmol/L of EDTA, 1 mmol/L of DTT, and protease inhibitor cocktail tablets (Roche Diagnostics GmbH). After sonication, the whole cell lysate was collected by centrifugation at 15,000 × g, 4°C for 10 min to remove cell debris, and stored in aliquots at −80°C until use. The protein concentration in the cell extracts were determined by the Bio-Rad protein assay (Bio-Rad). Thirty micrograms of cellular protein was subjected to SDS-PAGE on 4% to 20% Tris-glycine gels for PARP, β-catenin, GSK-3β, phosphorylated GSK-3β, Akt, phosphorylated Akt, c-Met, Axin, cytochrome c, caspase-3, caspase-9, COX-2, 15-PGDH, or β-actin. The separated proteins were electrophoretically transferred onto the nitrocellulose membrane (Bio-Rad). Nonspecific binding was blocked with PBS-T (0.5% Tween 20 in PBS) containing 5% nonfat milk for 1 h at room temperature. The membranes were then incubated overnight at 4°C with individual primary antibodies in PBS-T containing 5% nonfat milk at the dilutions specified by the manufacturers. Following three washes with PBS-T, the membranes were then incubated with the horseradish peroxidase–conjugated secondary antibodies at 1:10,000 dilution in PBS-T containing 5% nonfat milk for 1 h at room temperature. The membranes were then washed thrice with PBS-T and the protein bands were visualized with the ECL Western blotting detection system according to the manufacturer's instructions.
Preparation of Wnt3a-CM. Wnt3a-CM and the control L cell–conditioned medium were prepared as described in the American Type Culture Collection protocol. Briefly, the cells were split 1:10 in 10 mL culture medium from 10 cm Petri dishes and cultured at 37°C. After 4 days of culture, the medium was collected, centrifuged, and sterilized through a filter to obtain the first batch of Wnt3a-CM. An additional 10 mL of fresh medium was subsequently added to the cells and the cultures were continued for an additional 3 days; the medium was then removed, centrifuged, and sterilized through a filter to obtained the second batch of Wnt3a-CM. The first and second batches of the medium were mixed 1:1 and then used for experiments of Wnt3a-CM. The control L cell–conditioned medium was prepared following the same procedure. They were stored at −80°C until use.
Immunohistochemical stains for COX-2 and β-catenin in human cholangiocarcinoma tissues. Twenty-seven archival formalin-fixed, paraffin-embedded specimens of human cholangiocarcinoma and adjacent nonneoplastic liver tissues were obtained from the University of Pittsburgh Medical Center. The tissue specimens were used for immunohistochemical analysis for COX-2 and β-catenin according to standard procedures. Briefly, the slides were incubated with 1:100 diluted monoclonal antibody against human COX-2 or β-catenin, followed by probing with biotin-conjugated secondary antibody (1:200), incubation with the ABC complex, and addition of a 3,3′-diaminobenzidine substrate. The staining intensity for COX-2 was scored in each specimen on a scale of 0 to 3, in which 0, negative staining; 1, weakly positive staining; 2, moderately positive staining; and 3, strongly positive staining. For each sample, 10 random high-power fields were scored. The immunoreactivity for β-catenin was determined in all the sections, with special attention to the staining patterns (nuclear versus plasma membrane). The protocol for using human tissue specimens was approved by the University of Pittsburgh (Institutional Review Board no. 020134). None of the cases used in this study had patient identifiers and strict confidentiality was maintained in accordance with the approval granted by the institutional review board.
Results and Discussion
We first did immunohistochemical stains for β-catenin and COX-2 in human cholangiocarcinoma tissues. Twenty-seven paired human cholangiocarcinomas and their matched nonneoplastic/nondysplastic bile duct tissues were analyzed by immunohistochemistry using antibodies against β-catenin and COX-2. Increased cytoplasmic staining for COX-2 and nuclear staining for β-catenin was observed in cholangiocarcinoma cells when compared with the nonneoplastic bile duct epithelium (Fig. 1). COX-2 is expressed exclusively in the cytoplasm of cholangiocarcinoma cells, and to a lesser degree, in BECs. The average staining intensity for COX-2 in cholangiocarcinoma cells is 2.04 ± 0.52, which is significantly higher than that in nonneoplastic bile duct epithelium (0.48 ± 0.12; n = 27, P < 0.01, Student's t test). Different β-catenin expression patterns were observed between cholangiocarcinoma cells and interlobular bile ducts. In the interlobular BECs, β-catenin is expressed exclusively in the plasma membrane with no significant cytoplasmic staining and the absence of nuclear staining. In cholangiocarcinoma cells, there is evident cytoplasmic staining with decreased plasma membrane. Nuclear staining for β-catenin was observed in 9 of 27 cholangiocarcinoma tissues, but not in nonneoplastic BECs. These findings indicate the activation of β-catenin and overexpression of COX-2 in human cholangiocarcinomas.

Representative immunohistochemical stains for β-catenin and COX-2 in human cholangiocarcinoma and nonneoplastic BECs. Twenty-seven archival formalin-fixed, paraffin-embedded specimens of human cholangiocarcinoma and adjacent nonneoplastic liver tissues were used for immunohistochemical analysis of COX-2 and β-catenin protein. Note the plasma membrane staining of β-catenin in the nonneoplastic BECs and the nuclear staining of β-catenin in cholangiocarcinoma cells. COX-2 is expressed exclusively in the cytoplasm, with higher expression in cholangiocarcinoma cells than in nonneoplastic BECs. No stain was seen when the primary antibodies were substituted with nonimmunized serum. CC, cholangiocarcinoma; BD, bile duct.
Representative immunohistochemical stains for β-catenin and COX-2 in human cholangiocarcinoma and nonneoplastic BECs. Twenty-seven archival formalin-fixed, paraffin-embedded specimens of human cholangiocarcinoma and adjacent nonneoplastic liver tissues were used for immunohistochemical analysis of COX-2 and β-catenin protein. Note the plasma membrane staining of β-catenin in the nonneoplastic BECs and the nuclear staining of β-catenin in cholangiocarcinoma cells. COX-2 is expressed exclusively in the cytoplasm, with higher expression in cholangiocarcinoma cells than in nonneoplastic BECs. No stain was seen when the primary antibodies were substituted with nonimmunized serum. CC, cholangiocarcinoma; BD, bile duct.
Given the presence of active COX-2 and β-catenin signaling pathways in cholangiocarcinoma, we next examined the potential effect of COX-2 on β-catenin activation in cultured human cholangiocarcinoma cells. Our results show that overexpression of COX-2 increased the TCF/LEF reporter activity in the human cholangiocarcinoma cell line, CCLP1 (Fig. 2A). Because PGE2 is the predominant COX-2 metabolite in cholangiocarcinoma cells (4), its effect on β-catenin activation was next examined. As shown in Fig. 2B, PGE2 treatment of CCLP1 cells also increased TCF/LEF reporter activity. In contrast, PGE3, a ω3-PUFA metabolite, showed no significant effect. Moreover, PGE2 treatment also increased GSK-3β phosphorylation (and thus inactivation; Fig. 2C) and caused GSK-3β dissociation from Axin (thus preventing the formation of the β-catenin destruction complex) in a time- and dose-dependent manner (Fig. 2D). These findings suggest that PGE2 activates the β-catenin signaling pathway through inactivation of GSK-3β and dissociation of the β-catenin destruction complex in human cholangiocarcinoma cells.

The effect of COX-2 and PGE2 on β-catenin activation in human cholangiocarcinoma cells (CCLP1). A, overexpression of COX-2 increases TCF/LEF reporter activity. CCLP1 cells were transiently transfected with the COX-2 expression plasmid or pcDNA control plasmid with cotransfection of the TCF/LEF-Luc reporter construct. After transfection, the cell lysates were obtained to determine the luciferase activity. Top, the TCF/LEF reporter activity. Columns, mean of six independent experiments; bars, SD (*, P < 0.01 compared with pcDNA control). Bottom, COX-2 protein level in the transfected cells. B, PGE2 treatment increases TCF/LEF transcription activity. CCLP1 cells transfected with the TCF/LEF-Luc reporter construct were treated with 10 μmol/L of PGE2 or PGE3 or vehicle in serum-free medium for 24 h. The cell lysates were obtained to determine the luciferase activity. Columns, mean of six independent experiments; bars, SD. PGE2 treatment significantly increased COX-2 reporter activity (*, P < 0.01), whereas PGE3 had no effect. C, PGE2 induces GSK-3β phosphorylation in CCLP1 cells. The cells were treated with different concentrations of PGE2 (1–20 μmol/L) in the serum-free medium for 1 h. The cell lysates were obtained for Western blot analysis of phosphorylated and nonphosphorylated GSK-3β. D, the dose- and time-dependent effect of PGE2 on Axin/GSK-3β dissociation. CCLP1 cells were treated with different concentrations of 1 to 20 μmol/L of PGE2 for 1 h (left) or treated with 10 μmol/L of PGE2 at different time points (30, 60, 90, and 120 min; right). The cell lysates were obtained for immunoprecipitation with Axin antibody followed by immunoblotting with antibodies against GSK-3β and Axin.
The effect of COX-2 and PGE2 on β-catenin activation in human cholangiocarcinoma cells (CCLP1). A, overexpression of COX-2 increases TCF/LEF reporter activity. CCLP1 cells were transiently transfected with the COX-2 expression plasmid or pcDNA control plasmid with cotransfection of the TCF/LEF-Luc reporter construct. After transfection, the cell lysates were obtained to determine the luciferase activity. Top, the TCF/LEF reporter activity. Columns, mean of six independent experiments; bars, SD (*, P < 0.01 compared with pcDNA control). Bottom, COX-2 protein level in the transfected cells. B, PGE2 treatment increases TCF/LEF transcription activity. CCLP1 cells transfected with the TCF/LEF-Luc reporter construct were treated with 10 μmol/L of PGE2 or PGE3 or vehicle in serum-free medium for 24 h. The cell lysates were obtained to determine the luciferase activity. Columns, mean of six independent experiments; bars, SD. PGE2 treatment significantly increased COX-2 reporter activity (*, P < 0.01), whereas PGE3 had no effect. C, PGE2 induces GSK-3β phosphorylation in CCLP1 cells. The cells were treated with different concentrations of PGE2 (1–20 μmol/L) in the serum-free medium for 1 h. The cell lysates were obtained for Western blot analysis of phosphorylated and nonphosphorylated GSK-3β. D, the dose- and time-dependent effect of PGE2 on Axin/GSK-3β dissociation. CCLP1 cells were treated with different concentrations of 1 to 20 μmol/L of PGE2 for 1 h (left) or treated with 10 μmol/L of PGE2 at different time points (30, 60, 90, and 120 min; right). The cell lysates were obtained for immunoprecipitation with Axin antibody followed by immunoblotting with antibodies against GSK-3β and Axin.
Because the COX-2/PGE2 and Wnt/β-catenin signaling pathways are active in human cholangiocarcinoma cells, we postulate that therapies aimed at simultaneous disruption of the COX-2/PGE2 and Wnt/β-catenin pathways might produce effective chemopreventive and antitumorigenic effects. The results presented below in this article describe a strong inhibition of cholangiocarcinoma cell growth by ω3-PUFAs through blocking of Wnt/β-catenin and COX-2 signaling pathways.
We next examined the effect of ω3-PUFAs on the growth of three human cholangiocarcinoma cell lines (CCLP1, SG231, and HuCCT1). Incubation of CCLP1 cells with two ω3-PUFAs (20 μmol/L), DHA and EPA, resulted in a time-dependent reduction of cell viability; in contrast AA, a ω6-PUFA, had no significant effect (Supplementary Fig. S1). Treatment with 20 μmol/L of DHA for 12, 24, 48, and 72 h induced 20.53 ± 1.8%, 16.77 ± 2.56%, 5.1 ± 1.49%, and 4.34 ± 0.3% reduction of viable cells, respectively. DHA seems to have more effect, with ∼80% reduction of viable cells at 12 h and >95% reduction at 48 and 72 h. The effects of DHA and EPA were dose-dependent in all three human cholangiocarcinoma cell lines. The cells treated with DHA and EPA exhibited morphologic features of cell death, characterized by shrunken cells, rounded cells, and detachment (Supplementary Fig. S1). The inhibitory effect of ω3-PUFAs on cholangiocarcinoma cell growth is specific, given that DHA and EPA did not affect the growth of differentiated nonneoplastic primary biliary epithelial cells (Supplementary Fig. S2).
We next examined the proapoptotic molecules in cells treated with DHA. As shown in Fig. 3, incubation of CCLP1 cells with 25 μmol/L of DHA for 24 h resulted in the activation of caspase-3 and caspase-9 with PARP cleavage and concomitant release of cytochrome c from the mitochondria to the cytosol. These findings indicate that ω3-PUFAs inhibit human cholangiocarcinoma cell growth through the induction of apoptosis.
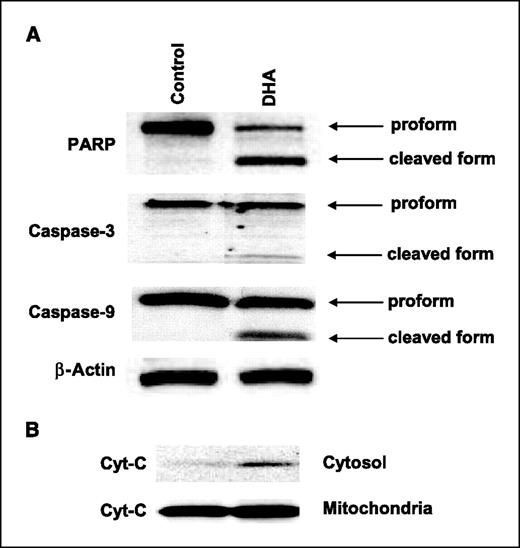
DHA treatment induces apoptosis in human cholangiocarcinoma cells. A, DHA induces activation of caspase-3, caspase-9, and cleavage of PARP. CCLP1 cells were treated with DHA (20 μmol/L) for 24 h and the cell lysates were obtained for Western blot analysis. Equal amounts of cellular proteins were subjected to SDS-PAGE followed by Western blotting using antibodies against PARP, caspase-3, and caspase-9 as described in Materials and Methods. B, DHA induces the release of cytochrome c in CCLP1 cells. The levels of cytochrome c in the cytosolic and mitochondrial fractions were determined by Western blotting analysis.
DHA treatment induces apoptosis in human cholangiocarcinoma cells. A, DHA induces activation of caspase-3, caspase-9, and cleavage of PARP. CCLP1 cells were treated with DHA (20 μmol/L) for 24 h and the cell lysates were obtained for Western blot analysis. Equal amounts of cellular proteins were subjected to SDS-PAGE followed by Western blotting using antibodies against PARP, caspase-3, and caspase-9 as described in Materials and Methods. B, DHA induces the release of cytochrome c in CCLP1 cells. The levels of cytochrome c in the cytosolic and mitochondrial fractions were determined by Western blotting analysis.
Additional experiments were performed to assess the potential effect of ω3-PUFAs on β-catenin protein level and activity. As shown in Fig. 4A and B, treatment with DHA or EPA reduced the level of β-catenin protein as well as c-Met, a β-catenin–controlled downstream gene. Because β-catenin regulates gene expression via binding as a transcription factor in complex with the TCF/LEF transcription factor family to the promoter region of target genes, we also determined the effect of DHA on TCF/LEF reporter activity. The TCF/LEF transcription activity was assayed after transient transfection of a luciferase reporter construct under the control of the TCF/LEF response element. As shown in Fig. 4C, DHA treatment significantly inhibited the TCF/LEF reporter activity (∼3.5-fold inhibition, P < 0.01).
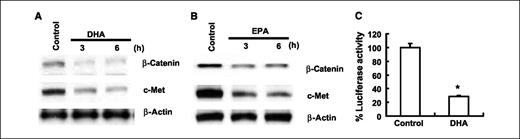
ω3-PUFAs reduce β-catenin protein level and TCF/LEF transcription activity in human cholangiocarcinoma cells. A and B, DHA and EPA decrease β-catenin and c-Met expression in CCLP1 cells. The cells were treated with 60 μmol/L of DHA or EPA in serum-free medium for 3 or 6 h. The cell lysates were obtained for Western blot analysis using antibodies against β-catenin and c-Met as described in Materials and Methods. C, DHA treatment decreases TCF/LEF reporter activity. CCLP1 cells were transiently transfected with the pTCF/LEF-Luc reporter vector. After transfection, the cells were cultured in serum-free medium with DHA (20 μmol/L) for 24 h and then the cell lysates were obtained to determine the luciferase activity. Columns, mean of six independent experiments; bars, SD (*, P < 0.01 compared with control).
ω3-PUFAs reduce β-catenin protein level and TCF/LEF transcription activity in human cholangiocarcinoma cells. A and B, DHA and EPA decrease β-catenin and c-Met expression in CCLP1 cells. The cells were treated with 60 μmol/L of DHA or EPA in serum-free medium for 3 or 6 h. The cell lysates were obtained for Western blot analysis using antibodies against β-catenin and c-Met as described in Materials and Methods. C, DHA treatment decreases TCF/LEF reporter activity. CCLP1 cells were transiently transfected with the pTCF/LEF-Luc reporter vector. After transfection, the cells were cultured in serum-free medium with DHA (20 μmol/L) for 24 h and then the cell lysates were obtained to determine the luciferase activity. Columns, mean of six independent experiments; bars, SD (*, P < 0.01 compared with control).
The level of β-catenin in cells is tightly controlled by its degradation complex composed of Axin, GSK-3β, and β-catenin in which GSK-3β phosphorylates β-catenin and thus triggers its ubiquitination and subsequent proteosomal degradation. The activity of GSK-3β is regulated by its phosphorylation status, with GSK-3β phosphorylation at Ser9 being functionally inactive. To determine whether ω3-PUFAs might induce β-catenin degradation through inhibition of GSK-3β phosphorylation, we examined the phosphorylated Ser9-GSK-3β and total GSK-3β protein levels in CCLP1 cells treated with DHA. DHA treatment reduced GSK-3β phosphorylation, whereas it had no effect on the protein level of total GSK-3β (Supplementary Fig. S3A). Because the phosphorylation of GSK-3β is controlled by Akt, we also examined the potential effect of DHA on Akt phosphorylation. Our data showed that DHA had no effect on Akt phosphorylation (Supplementary Fig. S3A). Thus, DHA most likely inhibited GSK-3β phosphorylation through mechanisms independent of Akt. These findings provide evidence for GSK-3β dephosphorylation (activation) in ω3-PUFA–induced degradation of β-catenin in cholangiocarcinoma cells.
To further determine the role of GSK-3β in DHA-induced β-catenin degradation, CCLP1 cells were pretreated with the GSK-3β inhibitor, SB216763 (5 μmol/L), for 1 h prior to DHA treatment and the cells were analyzed for β-catenin protein level, TCF/LEF reporter activity, and apoptosis. Inhibition of GSK-3β by SB216763 prevented DHA-induced reduction of β-catenin protein and TCF/LEF reporter activity as well as c-Met expression (Supplementary Fig. S3B and C). SB216763 also prevented DHA-induced PARP and caspase-9 cleavage in a dose-dependent manner (Supplementary Fig. S3D). Accordingly, inhibition of GSK-3β by SB216763 also partially restored cell survival (Supplementary Fig. S3E). These findings further support the role of GSK-3β dephosphorylation (activation) in DHA-induced β-catenin degradation in cholangiocarcinoma cells.
The degradation of β-catenin depends on β-catenin phosphorylation, which occurs in a multiprotein complex containing Axin, GSK-3β, and β-catenin. It is believed that in this complex assembled by Axin, GSK-3β phosphorylates the β-catenin primarily when it is bound to Axin. To determine whether DHA alters the assembly of the Axin/GSK-3β/β-catenin complex, immunoprecipitation and Western blot experiments were performed to detect the Axin/GSK-3β/β-catenin binding complex. As shown in Fig. 5A–C, treatment of CCLP1 cells with DHA induced the association of Axin with GSK-3β as well as β-catenin. This effect was observed within 1 h and persisted at 5 h. In contrast, AA, an ω6-PUFA, decreased the association of Axin with β-catenin and GSK-3β (Fig. 5D). These findings indicate that DHA induces the formation of the β-catenin destruction complex. Thus, ω3-PUFAs induce β-catenin degradation through dephosphorylation of GSK-3β and the formation of the β-catenin destruction complex in human cholangiocarcinoma cells.

DHA induces the formation of the Axin/GSK-3β/β-catenin complex in human cholangiocarcinoma cells. A, the effect of DHA on the association of Axin with β-catenin and GSK-3β. CCLP1 cells were treated with 60 μmol/L of DHA for 1 h. The cell lysates were immunoprecipitated with anti-Axin antibody followed by immunoblotting with antibodies against GSK-3β, β-catenin, and Axin. B, the time course effect of DHA. CCLP1 cells were treated with 60 μmol/L of DHA at different time points (1, 2, 3, 4, and 5 h). The cell lysates were immunoprecipitated with anti-Axin antibody followed by immunoblotting with antibodies against GSK-3β, β-catenin, and Axin. C, the dose-dependent effect of DHA. CCLP1 cells were treated with different concentrations of DHA for 1 h. The cell lysates were immunoprecipitated with anti-Axin antibody followed by immunoblotting with antibodies against GSK-3β and Axin. D, AA inhibits the association of the Axin/GSK-3β/β-catenin complex. CCLP1 cells were treated with 60 μmol/L of AA, DHA, or vehicle for 1 h. The cell lysates were immunoprecipitated with anti-Axin antibody followed by immunoblotting with antibodies against GSK-3β, β-catenin, and Axin.
DHA induces the formation of the Axin/GSK-3β/β-catenin complex in human cholangiocarcinoma cells. A, the effect of DHA on the association of Axin with β-catenin and GSK-3β. CCLP1 cells were treated with 60 μmol/L of DHA for 1 h. The cell lysates were immunoprecipitated with anti-Axin antibody followed by immunoblotting with antibodies against GSK-3β, β-catenin, and Axin. B, the time course effect of DHA. CCLP1 cells were treated with 60 μmol/L of DHA at different time points (1, 2, 3, 4, and 5 h). The cell lysates were immunoprecipitated with anti-Axin antibody followed by immunoblotting with antibodies against GSK-3β, β-catenin, and Axin. C, the dose-dependent effect of DHA. CCLP1 cells were treated with different concentrations of DHA for 1 h. The cell lysates were immunoprecipitated with anti-Axin antibody followed by immunoblotting with antibodies against GSK-3β and Axin. D, AA inhibits the association of the Axin/GSK-3β/β-catenin complex. CCLP1 cells were treated with 60 μmol/L of AA, DHA, or vehicle for 1 h. The cell lysates were immunoprecipitated with anti-Axin antibody followed by immunoblotting with antibodies against GSK-3β, β-catenin, and Axin.
Because Wnt3a is known to activate β-catenin signaling in cells, additional experiments were carried out to determine whether Wnt3a might protect cholangiocarcinoma cells from DHA-induced apoptosis. Treatment of CCLP1 cells with Wnt3a-CM partially prevented DHA-induced reduction of TCF/LEF transcription activity and restored cell survival (Supplementary Fig. S4). These results indicate that DHA inhibits cholangiocarcinoma growth at least in part through down-regulation of Wnt/β-catenin signaling pathway.
A prominent mechanism for the chemopreventive action of ω3-PUFAs is their suppressive effect on the production of AA-derived prostanoids, particularly PGE2 (28, 32). This is important because PGE2 is implicated in multiple stages of tumorigenesis, including modulation of inflammation, cancer cell proliferation, differentiation, apoptosis, angiogenesis, metastasis, and host immune response to cancer cells (4). Therefore, we next examined whether DHA might also affect the expression of COX-2 in our system. As shown in Fig. 6A and B, treatment of CCLP1 cells with DHA inhibited COX-2 promoter activity in a time- and dose-dependent manner. DHA treatment also inhibited the expression of COX-2 protein in cholangiocarcinoma cells (Fig. 6C). These findings suggest that DHA inhibits the expression of COX-2 at least in part through the suppression of gene transcription.
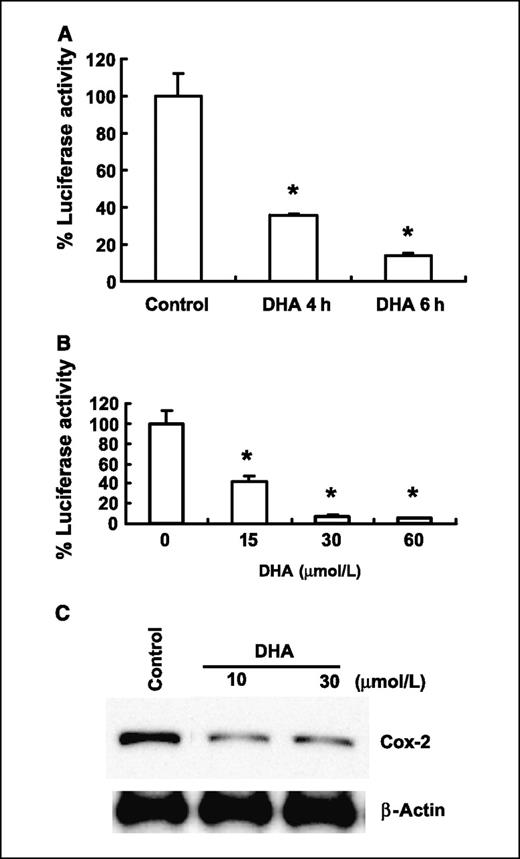
DHA inhibits COX-2 gene expression in human cholangiocarcinoma cells. A and B, DHA reduces COX-2 promoter activity. CCLP1 cells were transiently transfected with a luciferase reporter construct controlled by the COX-2 promoter. After transfection, the cells were treated with vehicle or DHA in serum-free medium at the indicated concentrations and time periods. The cell lysates were obtained to determine the luciferase reporter activity. Columns, mean of six independent experiments; bars, SD (*, P < 0.01 compared with control). C, DHA inhibits COX-2 protein expression. HuCCT1 cells were treated with 10 to 30 μmol/L of DHA in serum-free medium for 24 h and then the cell lysates were obtained for Western blot analysis using COX-2 antibody as described in Materials and Methods.
DHA inhibits COX-2 gene expression in human cholangiocarcinoma cells. A and B, DHA reduces COX-2 promoter activity. CCLP1 cells were transiently transfected with a luciferase reporter construct controlled by the COX-2 promoter. After transfection, the cells were treated with vehicle or DHA in serum-free medium at the indicated concentrations and time periods. The cell lysates were obtained to determine the luciferase reporter activity. Columns, mean of six independent experiments; bars, SD (*, P < 0.01 compared with control). C, DHA inhibits COX-2 protein expression. HuCCT1 cells were treated with 10 to 30 μmol/L of DHA in serum-free medium for 24 h and then the cell lysates were obtained for Western blot analysis using COX-2 antibody as described in Materials and Methods.
15-PGDH catalyzes the rate-limiting step of prostaglandin catabolism and thus represents a physiologic antagonist of COX-2 (33, 34). Recent emerging evidence suggests that elevated PGE2 in cancers may be the result of enhanced COX-2–mediated PGE2 synthesis as well as reduced 15-PGDH–mediated degradation of PGE2. Therefore, we sought to further determine whether DHA might affect 15-PGDH expression in human cholangiocarcinoma cells. DHA treatment enhanced the expression of 15-PGDH in CCLP1 cells, whereas AA exhibited no significant effect (Supplementary Fig. S5A). In fact, DHA treatment induced a dose-dependent induction of 15-PGDH in all three human cholangiocarcinoma cells (CCLP1, SG231, and HuCCT1; Supplementary Fig. S5B–D). The latter observations are noteworthy, because 15-PGDH is a prostaglandin-degrading enzyme that physiologically antagonizes COX-2 and suppresses tumor growth.
Because DHA reduces PGE2 levels through concomitant inhibition of COX-2 and induction of 15-PGDH, we postulate that DHA might also inhibit β-catenin through the inhibition of PGE2. To evaluate this hypothesis, further experiments were performed to determine whether DHA might prevent PGE2-induced β-catenin activation. DHA prevented PGE2-induced phosphorylation of GSK-3β as well as dissociation of the GSK-3β/Axin complex (Supplementary Fig. S6). Accordingly, DHA treatment also inhibited PGE2-induced TCF/LEF reporter activity. These findings suggest that suppression of PGE2 by DHA represents another mechanism for β-catenin degradation.
Taken together, our data suggest that ω3-PUFAs induce β-catenin degradation through three interrelated mechanisms. First, we show that DHA and EPA induce a rapid dephosphorylation of GSK-3β in human cholangiocarcinoma cells, suggesting that GSK-3β activation is involved in ω3-PUFA–induced β-catenin degradation. This assertion is further supported by the observations that the GSK-3β inhibitor, SB216763, prevents DHA-induced reduction of β-catenin protein and transcription activity and restored cell survival. Second, DHA treatment induces the association of Axin with GSK-3β forming the β-catenin destruction complex. Third, ω3-PUFAs suppress PGE2 signaling through concomitant inhibition of COX-2 and induction of 15-PGDH, thus preventing PGE2-induced β-catenin accumulation. The involvement of β-catenin degradation in ω3-PUFA–induced inhibition of tumor growth is further supported by the observation that Wnt3a-CM partially protects cholangiocarcinoma cells from DHA-induced inhibition of cell growth.
In summary, this study presents the first evidence that COX-2–derived PGE2 activates β-catenin in human cholangiocarcinoma cells and that ω3-PUFAs inhibit cholangiocarcinoma cell growth by simultaneously blocking β-catenin and COX-2 signaling pathways. Our findings provide important preclinical evidence and molecular insight for potential utilization of ω3-PUFAs in the chemoprevention and treatment of cholangiocarcinoma.
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
Acknowledgments
Grant support: NIH R01 grants CA102325 and CA106280 (T. Wu) and DK49615 (A.J. Demetris).
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.