Applications of Sesquiterpene Lactones: A Review of Some Potential Success Cases
by
 ,
and
,
and
Laila Moujir
1,*,

Oliver Callies
2 ,
,
Pedro M. C. Sousa
3,
Farukh Sharopov
4 and
Ana M. L. Seca
5,6,*
1
Department of Biochemistry, Microbiology, Genetics and Cell Biology, Facultad de Farmacia, Universidad de La Laguna, 38206 San Cristóbal de La Laguna, Spain
2
AbbVie Deutschland GmbH & Co. KG, Knollstrasse, 67061 Ludwigshafen, Germany
3
Faculty of Sciences and Technology, University of Azores, 9500-321 Ponta Delgada, Portugal
4
Research Institution “Chinese-Tajik Innovation Center for Natural Products”, Academy of Sciences of the Republic of Tajikistan, Ayni 299/2, Dushanbe 734063, Tajikistan
5
cE3c—Centre for Ecology, Evolution and Environmental Changes/Azorean Biodiversity Group & Faculty of Sciences and Technology, University of Azores, Rua Mãe de Deus, 9500-321 Ponta Delgada, Portugal
6
LAQV-REQUIMTE, University of Aveiro, 3810-193 Aveiro, Portugal
*
Authors to whom correspondence should be addressed.
Appl. Sci. 2020, 10(9), 3001; https://doi.org/10.3390/app10093001
Submission received: 25 March 2020 / Revised: 17 April 2020 / Accepted: 19 April 2020 / Published: 25 April 2020
(This article belongs to the Special Issue Biological Activity and Applications of Natural Compounds)
Abstract
:
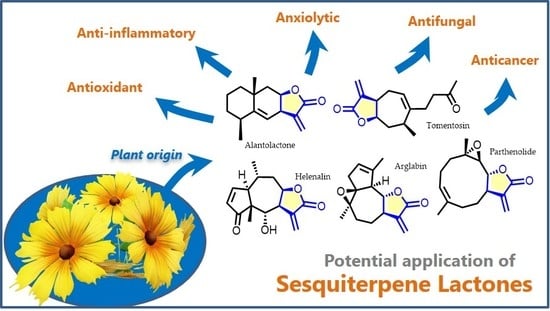
Sesquiterpene lactones, a vast range of terpenoids isolated from Asteraceae species, exhibit a broad spectrum of biological effects and several of them are already commercially available, such as artemisinin. Here the most recent and impactful results of in vivo, preclinical and clinical studies involving a selection of ten sesquiterpene lactones (alantolactone, arglabin, costunolide, cynaropicrin, helenalin, inuviscolide, lactucin, parthenolide, thapsigargin and tomentosin) are presented and discussed, along with some of their derivatives. In the authors’ opinion, these compounds have been neglected compared to others, although they could be of great use in developing important new pharmaceutical products. The selected sesquiterpenes show promising anticancer and anti-inflammatory effects, acting on various targets. Moreover, they exhibit antifungal, anxiolytic, analgesic, and antitrypanosomal activities. Several studies discussed here clearly show the potential that some of them have in combination therapy, as sensitizing agents to facilitate and enhance the action of drugs in clinical use. The derivatives show greater pharmacological value since they have better pharmacokinetics, stability, potency, and/or selectivity. All these natural terpenoids and their derivatives exhibit properties that invite further research by the scientific community.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
Traditional or folk medicine relies heavily on the use of compounds-rich plants, like those of the Asteraceae family, of which many such species are commercially available in the form of herbal preparations. These are particularly rich in a wide range of natural terpenoids named sesquiterpene lactones [1] that, in some cases, are considered the active principles of such therapeutic plants [2]. Structurally speaking, sesquiterpene lactones are terpenes that have in common a basic structure of 15 carbons (thus the prefix sesqui-) resulting from biosynthesis involving three isoprene units with a cyclical structure along with a fused α-methylene-γ-lactone ring [3]. Sesquiterpene synthases catalyze a common biosynthesis route for sesquiterpene lactones, based on the cyclization of farnesyl phosphate resulting from the 2-C-methyl-D-erythritol-4-phosphate (MEP) and mevalonate (MVA) pathways of dimethylalyl diphosphate and isopentenyl diphosphate precursors in chloroplasts and cytosol, respectively [4]. However, sesquiterpene lactones have very different chemical structures regarding the type and position of the substituents, as well as the size of the non-lactone ring [5,6]. For this reason, in structural terms, sesquiterpene lactones are organized into several subclasses: eudesmanolide (a 6/6 bicyclic structure), guaianolide and pseudoguaianolide (both 5/7 bicyclic compounds), germacranolide (with a 10-membered ring) and xanthanolide (containing a non-cyclic carbon chain and a seven-membered ring) [7,8] (Figure 1).
Some of these subclasses have compounds that exhibit a wide range of biological activities. These range from antitumor [9] to anti-inflammatory, including antimalarial, antimicrobial, antioxidant [10], neuroprotective [11], hepatoprotective, and immune-stimulant properties [12,13]. Regarding the structure/activity relationship of these compounds, it appears that the α-methylene-γ-lactone nucleus has a crucial role in almost all their observed biological effects, such as cytotoxic, antitrypanosomal, and anti-inflammatory actions [12,14]. Other specific structural moieties of sesquiterpene lactone seem to influence their activity. For example, the presence of electrophilic sites associated with medium/high lipophilicity increase antimycobacterial activity, while a double bond exo to the cyclopentenone ring seems to favor anti-inflammatory activity [7,8,14]. The interaction of the α,β-unsaturated cyclopentenone nucleus with the target depends largely on the geometry of the molecule, which is also a factor that influences the level of activity exhibited by sesquiterpene lactones [7,15]. Moreover, the number of alkylating groups in the structure of sesquiterpene lactones contributes to the level of activity they display, two groups being the optimal number [7,15]. The structure/activity relationship specific to each sesquiterpene lactone presented in this review will be discussed in detail throughout Section 2. Scientific evidence shows an action mechanism common to sesquiterpene lactones. The structural elements α-methylene-γ-lactone and α,β-unsaturated cyclopentenone act as alkylating groups on proteins found in cells through Michael addition, especially their thiol groups. They thereby affect cell functionality, i.e., gene regulation, protein synthesis, and cell metabolism [15,16,17].
In recent years, the scientific community has already shown an oustanding growth of interest in the sesquiterpene lactones, largely due to the success of artemisinin—one of the best known—and the broad spectrum of activities exhibited by this compound’s chemical family [18,19,20]. Thus, the high number of studies published concerning their isolation from new natural sources, total and semi-synthetic syntheses, and evaluation of pharmacological potential, is not surprising. Natural sesquiterpene lactones exhibit poor pharmacokinetic properties due mainly to their low bioavailability, deriving from low solubility in water. As a result, in order to overcome these limitations, research interest in the synthesis of their derivatives has increased [21,22]. In addition, structural modification and synthesis has also allowed for an in-depth knowledge of their chemical properties, as well as the establishment of structure-activity relationships.
In terms of health promotion, the applications of sesquiterpene lactones and their derivatives are a key research area. From this point of view, in vivo, preclinical and clinical studies are those that allow a more realistic assessment of their medicinal potential [23].
The literature on this topic is extensive, especially for the most successful compounds; however, studies on less known sesquiterpenes and their derivatives show that these compounds deserve more attention, since they could play an important part in future human health maintenance. Therefore, this review aims to point out the results of the most impactful and recent in vivo and (pre)clinical studies of these insufficiently explored compounds and derivatives, which could be a valuable alternative in the development of new therapeutic drugs.
2. Sesquiterpene Lactones with Significant In Vivo Activity
2.1. Alantolactone
The eudesmanolide alantolactone (1) (C15H20O2) (Figure 2) was first isolated from Inula helenium L. roots, and later was also found in other Inula species [24], as well as non-Inula spp. such as Saussurea lappa C.B. Clarke [25] and Aucklandia lappa DC. [26].
Alantolactone (1) is obtained mainly by extraction and purification from natural sources, although its total synthesis is possible [27,28]. Modern laboratory techniques have resulted in a significant increase in its extraction yield [29,30,31]. For example, ~3% pure alantolactone (1) yields were obtained from Inula helenium roots by Zhao et al. [30], which meant a significant improvement over the ~1% yields from the roots of Inula magnifica Lipsky [32].
Although alantolactone is associated with allergic contact dermatitis triggered by Inula species [33,34,35], this compound was first described as anti-helminthic [36], with subsequent studies demonstrating its tremendous potential, mainly as antitumor [37,38,39,40], anti-inflammatory [26,41], and antioxidant [42] agent.
Regarding anticancer activity, the effect of alantolactone (1) on leukemia is well documented and was recently reviewed by Da Silva Castro et al. [43]. Xu et al. [44] reported a positive and significant effect (1) on B-cell acute lymphoblastic leukemia. In this study, 100 mg/kg (b.w.) doses were intravenously administered every two days in leukemia xenografted NOC/SCID mice. Results showed that treated mice lived an average of eight days more than non-treated mice (31.5 days after xenografting in treated compared to 23.5 days in non-treated mice), without significant weight loss. Before this study, Chun et al. [45] had also reported an in vivo antitumor effect on triple negative breast cancer in mice. The researchers used only a 2.5 mg/kg (b.w.) dose every two days to treat athymic nude mice xenografted with MDA-MB-231 cells. In this assay, alantolactone (1) reduced tumor size by over half, and significantly reduced tumor weight by about half after 24 days. Alantolactone (1) also exhibits in vivo activity against gastric cancer. Human gastric cancer cells (SGC-7901) were xenografted onto nude athymic mice, which were treated with 15 mg/kg (b.w.) of alantolactone (1) injected every two days [46]. Alantolactone (1) caused significant tumor growth inhibition and reduced Ki-67 and Bcl-2 expression (tumor associated proteins), without significant liver and kidney toxicity or impact on mouse weight [46].
Alantolactone (1) has also proven to be an especially potent sensitizing agent. In fact, it exhibits a synergistic effect with known chemotherapy drugs, such as oxaliplatin. Cao et al. [47] showed that a 10:2 mg/kg (b.w.) dose of alantolactone: oxaliplatin reduced tumor volume and weight by more than 50% in athymic mice xenografted with colorectal tumor cells (HCT116). The anticancer effect increased substantially when both compounds were used together, as opposed to alone. This result is in line with that obtained by He et al. [48], according to which, using a small weekly injected dose of 3 mg/kg (b.w.), tumor volume significantly decreased by around 75% in xenografted pancreatic cancer cells (PANC-1), while increasing cancer cell chemosensitivity to oxaliplatin, revealing a synergistic action.
Alantolactone’s anticancer mechanism was also the main focus of several studies, some of them mentioned in recent reviews [49,50] The authors attribute its activity to the multiple pathways it activates. Alantolactone (1) acts as an alkylating agent leading to inhibition of key enzymes and proteins, and as an apoptosis inducer in cancer cells at mitochondrial level by interacting with cytochrome c. It promotes overproduction of reactive oxygen species (ROS) due to specific caspase activation, by inhibiting autophagic deregulation, among other processes.
Beyond its anticancer activity, compound 1 also exhibits interesting anti-inflammatory activity. Ren et al. [51] used the DSS-induced colitis mouse model to test alantolactone (1) anti-inflammatory activity. A 50 mg/kg dose reversed colitis symptoms (bloody diarrhea, colon shortening and weight loss), besides significantly reducing pro-inflammatory cytokine TNF-α expression (to about half that of positive control) and IL-6 (by over 2.5 times compared to positive control) [51]. Wang et al. [41], showed that a 10 mg/kg (b.w.) dose of alantolactone (1) significantly improved neurological function and reduced cerebral edema in a traumatic brain injury mouse model. This neuroprotective effect was attributed to alantolactone’s capacity to inhibit the NF-κB inflammatory pathway and the cytochrome c/caspase-mediated apoptosis pathways [41]. To the best of our knowledge, no other authors have elaborated on this interesting double action: the fact that alantolactone (1) can simultaneously activate apoptotic pathways in cancer cells and inhibit these pathways in a cerebral edema model.
Seo et al. [52] described the neuroprotective effect of alantolactone (1) using amyloid β25–35-induced ex vivo neuronal cell death and scopolamine-induced amnesia in mouse models meant to emulate conditions common to neurodegenerative conditions like Alzheimer’s disease. A 1 µM alantolactone (1) treatment increased cortical neuron viability to almost baseline control readings, and 1 mg/kg (b.w.) significantly decreased scopolamine-induced cognitive impairments. It is a interesting to note that this particular neuroprotective effect is attributed to a drop in ROS levels. High ROS concentrations are associated with the neurodegenerative damage of Alzheimer’s disease. Again, alantolactone (1) exhibits a double action: it raises ROS levels to induce apoptosis in murine models of neurodegenerative damage.
The interesting advances with alantolactone (1) derivatives are also worth mentioning. Kumar et al. [53] assayed three of the 17 thiol derivatives synthesized, at 10 mg/kg (b.w.) doses, in mice showing that they have in vivo anti-inflammatory activity comparable to alantolactone (1). The novel compounds shared alantolactone’s anti-inflammatory mechanisms. Another noteworthy study involving alantolactone (1) derivatives was published in 2018 by Li et al. [54], testing 44 derivatives for their ability to inhibit induced pulmonary fibrosis in mice. The results showed that 2 of these compounds are particularly active at 100 mg/kg (b.w.), reducing the fibrotic area by more than 60%. This is achieved by inhibition of the TGF-β1 pathway of myofibroblast differentiation. It should be noted that no toxic effects were observed for either compound in the chronic toxicity test (seven days), using a dose of 2 g/kg (b.w.) administered orally. Finally, a patent involving an alantolactone (1) spiro-isoxazoline derivative was filed in June 2019 (US patent N° 20190185487) for the development and production of these compounds that exhibit significant anti-inflammatory activity.
One factor which contributes to making alantolactone (1) very interesting as a future medicine is its low toxicity. In work by Khan et al. [55], Kunming mice treated with 100 mg/kg (b.w.) alantolactone (1) showed no significant signs of hepatotoxicity or nephrotoxicity, in line with the previously cited work by He et al. [38]. This is especially important for alantolactone (1) as an anticancer drug, because it is well known that the liver and kidneys are particularly susceptible to negative side effects from chemotherapy approaches [56,57].
Another engaging finding obtained by Khan et al. [55] is alantolactone’s ability to cross the blood-brain barrier. It may thus become useful in the treatment of brain tumors or other conditions involving the central nervous system (CNS), since the blood-brain barrier is the greatest obstacle for drug delivery to those areas [58].
The metabolism and pharmacokinetics of alantolactone (1) have also been studied in vivo, with future pre-clinical testing in mind. Research shows that alantolactone (1) exhibits low absorption and is rapidly eliminated after intravenous and oral administration. Its metabolism involves conjugation with thiol, and α, β-unsaturated carbonyl is the preferential structural metabolic site. The low aqueous solubility of alantolactone (1) causes low oral bioavailability [59,60,61].
All these recent reports show there is great interest in alantolactone (1), and that it has potent proven in vivo activities, mainly several different types of anticancer activity. This broad-spectrum activity, combined with its synergistic action with known cancer therapy agents, shows alantolactone (1) has great potential for future drug development. However, further research is necessary, especially clinical trials, to identify its intracellular action sites and secondary targets and thereby elucidate its mode of action.
2.2. Arglabin
Arglabin (2) (Figure 3), is a guaianolide sesquiterpene lactone with the chemical formula C15H18O3, isolated from several species including Artemisia myriantha Wall. ex Besser [62,63], Artemisia jacutica Drob [64], and Artemisia glabella Kar. and Kir. species [65] where the extraction yield was 0.27% [66]. Fortunately, several synthetic and hemisynthetic methods for preparing compound 2 have been reported [66,67,68,69,70], allowing it to be provided in the quantities necessary for research and medicinal applications.
The arglabin (2) molecule contains a 5,7,5-tricyclic ring structure with contiguous stereo centers [70]. Furthermore, it contains both an epoxide and an α,β-unsaturated Michael acceptor, two functional groups that have important roles in its pharmacological activities [71]. There are many research lines advancing towards more effective drugs on the basis of the arglabin (2) molecule. New arglabin (2) derivatives have been obtained by chemical modification, the most successful being obtained via amination followed by treatment with gaseous hydrochloric acid. This converts arglabin (2) into the hydrochloride salt of the dimethyl amino adduct, which is very soluble in water [66,72]. This arglabin (2) salt form was used in the therapy of several cancer types such as breast, lung, liver and esophageal tumors in oncological clinics of Kazakhstan [73,74,75]. Treatment of an esophageal carcinoma patient with compound 2 contributed to a significant reduction in tumor volume, favoring its regression and a lower incidence of leucopenia [75].
In terms of mechanism of action, arglabin (2) inhibits farnesyl protein transferase enzyme [65,72]. It influences DNA synthesis in murine P388 lymphocytic leukemia cells [74]. Recently, Schepetkin and co-authors [76] have reported that compound 2 inhibits T-cell receptor activation and anti-CD3-induced movement of intercellular Ca2+ ions ([Ca2⁺]i), blocks ERK1/2 phosphorylation and depletes [GSH] in Jurkat T cells.
The anticancer effect of Arglabin (2) was also demonstrated by He and colleagues [77] on xenografted oral squamous cell carcinomas. They elucidated that tumor growth is inhibited via downregulation of relevant protein expression in the mTOR/PI3K/Akt signaling pathway and impairment of mitochondrial membrane potential, leading to apoptosis.
Recently, the pharmacokinetic properties of arglabin (2) have been reviewed [72], highlighting that it mainly accumulates in the liver, quickly reaches peripheral tissues and penetrates the blood-brain barrier. Furthermore, according to the literature, arglabin (2) has no described adverse effects, since it does not affect normal liver and kidney function or cause local irritative/allergenic reactions, nor mutagenic or embryotoxic effects [66,72,78].
Besides anticancer activity, arglabin (2) demonstrated in vivo anti-arthritis activity in a rat model. Arglabin (2) lowers the levels of inflammatory mediators and cytokines, and reduces expression of NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells), COX-2 (cyclooxygenase-2) and iNOS (inducible nitric oxide synthase) [79]. Abderrazak and co-authors [80] suggest that arglabin (2) is a compound with great potential in inflammation and atherosclerosis therapy. Using a high-fat diet ApoE2.Ki mouse model, experimental results [80] showed that arglabin (2) reduces inflammation by decreasing IL-1β and IL-18 levels and increases autophagy apoptosis [81].
Studies carried out with arglabin (2) show its potential for developing new anticancer and/or anti-inflammatory drugs, the main limiting factor being its low bioavailability. The preparation of soluble active versions of compound 2, and/or new delivery systems may attract further interest in this sesquiterpene lactone.
2.3. Costunolide
Costunolide (3) (Figure 4), has the same chemical formula as alantolactone (1) (C15H20O2). It is a member of the germacranolide subclass and was first isolated from Saussurea costus (Falc.) Lipsch. roots in 1960 [82]. It is present in many Asteraceae genera such as Inula [24], Lactuca and Helianthus [83], but also those from other families like Magnoliaceae [84].
Biosynthesis of costunolide (3) is well documented and occurs through the mevalonate pathway [85]. Briefly, the process starts with the cyclization of farnesyl pyrophosphate, forming germacrene A. Next, the isoprenyl side chain of germacrene A undergoes hydroxylation by germacrene A hydroxylase, followed by oxidation to germacrene acid. It is finally synthesized after oxidation and cyclization of germacrene acid [85]. Some species are rich sources of costunolide (3), such as essential oil of Saussurea lappa roots (yield ~3%), of which 52% was identified as costunolide (3) [86]. The total synthesis of costunolide (3) has been described using different methods/strategies [87,88,89].
In a recent review [90], Kim and Choi detail the activities of custonolide (3) and discuss its therapeutic potential. Like alantolactone (1), compound 3 also exhibits anticancer bioactivity against different cancer cells via various routes. In fact, it acts as an apoptosis inducer, cell cycle regulator, angiogenesis and metastasis inhibitor and can also reverse the drug resistance mechanism [91].
There are several recent publications describing in vivo costunolide (3) studies, such as the work by Jin et al. [92] on costunolide’s anticancer effect on osteosarcoma xenografted mice. Results showed a daily 20 mg/kg (b.w.) dose was enough to significantly reduce tumor weight by about half, as well as reduce the number of lung metastases to around one third of that in the control group. Western blot analysis of the tissues revealed this effect could be attributed to costunolide (3) inhibition of STAT3 transcription, a factor that is widely known to be linked to oncogenesis and cancer proliferation [93]. It is interesting to note the similarity between previously mentioned STAT3 inhibitory activity by alantolactone (1) [45]. To highlight another similarity, costunolide (3) also exhibits activity against gastric adenocarcinoma in xenografted mice [94]. Results from this study showed costunolide (3) induced caspase-mediated mitochondrial apoptosis in cancer cells and a 50 mg/kg (b.w.) dose on alternate days significantly reduced tumor size to about half. It achieved very similar results to the positive control cisplatin, a chemotherapeutic agent used in clinical treatment [94]. Interesting to note is costunolide’s anticancer effect through telomerase reverse transcriptase inhibition [95], a mechanism not known in alantolactone (1). As reported by the researchers, a 5 mg/kg (b.w.) dose injected on alternate days in glioma xenografted mice significantly reduced tumor size by over 50%. This inhibition was associated with reduced telomerase activity, which leads to ROS-associated apoptosis [95]. A subsequent work by the same research group also showed the same dose affects lipid metabolism in glioma xenograft tumors, by lowering expression of FASN, SREBP-1, and PGC-1α [96], which are key genes targeted for cancer treatment [97,98,99].
Beyond costunolide’s anticancer effect, it has also proven to be a potent anti-inflammatory agent [90], as well as an antidiabetic, antihelminth, antimicrobial, antiulcer and antioxidant [91]. Its anti-inflammatory action has been well documented in vitro [84,100] and in vivo, appearing to be linked to NF-κB pathway inhibition [101]. For example, costunolide (3) is able to suppress inflammatory angiogenesis [102], alleviating gastric ulcers [103], and acute lung and liver damage [104,105,106,107].
Recently, studies have shown that costunolide (3) also exhibited anti-osteoarthritic effects [108]. In fact, treatment in rats with osteoarthritis causes attenuation of cartilage degeneration compared to the control osteoarthritic group. The observed effect has been attributed to the inhibitory action of this compound on the Wnt/β-catenin and NF-κB signaling pathways, and on the expression of matrix metalloproteinases [108].
Costunolide (3) seems to be also a powerful antiasthmatic [109]. In this work, the researchers treated asthma-induced mice with 10 mg/kg (b.w.) before an immune challenge. Results showed a 61.8% inhibition of asthma-associated eosinophil increase, as well as significantly reducing lung inflammation scores and mucin production [109].
Recently, it has been shown that costunolide (3) is an effective inducer of hair growth in mice [110]. For this assay, the researchers implanted mouse dermal cells, treated with 3 mM costunolide (3) for two days. Results showed a 2.5-fold increase in induced hair follicles in the implanted treated cells, and topically applied costunolide (3) significantly and visibly improved hair growth. The authors claim this might be due to activation by compound 3 of key follicle-cell cycle pathways, including the Wnt/b-catenin, Shh/Gli, and TGF-β/Smad pathways. It is worth mentioning the TGF-β/Smad pathway was also mentioned in previous studies related to anti-inflammatory action of costunolide (3) [106].
Costunolide (3) derivatives have also proven to be highly interesting, with remarkable in vivo effects. In recent work by Cala et al. in an agro-research context [111], many different functional groups were added to the sesquiterpene backbone, yielding, among others, two amino and two methyl ether derivatives with strong herbicidal activity. The etiolated wheat coleoptile assay indicated treatment with these costunolide (3) derivatives had dose-dependent growth inhibition responses equivalent to a widely used synthetic herbicide used as positive control. These results show there is potential for some of these derivatives as bio-herbicides, but further studies are necessary. There was also another interesting result of this work which the authors did not address, but is worth pointing out: two tested thiol derivatives boosted coleoptile growth instead of inhibiting it, with one compound increasing coleoptile length by ~30% with a 30 µM treatment. The general implication of these results is that costunolide (3) modification can be powerful and versatile, capable of yielding diametrically opposed bioactive compounds depending on the functional group added.
In conclusion, costunolide (3) is a highly multitalented and potent bioactive compound. Similar to other sesquiterpene lactones such as the previously discussed alantolactone (1), much of its potential for future drug development lies in its anticancer and anti-inflammatory effects. It is interesting that there appears to be more work done with costunolide (3) derivatives than alantolactone (1) derivatives, which in general suggests greater interest of the scientific community in costunolide (3) as opposed to alantolactone (1). With so many relevant publications in 2019 alone, we expect to see costunolide (3) research ramping up to a pre-clinical stage very soon.
2.4. Cynaropicrin
Cynaropicrin (4) (Figure 5) is a guaianolide type sesquiterpene lactone with a chemical formula of C19H22O6 and a 5,7,5 fused tricyclic skeleton [112].
The sesquiterpene lactone cynaropicrin was isolated from artichoke (Cynara scolymus L.) in 1960 for the first time [113] and later it was found in Cynara cardunculus L. [114,115] and Cynara scolymus L. species [116], being considered as a chemotaxonomic marker for artichoke plant species [112]. Cynaropicrin (4) was also found in many species of Asteraceae family such as Centaurea drabifolia subsp. floccosa (Boiss.) Wagenitz and Greuter [117], Psephellus sibiricus (L.) Wagenitz [118], Rhaponticum pulchrum Fisch. and C.A.Mey. [119], Moquinia kingii (H.Rob.) Gamerro [120], Saussurea calcicola Nakai [121], Saussurea costus (Falc.) Lipsch. [122,123], Tricholepis glaberrima DC. [124], and many others [112]. Yields of cynaropicrin (4) extraction from Cynara cardunculus L. leaves using ethanol, ethyl acetate, dichloromethane and water were 56.9, 37.5, 40.3 and 13.6 mg/g dry weight, respectively [125].
The biological activities of cynaropicrin (4), as with other sesquiterpenic lactones, are related to its pharmacophore γ-butyrolactone ring [112]. There are many studies reporting on the important pharmacological activities of cynaropicrin (4), and plants rich in cynaropicrin (4), such as antitumor, anti-inflammatory, antitrypanosomal, and antihepatitis C virus, among many others. Due to these notable effects, cynaropicrin (4) will be suitable for the development of medicinal compounds [123].
Cynaropicrin (4) is the first natural product that in vivo potently inhibits the African trypanosome diseases [126]. Using the acute model of mice infected with Trypanosoma brucei rhodesiense STIB 900, when treated with two doses of 10 mg/kg (b.p.) per day, on the 7th day after infection, there was a 92% reduction in parasitemia when compared to the untreated group. Additionally, selectivity indices of 7.8 were obtained for cynaropicrin (4) against L6 cells of rat myoblasts [127]. The action mechanism is still under study to date, but is thought to be related to the interaction of compound 4 with the trypanothione redox system in Trypanosoma brucei [127]. However, Da Silva et al. [128] demonstrated that cynaropicrin (4) at a dose of up to 50 mg/kg (b.w.) per day has no effect in mice infected with Trypanosoma cruzi, in either Y or Colombiana strains. The synthesis and semi-synthesis of several cynaropicrin (4) derivatives allowed the structure/antitrypanosomal activity of these compounds to be evaluated. It was concluded that the α-methylene-γ-lactone structure is indispensable to maintain the biological effect, whereas 3-OH and 19-OH derivatization does not change the activity and some types of side-chain promote the selectivity of the compound [129,130]. The in vivo evaluation of some derivatives, using the Trypanosoma brucei rhodesiense acute mouse model, indicated that the dimethylamino derivative exhibits much less toxicity than cynaropicrin (4), but also less activity [129].
Cynaropicrin (4) also showed the ability in vivo to delay the effects of skin photoaging, promoting the proliferation of melanocytes and keratinocytes, by acting as inhibitor of NF-ĸB transcription activity [131].
2.5. Helenalin
Helenalin (5) (C15H18O4) (Figure 6), a 5/7-fused bicyclic sesquiterpene lactone that belongs to the pseudoguaianolide subclass. It is very abundant in Arnica montana L. but is also identified in other species from Arnica and Helenium genera and from Centipeda minima (L.) A.Braun and Asch. [132,133,134,135,136].
Alcohol extracts containing helenalin (5) and its derivatives have been used as a staple of traditional herbal anti-inflammatory medicine for many decades [134]. For instance Arnica montana L. solutions are used to treat rheumatism, arthritis, inflammation, hematoma, soreness, sprains, swelling and muscle spasms from athletic activity etc., seasonally triggered arthritis, arteriosclerosis, angina pectoris, postoperative conditions, and joint pain. The plant is used externally in creams, alcoholic tincture, and ointment form but also taken highly diluted in homeopathic remedies [134]. Many such preparations, even hair oil and shampoo, are commercially available from a range of suppliers in healthfood shops and pharmacies almost worldwide [137]. Likewise, clinical trials have aimed to assess topical Arnica applications, regarding possible reduction of laser-induced minor hematomas and osteoarthritis-type symptoms. Orally administered homeopathic formulations are also widely employed in the clinical setting to treat and manage conditions such as carpal tunnel, slow knee-surgery recovery, tonsil and wisdom tooth extractions, facelifts, neuralgia, hysterectomy, venous surgery, hallux valgus, heart-valve surgery, hemarthrosis, prolonged intravenous perfusion, joint sprains and strains, muscle pain, etc. [133].
Helenalin (5) inhibits NF-κB transcription of inflammatory cytokines, which have an essential role in both inflammation and cancer [138]. It also efficiently inhibits cancer cell proliferation through a variety of action modes, e.g., telomerase inhibition [139], DNA and protein synthesis attenuation, apoptosis induction and promoting reactive oxygen species generation [140]. It is noteworthy that inhibition of NF-κB activation associated with another sesquiterpene, helenin, occurs in T-cells, B-cells and epithelial cells in response to four different stimuli, nullifying kB-driven gene expression. Since this activity does not affect transcription factors Oct-1, TBP, Sp1 or STAT 5, this NF-κB activation is probably inhibited selectively [141,142]. Lyss et al. [141] described how helenalin modifies the NF-κB/IκB complex, preventing IκB release. They proposed a molecular action mode for the anti-inflammatory influence, which is different from other nonsteroidal anti-inflammatory drugs (NSAIDs) such as aspirin and indomethacin. Furthermore, it targets Cys38 on p65, ablating DNA-binding [143], inhibiting neutrophil chemotaxis and migration as well as 5-lipoxygenase and leukotriene C4 synthase activities [144].
Experiments performed by Schröder et al. [145] demonstrate that compound 5 inhibits platelet aggregation induced by collagen, 5-hydroxytryptamine secretion and thromboxane formation, depending its on concentration (between 3–300 µM). They concluded that helenalin inhibits platelet function via interaction with platelet sulfhydryl groups in a way most likely associated with reduced phospholipase A2 activity.
Helenalin (5) inhibited complete Freund’s adjuvant-induced arthritis and carrageenan-induced paw edema in the rat [146]. Topical application of Arnica 3D gel (10%), combined with a 10 mA microcurrent for 2 min also significantly improved wound healing in a linear incision wound model in the rat back [147]. The evidence was consistent with a higher percentage of mature collagen fibers and a significantly larger total number of cells in the wound, as assessed by structural and morphometric analysis. According to this, different proportions of Arnica montana extracts alone or combined with other plants have been patented for their therapeutic potential [137]. Widrig et al. [148] performed a randomized, double-blind study in 204 patients with osteoarthritis (OA) to compare the effects of ibuprofen (5%) and Arnica montana gel (50 g tincture/100 g) containing helenalin (5), 11α,13-dihydrohelenalin and its ester. The results show that short-term use, up to three weeks, of Arnica gel improves pain and hand function in OA, indistinguishably from ibuprofen gel. Substantial antiosteoarthritic activity by blocking transcription factors NF-κB and NF-AT is attributable to helenalin and derivatives.
Boulanger et al. [149] demonstrated that helenalin (5), intraperitoneally delivered at 20 mg/kg in lactating-Balb/C mice 9 and 3 h prior to infection, reduced intracellular growth of Staphylococcus aureus in mouse mammary glands This suggests the compound interferes with host molecular mechanisms and not directly with Staphylococcus aureus growth. The authors conclude helenalin might be worth investigating as a potential treatment for Staphylococcus aureus-induced mastitis in bovine species. There is however some concern regarding this treatment, mainly regarding helenin persistence in the animal’s milk, and whether or not the therapeutic doses pose short- and long-term toxicity risks. It is therefore imperative to further study and characterize its safety and pharmacological properties.
Valan et al. [150] showed that helenalin (5) has a significant biphasic positive inotropic effect on the myocardium of guinea pigs at doses of 10−5–10−3 mol. Nevertheless, concentrations above 10−3 mol. cause an irreversible negative inotropic action leading to a blocking of muscle contraction.
The skin is susceptible to environmental damage by multiple agents, particularly solar ultraviolet (UV), which induces skin hyperpigmentation disorders. Expression of heat shock proteins (HSPs, particularly HSP70) is receiving consideration in the field of cosmetics, to reduce skin damage and signs of aging. Usui et al. [151] isolated AM-2 (helenalin 2-methylbutyrate) from Arnica montana as a good inductor of HSP70, with low cytotoxicity. Treatment of cultured mouse melanoma cells with AM-2 or Arnica montana extract up-regulated the expression of HSP70 in a dose-dependent manner. It also activated the transcription factor for hsp genes, i.e., heat shock factor-1. They concluded that both Arnica montana extract and AM-2 are likely to show beneficial effects if incorporated in hypopigmenting cosmetics.
Acute liver injury is a life-threatening syndrome frequently associated with hepatocyte damage and characterized by oxidative and inflammatory responses. Li et al. [136] recently observed that intragastric administration of helenalin (5) for 10 days significantly ameliorated hepatic injury induced previously in mice with LPS/D-GalN. These results were evidenced by the attenuation of histopathological changes and the decrease in serum aminotransferase and total bilirubin activities. Therefore, helenalin (5) shows a hepatoprotective effect against damage induced by LPS/D-GalN. This in turn may be associated with reduced hepatocyte apoptosis, by protection of mitochondrial function and oxidative stress inhibition by Nrf2 pathway activation, as well as attenuating inflammation by inhibiting NF-κB activation. The co-authors of that study [152] submitted Patent No. CN 110283151 in 2019 as a method for isolation of helenalin from Centipeda minima and its application for treating hepatic fibrosis and inflammation. Furthermore, the same authors [153] patented it for inhibiting hepatic stellate cell activation, showing the advantages of reducing collagen deposition and synthesis of inflammation-related proteins, promoting death of stellate cells, and its application in liver fibrosis treatment.
2.6. Inuviscolide
The guaianoline-type sesquiterpene lactone inuviscolide (6) (C15H20O3) (Figure 7) was identified in Ferula communis (Apiaceae) and described to be the main metabolite in Inula viscosa (L.) Ait. [154].
Inuviscolide (6) inhibited in vivo inflammation in mice, as shown in the investigation performed by Hernández et al. [155]. The authors used inuviscolide (6) and ilicic acid from Inula viscosa and found that compound 6 influenced cell degranulation and leukotriene biosynthesis, as well as neurogenic drive and glucocorticoid-like interactions. During the testing, ear and paw edema were induced in Swiss female mice using phorbol esters or ethyl phenylpropiolate (EPP), and phospholipase A2 (PLA2) or serotonin, respectively. The drug dose was applied topically in the ear models but as subcutaneous or intraperitoneal injections in the paw models. For quantitation of leukotriene B4 (LTB4) formation, high-performance liquid chromatography (HPLC) was performed on rat peritoneal neutrophils. The results showed that compound 6 reduced PLA2-induced edema with an ID50 of 98 µmol/kg. The results did not indicate that glucocorticoid response modifiers had an influence on the edema induced by serotonin. In intact cells, inuviscolide (6) resulted in reduced generation of LTB4 (IC50 value of 94 µM). The overall results indicated that compound 6 has an important role in the anti-inflammatory activity of Inula viscosa, being more active than ilicic acid. The action mechanism was suggested to be related to an interference with leukotriene synthesis and to PLA2-induced mastocyte release of inflammatory mediators [156].
2.7. Lactucin and Its Derivatives Lactupicrin, and Lactucopicrin
Other compounds from the guaianolide subclass are lactucin (7) (C15H16O5) and its 8- and 15- (4-hydroxyphenylacetate) derivatives lactupicrin (8) and lactucopicrin (9), respectively (Figure 8). Dolejš et al. [156] and Ruban et al. [157] had previously described the chemical structure of lactucin (7). These compounds are distributed within Asteraceae like Lactuca serriola [158], especially plants commonly used in salads. Lactuca sativa (lettuce), Cichorium intybus (chicory and radicchio), and Cichorium endivia (endive) have been reported to contain lactucin (7) and lactucin-related substances [159,160,161,162,163,164]. The content depends largely on the species, variety and the part of the plant analyzed. In fact, lettuce guaianolide content was high and reached concentrations between 61.7 mg/mL and 147.1 mg/mL in its latex [160] and 2.9 mg/g to 36.1 mg/g in the overall plant, expressed as dry weight [162].
The biological activities of lactucin (7) and lactucin-related compounds had already attracted interest at the end of the 19th century, as they were credited to be responsible for the bitter taste and pharmacological effect of lactucarium or lettuce opium [165]. This is the condensed latex of wild lettuce Lactuca virosa. This dried exudate was used in Europe for centuries with similar applications to opium as an analgesic, antitussive, and sedative [163]. Due to its widespread use, it was described and standardized in the United States Pharmacopoeia (USP) and the British Pharmaceutical Codex as a sedative for irritable cough and as a mild hypnotic for insomnia [166,167,168]. Lactucopicrin (9) and lactucin (7) were identified as its major active compounds, although compound 7 was suggested to be one of the main metabolites related to the sedative and the sleep-promoting effects [163]. In an in vivo study in mice, both substances were confirmed to be analgesics, with an activity equal to or greater than ibuprofen. In addition, when lactucin (7) and lactucopicrin (9) were administered to mice on the hot plate, both compounds were revealed to have analgesic and sedative effects at concentrations of 15 and 30 mg/kg, respectively. Furthermore, in a spontaneous locomotor activity test, these compounds were active at concentrations of 30 mg/kg [168].
Although the pure compounds currently are not easily available on the market, several lettuce extracts and seed oils containing these sesquiterpene lactones as main active compounds are commercialized, e.g., Sedan® (Pharco Pharmaceutical Company, Egypt). The sedative effects were addressed in the pilot study by Yakoot et al. [169]. The authors investigated if lettuce seed oil was efficient and safe to treat patients with sleep disorders. The results showed that the seed oil of L. sativa was a potentially hazard-free agent, able to reduce sleeping difficulties and alleviate mild to moderate forms of anxiety in geriatric patients, without showing side effects [169,170]. Additionally, the study performed by Kim et al. [163] examined the sleep-inducing and sleep-prolonging effect of four lettuce varieties. The seed and leaf extracts were evaluated using a four-week-old ICR mouse model to analyze their effects on pentobarbital-induced sleep. The results showed that both extract types lengthened sleep duration and significantly reduced sleep latency at both evaluated doses of 80 mg/kg and of 160 mg/kg [163]. Although these studies in patients were performed with plant extracts reported to contain lactucin (7) and its derivatives as principal active pharmacological compounds, they confirm that their application for treatment of anxiety and sleep disorders is worthy of further evaluation. Additionally, the results showed that these sesquiterpenes might be assessed as potential alternatives to currently used sleep-promoting, sedative, and anxiolytic agents, with their varied negative side-effects and addiction-potential.
2.8. Parthenolide
Parthenolide (10) (Figure 9) has the same chemical formula as inuviscolide (6) (C15H20O3) and is the best known sesquiterpene lactone in the germacranolide class. It was isolated for the first time from feverfew leaves and flowerheads (Tanacetum parthenium (L.) (Sch.Bip.), a plant known in traditional Chinese medicine for centuries to treat various ailments. Among them, it is used to relieve fever, pain of different etiologies such as migraine and rheumatoid arthritis and even to treat insect bites [171].
Compound 10 has been applied as an anti-inflammatory through specific inhibition of the signal proteins IKK2, STAT3, and MAPK, along with the activity and expression of many inflammatory mediators including COX, which is involved in the NF-κB mediated proinflammatory signal transduction pathway [172]. It also exhibits antitumoral activity by proliferation inhibition in various cancer cell types, including prostate, pancreas, cervical, breast, lung, colorectal, glioblastoma, multiple myeloma, and leukemia [8,173]. There are numerous reviews describing the outstanding anti-inflammatory and antitumor activities of parthenolide, its analogs, and derivatives upon different pathways in human cancer cells [8,174,175,176,177,178].
Parthenolide (10) has a multitarget action mechanism. It triggers EGF receptor phosphorylation, interferes with AP-1 [179] and the signal transducer and activator of transcription 3 (STAT3) [180] and induces activation of c-Jun N-terminal kinase (JNK) [181]. Furthermore, the molecular mechanisms of parthenolide action are strongly associated with DNA-binding inhibition of two transcription factors NF-κB [182,183], as well as the proapoptotic activation of p53, along with reduced glutathione (GSH) depletion [184]. Parthenolide rapidly induces ROS generation [185,186], and lowers histone deacetylase activity (HDAC) [187] and DNA methyltranspherase 1 (DNMT1) [188]. Additionally, parthenolide can interfere with microtubule function through tubulin binding [189].
Recent advances regarding in vivo therapeutic applications of parthenolide (10), mainly focused but not limited to anticancer and anti-inflammatory activities, are discussed below, together with the synergistic effects and toxicity of compound 10 and some derivatives.
One of the advantages of parthenolide (10) is its cancer stem-cell selectivity while remaining non-cytotoxic to non-tumor cells [190,191]. In this case, the molecular mechanism of parthenolide involves induction of apoptosis through mitochondrial and caspase signaling pathways, and also an increase in the cytosolic concentration of calcium, cell cycle arrest, and inhibition of metastasis [178,192].
Cholangiocarcinoma (CC) is an intrahepatic bile duct carcinoma with a poor prognosis due to being chemoresistant. Parthenolide induced oxidative stress-mediated apoptosis in CC cells and the Bcl-2-related family of proteins modulated that susceptibility [193]. Moreover, intraperitoneal injection of parthenolide at 4 mg/kg caused significant inhibition of tumor growth and angiogenesis in the xenograft model [194]. Recently, Yun et al. [195] demonstrated that low concentrations of parthenolide (5–10 µM) suppressed HO-1 expression, enhancing oxidative stress by the PKC-α inhibitor Ro317549 (Ro) through inhibition of Nrf2 expression and its nuclear translocation. The effects of parthenolide (10) and Ro at 2.5 mg/kg on tumor growth were tested using a xenograft nude mouse tumor model with subcutaneously implanted ChoiCK and SCK cells. This assay indicated that their combined application more effectively inhibited cancer cell growth inhibition as compared to treatment with either compound by itself. Furthermore, the effect of parthenolide on the development of colitis-associated colon cancer (CAC) was investigated using a murine model of azoxymethane (AOM)/dextran sulfate sodium (DSS) induced CAC. This study showed that parthenolide administration (10) at 2 and 4 mg/kg can significantly inhibit the inflammation-carcinoma sequence and be crucial in experimental CAC regulation. The mechanism of action involves decreased NF-κB p65 expression levels blocking phosphorylation and subsequent degradation of κB-α inhibitor (IκBα) [196]. The authors conclude that parthenolide (10) could be a novel chemopreventive agent for CAC treatment [196].
Oral cancer is one of the five most common cancers worldwide. Chemoprevention is a new approach to cancer research, focusing more on the prevention, suppression, and reversal of the carcinogenic process by the use of natural plant products and/or synthetic chemical compounds. Thus, Baskaran et al. [197] tested the chemopreventive potential of parthenolide in DMBA-induced hamster buccal pouch carcinogenesis (DMBA, 7,12-dimethylbenz[a]anthracene). Oral administration of parthenolide (10) at 2 mg/kg b.w. completely prevented tumor formation and significantly reduced the nefarious histopathological changes. In addition, the parthenolide treated group showed significant improvement regarding antioxidants, detoxification enzymes and lipid peroxidation.
Glioblastoma, or glioblastoma multiforme (GBM), is the most aggressive type of brain cancer and is very difficult to treat. Nakabayashi and Shimizu [198] examined the effect of compound 10 on tumor growth using a xenograft mouse model of glioblastoma, administering it intraperitoneally (10 mg/kg/day) for 22 days. It significantly inhibited the growth of transplanted glioblastoma cells with respect to the control group.
Zhang et al. [199] demonstrated that a high parthenolide dose (8 mg/kg/day) impedes initiation of experimental autoimmune neuritis (EAN), an animal model for peripheral nervous system acute inflammatory disease. This is achieved by parthenolide suppressing TNF-α, IFN-γ, IL-1β and IL-17 pathways and quickly decreasing Th1 and Th17 cells in the early stages. Although this anti-inflammatory effect is short-lived, compound 10 also suppresses late-stage recovery of EAN models, along with inhibiting the apoptosis of inflammatory cells. Such results suggest that parthenolide is not suitable for nervous system autoimmune disease treatment.
Nitric oxide (NO) plays a key role in the etiopathology of central nervous system (CNS) diseases like multiple sclerosis (MS). It has been proposed that inhibition of NO synthesis could prove a relevant mechanism of action in treating multiple sclerosis and migraine. Accordingly, Fiebich et al. [200] investigated the effect of parthenolide (10) on iNOS synthesis and NO release using primary rat microglia. The results indicated that compound 10 prevents iNOS/NO synthesis and inhibits the activation of p42/44 mitogen-activated protein kinase (MAPK), but not IkBα degradation or NF-kB p65 activation. These results show parthenolide may be a potential therapeutic agent in the treatment of CNS diseases.
Mechanisms of axon regeneration and optimal functional recovery after nerve injury are key to in higher animals. However, insufficient growth rates of injured axons often lead to incomplete peripheral nerve regeneration. Gobrecht et al. [201], demonstrated that a single parthenolide injection at 5 nM into the injured sciatic nerve or its systemic intraperitoneal application was already enough to significantly increase the number and length of regenerating axons in the distal nerve at three days post-lesion. This application of parthenolide (10) appears to act on the great instability of microtubules in promoting axonal growth, at least in the CNS [174]. For this reason, the efficacy of parthenolide is very promising for a therapeutic promotion of nerve regeneration, since compound application and recurrent treatments are facilitated, compared to invasive local nerve injections [202].
Pulmonary fibrosis (PF) in general and idiopathic pulmonary fibrosis (IPF) in particular, is a disease for which there is no effective therapy. In vivo studies have shown that parthenolide (10), via intragastric administration, inhibited the NF-κB/Snail pathway, attenuating bleomycin-induced pulmonary fibrosis. Moreover, there were significant improvements in body weight and other pathological changes associated with this disease [203].
NF-κB has been associated with the cardiovascular system; in fact, its function is related to the protection of cardiovascular tissues against injuries. However, its activation can also contribute to tissue pathogenesis, depending on the type of cells in which it is activated [204]. It is known that myocardial infarct size could be reduced up to 60% by antagonizing NF-κB activity [205]. To achieve this, parthenolide (10) at 250 or 500 mg/kg (b.w.) was administered intraperitoneally before reperfusion in rats, and caused a significant improvement in myocardial injury, with a reduction in the oxidative and inflammation state, consequently reducing infarct size [206]. However, Tsai et al. [207] reported that a prolonged treatment in bEND.3 cells affected Ca2þ signaling in the endothelial cells that regulate vascular tone; therefore, care should be taken on using this compound in experimental designs and clinically.
Parthenolide (10) has relatively poor pharmacological properties, derived from its low solubility in water and consequently reduced bioavailability, which limit its potential clinical use as anticancer drug. To increase its solubility, a series of parthenolide derivatives were obtained by diastereoselective addition of several primary and secondary amines to the exocyclic double bond [208,209]. N,N-Dimethylaminoparthenolide (DMAPT) (11) (Figure 9) was selected as a leader compound according to its pharmacokinetic, pharmacodynamic and bioavailability properties [209,210]. When formulated as a fumarate salt, DMPAPT is 1000-fold more soluble in water than parthenolide and maintained the anticancer activity because, DMAPT (11) is rapidly converted back to parthenolide in body fluids (10). Recently, molecular studies indicate that DMAPT has a similar action to parthenolide [192,211,212,213].
DMAPT has approximately 70% oral bioavailability and induces rapid death of primary human leukemia stem cells (LSCs) from both myeloid and lymphoid leukemias and is highly cytotoxic to bulk leukemic cell populations. Pharmacological studies carried out by Guzman et al. [210] using both mouse xenograft models and spontaneous acute canine leukemias demonstrate in vivo bioactivity. Indeed, DMAPT eliminates human AML stem and progenitor cells without harm to normal hematopoietic stem and progenitor cells, and eradicates phenotypically primitive blast-crisis chronic myeloid leukemia (bcCML) and acute lymphoblastic leukemia (ALL) cells. Moreover, it inhibited metastasis in a mouse xenograft model of breast cancer and enhanced the survival of treated mice [210].
DMAPT was assayed in a phase I trial against acute myeloid leukemia (AML), acute lymphoblastic leukemia (ALL) and other blood and lymph node cancers [12,176]. However, a year later the clinical trials were suspended [214].
Radiotherapy is widely used in cancer treatment; however, the benefits can be reduced by radiation-induced damage to neighboring healthy tissues. Morel et al. [215] demonstrated in mice that DMAPT (11) selectively induces radio-sensitivity in prostate cancer cell-lines, while protecting primary prostate epithelial cell lines from radiation-induced damage. Compound 11 has the advantage of being well-tolerated orally without the need to adjust the administration time to radiation exposure. Radiation-induced lung fibrosis is considered a critical determinant for late normal tissue complications. Therefore, the same group [216] examined the radioprotective effect of DMAPT (11) on fibrosis in normal tissues, according to the image-guided fractionated radiotherapy protocols used clinically. The results obtained show that DMAPT reduced radiation-induced fibrosis in the corpus cavernosum of the rat penis (98.1%) and in the muscle layer around the bladder (80.1%), and also the tendency towards reduced collagen infiltration into the submucosal and muscle layers in the rectum. They concluded that DMAPT could be useful in providing protection from the radiation-induced side effects such as impotence and infertility, urinary incontinence and fecal urgency resulting from prostate cancer radiotherapy [216].
On the other hand, radiation-resistant prostate cancer cells often overexpress the transcription factor NFκB. Mendonca et al. [217] suggest that DMAPT might have a potential clinical role as radio-sensitizing agent in prostate cancer treatment. This conclusion is based on the finding that combined treatment of PC-3 prostate tumor xenografts with oral DMAPT and radiation therapy significantly reduced tumor growth, when compared to those treated with either DMAPT or radiation therapy alone.
Li et al. [218], obtained a novel parthenolide derivative, HMPPPT, a 13-substituted derivative ((3R,3aS,9aR,10aR,10bS,E)-3-((4-(6-hydroxy-2-methylpyrimidin-4-yl)piperidin-1-yl)methyl)-6,9a-dimethyl-3a,4,5,8,9,9a,10a,10b-octahydrooxireno [2′,3′:9,10]cyclodeca[1,2-b]furan-2(3H)-one) (12) (Figure 9), with better bioavailability and pharmacological properties than DMAPT. In vitro studies pointed to compound 12 as the most promising derivative, from safety profile and ADME property standpoints. The newly identified compound was shown to have pro-oxidant activity and in silico molecular docking studies with components of the NF-κB pathway also supporting a pro-drug mode of action. This mechanism included release of parthenolide and covalent interaction with one or more proteins involved in that pathway [218]. The in vivo oral bioavailability study of compound 12 in murine PK at 10 mg/kg indicated that it has advantageous pharmacological properties and therefore can be considered an agent to be considered in therapy against drug resistant chronic lymphocytic leukemia [218].
In the last few years, nanotechnology has provided many selective strategies for the detection and treatment of cancer, overcoming the problems associated with conservative cancer diagnosis and therapy. In this context, the development and testing of parthenolide (10) nanoencapsulation and derivatives is a way to enhance its potential to provide effective pharmaceutical products for clinical use and resolve drawbacks such as low bioavailability [219,220,221,222]. Accordingly, several patents for elaboration of parthenolide and its derivatives in nanocarriers, and various pharmaceutical preparations combined with other products, have been recently registered in China: patents N° CN 110292640, 2019; N° CN108721276, 2018; N° CN1087211330, 2018; N° CN106366068, 2017; N° CN 109276553, 2017.
In recent years, parthenolide has been suggested for use in combination therapy with other anticancer agents, to overcome obstacles in the treatment of cancer, such as a) different types of cancer cells, b) resistance to chemotherapy, and c) drug toxicity to normal cells.
TRAIL (tumor necrosis factor (TNF)-related apoptosis inducing ligand) is now being developed as a promising natural immunity-stimulating molecule for clinical trials in cancer patients. However, various malignant tumors are currently resistant. Kim et al. [223] investigated how parthenolide (10) sensitizes colorectal cancer (CRC) cells to TRAIL-induced apoptosis. For this, HT-29 (TRAIL-resistant) and HCT116 (TRAIL-sensitive) cells were treated with compound 10 and/or TRAIL. The results revealed that parthenolide (10) increases induced apoptosis and upregulates DR5 protein level and surface expression in both cell lines, suggesting that combined therapy with TRAIL is a good strategy to overcome the resistance of certain CRC cells.
Pancreatic cancer is a common malignancy with high occurrence worldwide, and a poor survival rate. Recent research indicates that combination therapy with DMAPT (11) can enhance the antiproliferative effects of gemcitabine in pancreatic cancer cells in vitro and in vivo [224]. Yip-Schneider et al. [225] showed that celecoxib (a cyclooxygenase 2 (COX-2) inhibitor) at 50 mg/kg/day combined with DMPTA at 40 mg/kg/da has a significant inhibitory effect on tumor invasion of adjacent organs and metastasis in pancreatic cancer induced in Syrian golden hamsters. It reduced NFκB activity, and expression of prostaglandin E2 and its metabolite. They also demonstrated that compound 11 (40 mg/kg/day) in combination with sulindac (20 mg/kg/day) and gemcitabine (50 mg/kg twice weekly) can delay or prevent progression of premalignant pancreatic lesions) in a genetically engineered mouse model of pancreatic cancer [226]. Likewise, they demonstrated that DMAPT (11) (40 mg/kg/day) with gemcitabine (50 mg/kg/day) considerably improved average survival, lowering the frequency and multiplicity of pancreatic adenocarcinomas. This combination also significantly reduced tumor size and the incidence of metastasis into the liver [227].
Parthenolide exerted a cytotoxic effect on MDA-MB231 cells, a triple-negative breast cancer (TNBC) cell-line, however its success is scarce at low doses. In order to overcome this difficulty, Carlisi et al. [228] tested a histone deacetylase inhibitor, SAHA (suberoylanilide hydroxamic acid), which synergistically sensitized MDA-MB231 cells to the cytotoxic effect of parthenolide (10). It is noteworthy that treatment with parthenolide alone increased the survival of cell pathway Akt/mTOR and the consequent nuclear translocation of Nrf2, a protein regulating the expression of antioxidant proteins that protect against the oxidative damage triggered by injury and inflammation, while treatment with SAHA alone activated autophagy.
A phase 2 clinical trial indicated that actinomycin-D (ActD), a polypeptide antibiotic that intercalates to DNA and inhibits mRNA transcription in mammalian cells, could be a potent drug against pancreatic cancer. However, it is not a good candidate due to toxicity issues. Thus, given the modes of action of DMPTA and Actinomycin-D (ActD), Lamture et al. [229] postulated that combining both drugs would result in synergistic inhibition of Panc-1 pancreatic cancer cell growth, since the inhibitory activity of DMAPT on FkB would enhance apoptosis induction by ActD, through phosphorylation of c-Jun. Indeed, the combination of these two drugs produced a higher cell-death percentage than each drug alone.
2.9. Thapsigargin
In 1978, the structure of thapsigargin (13) (C34H50O12) (Figure 10) was elucidated [230] for the first time. It was found as the main component of Thapsia garganica L. [230,231], an umbelliferous Apiaceae species distributed in the Mediterranean area. This plant has long been used in folk medicine for treating common lung diseases (acute bronchitis and pneumonia) and dental pains [232]. The resin from Thapsia garganica root was described in the French Pharmacopoeia [232].
Thapsigargin (13) yield ranged between 0.2–4.91% of the dry weight of leaves and roots of Thapsia garganica and depends on the plant tissue, collection site (locality) and extraction methods [231,233]. For example, classical maceration was more efficient than other methods such as microwave-assisted extraction or simple extraction with liquid nitrogen [233].
Due to the importance of thapsigargin’s biological activities, there is great interest in its synthetic and semi-synthetic preparation, thus several studies have been published on this subject [234,235].
Thapsigargin (13) is an inhibitor of the sarco/endoplasmic reticulum (ER) calcium ATPase (SERCA) pump. The blockage of the SERCA pump results in malfunction of cellular calcium homeostasis and exerts a critical role in normal cell metabolism, leading to apoptosis [236,237]. Moreover, this sesquiterpene lactone causes apoptosis at all stages of the cell cycle.
Compound 13 strongly inhibited all the subtypes of SERCA. The inhibitory constants (Ki) of thapsigargin (13) were 0.21, 1.3, and 12 nm for SERCA1b, SERCA2b, and SERCA3a, respectively [238,239]. Its affinity with the SERCA1a pump is significantly reduced by removal of the acyl groups at O-3, O-8 and O-10 [240].
Thapsigargin (13) possesses interesting pharmacological properties; for instance, it induces expression of the L-histidine decarboxylase enzyme responsible for converting L-histidine to histamine in cells [241]. It also evokes ROS generation in cells through calcium-ion mediated changes (mitochondrial depolarization), which can result in cell dysfunction and damage [242,243]. In addition, compound 13 is known for its cytotoxic action against different cancer cell-lines, for instance melanoma [244], insulinoma [245,246], neuronal [247], and breast [248].
The thapsigargin LD100 value is 0.8 mg/kg in mice [249]. Zhong et al. [250] proposed that thapsigargin causes vomiting by triggering the phosphorylation of CaMKIIα (Ca2+/calmodulin kinase IIα) and ERK1/2 (extracellular signal-regulated protein kinase 1/2) cascade in the brainstem. Pharmacological preconditioning with the cell-stress inducer thapsigargin (0.3 mg/kg) protects against experimental sepsis in male KM (Kunming) mice [237].
Thapsigargin (13) is widely used as an experimental tool for endoplasmic reticulum stress inhibition and the discovery of new active therapeutic derivatives [232,251]. A new thapsigargin (13) derivative named mipsagargin, (8-O-(12-aminododecanoyl)-8-O-debutanoyl thapsigargin)-Asp-γ-Glu-γ-Glu-γ-GluGluOH (14) (Figure 10), has been developed for anticancer therapy, notably for the treatment of prostate cancer. Mipsagargin (14) has the ability to link the C-8 with prostate-specific membrane antigen (PSMA) peptide. It is a brand new example of a thapsigargin prodrug, which is currently undergoing preclinical evaluation as a targeted chemotherapeutic agent with selective toxicity against cancer cells [232,249]. Mahalingam and coauthors reported that mipsagargin (14) has an acceptable pharmacokinetic profile in patients with solid tumors [252]. It is relatively well tolerated, promoting prolonged disease stabilization in patients with advanced liver cancer [253].
2.10. Tomentosin
Tomentosin (15) (Figure 11), known also as xanthalongin, is a xanthanolide sesquiterpene lactone with the same chemical formula as inuviscolide (6) and parthenolide (10) (C15H20O3). It has been isolated from plants such as Dittrichia viscosa (L.) Greuter, Carpesium faberi C. Winkl. and Carpesium macrocephalum Franch. and Sav. [254], Leucophyta brownii Cass. [255], the sunflower (Helianthus annuus L.) [256] and Inula species [254,257]. Inula viscosa was widely used as a medicinal plant. A steam distilled extract of its leaves and flowers yielding 2% of tomentosin (15) [258] exhibited anti-inflammatory, antibacterial, and anticancer activities [257].
The biological activities of tomentosin (15) have been addressed in several studies focused on anticancer, antifungal, and insecticidal activities.
Anticancer activity observed in vitro tests was confirmed by in vitro and in vivo studies performed by Bar-Shalom et al. [259]. The authors found that application of an aqueous extract of Inula viscosa leaves at a concentration of 300 µg/mL to colorectal cancer cells induced apoptosis, through activation of caspases. During the subsequent in vivo study, mice transplanted with MC38 cells were treated intraperitoneally with the extract at concentrations of 150 and 300 mg/kg. The results indicated that tumor weight and volume were reduced significantly by this treatment after comparison with the untreated control group. Furthermore, staining the paraffin section of the tumors showed inhibition of cell proliferation, as well as induction of apoptosis. Interestingly, side effects, which often include weight loss, impact on fur, and changes in behavior or kidney or liver functions, were not observed. This suggests that the extract was non-toxic at the concentrations applied. However, this remains to be confirmed [259].
Fungal infection of grapevines causes severe economic damage to grape producers. As several fungicide-resistant strains of the pathogens have developed, efective fungicide application is becoming more and more difficult. Therefore, natural products such as sesquiterpene lactones and plant extracts may provide alternatives to current synthetic fungicides. The antifungal activity of six Inula viscosa leaf extracts on downy mildew under field conditions was addressed in the study performed by Cohen et al. [260]. This mildew is caused by the fungus Plasmopara viticola (Berk. and Curt.). The results established that the effective concentration of oily paste extract in water required to control 90% of the treated shoots in the field was lower than 0.125%. For whole vines, the required concentration ranged between 0.30% and 0.37%. Additionally, the results did not appear to depend on seasonal fluctuations. The evaluated leaf extracts contained 10.6% of tomentosin (15) and 10.6% of costic acid, which were identified as the active principles with antifungal activity. The overall results indicated that Inula viscosa extracts can be of great value as an alternative source for non-synthetic fungicidal preparations to treat downy mildew infestations of grapes [260].
In an in vivo investigation performed by Ahern et al. [261] on insecticidal activity, the effect of the stereochemistry of the lactone ring on feeding behavior of the herbivorous polyphagous grasshopper Schistocerca americana (Drury) was addressed. Schistocerca americana is locally related in the south of the U.S. to significant damage to crops when temporary mass populations occur. To evaluate the antifeedant activity of diastereomeric sesquiterpene lactones, the study focused on tomentosin (15) with a cis-configuration and the corresponding isomer xanthinosin with a trans-configuration. The results showed both compounds were able to reduce plant consumption by the insects. However, the trans-fused compounds were consumed less than the corresponding cis-fused compounds. The results of this study not only permitted conclusions on the importance of the stereochemistry-dependence of diastereomeric sesquiterpene lactones for insecticidal activity, but also might help to understand the geographical distribution and evolution of different clades among the Asteraceae family. The distribution of cis- and trans-fused compounds within plants of this family is still not fully understood, in this study, 12.5% of the sampled Asteraceae genera contained only cis-fused sesquiterpene lactones, 64% only trans-fused sesquiterpene lactones, and 23% both types [261].
3. Conclusions
Taking into account the research described above, the sesquiterpene lactones dealt with in this review have proved to be very useful compounds with promising anticancer and anti-inflammatory bioactivities in particular. For instance, alantolactone (1) was shown in vivo to be able to reduce tumor size by over 50%, while increasing life expectancy and reducing pathological indicators across several different cancer types. In addition, synergistic effects with well-known cancer therapeutics were also reported. Arglabin (2) in salt form could be used to treat several cancers and possesses valuable pharmacokinetic characteristics highly sought for in therapeutic drugs. It has no adverse effects described in the literature. Antitrypanosomal, antitumor and anti-inflammatory activities are reported for cynaropicrin (4). Another compound, helenalin (5), inhibits essential factors in both cancer and inflammation, i.e., the NF-κB pathway and the transcription of inflammatory cytokines. Inuviscolide (6) demonstrates potential as an anti-inflammatory therapeutic compound, being able to reduce rat ear and paw edema and LTB4 generation in intact cells. Lactucin (7) and derivatives lactupicrin (8) and lactucopicrin (9) have a strong potential as phytopharmaceutical agents for treatment of anxiety and sleep disorders, being the main active components of long commercialized substances, e.g., Sedan®, without observing side-effects.
Of all the sesquiterpene lactones reviewed in this work, parthenolide (10) and its derivatives DMAPT (11) and HMPPPT (12) are those most mentioned in the literature, with several studies particularly reporting their excellent anti-inflammatory and antitumor activities. Some work reveals synergistic effects of these compounds with current anticancer drugs, favoring more potent and selective antitumor action. Parthenolide derivatives have been designed and DMAPT (11) is 1000-fold more soluble in water than the natural product, while maintaining a similar mechanism of action with improved anticancer and anti-inflammatory activities.
Thapsigargin (13) has the particular capacity to cause endoplasmic reticulum stress and thus cell apoptosis, an interesting pharmacological property to be retained or potentiated in derivatives aimed at anticancer therapy. An example is mipsagargin (14), which is currently undergoing preclinical evaluation for selective toxicity against cancer cells in tumor sites, with minimal side-effects for the host. Another active compound is tomentosin (15), additionally with potential applications in cancer but also as antifungal and antifeedant.
In summary, given the activities described in this review these sesquiterpene lactones exhibit properties that justify more research by the scientific community, to drive preclinical and clinical studies leading to development of new drugs.
Author Contributions
Conceptualization, A.M.L.S. and L.M.; writing—original draft preparation, A.M.L.S., L.M., O.C., P.M.C.S., F.S.; writing—review and editing, L.M., O.C., A.M.L.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by FCT–Fundação para a Ciência e a Tecnologia, the European Union, QREN, FEDER, COMPETE, by funding the cE3c centre (UIDB/00329/2020), the LAQV-REQUIMTE (UIDB/50006/2020) and QOPNA (UID/QUI/00062/2019) research units, and by the Spanish Ministry Science and Research (MINECO RTI2018-094356-B-C21).
Acknowledgments
Thanks are due to the University of Azores and University of La Laguna. AbbVie participated in the revision of the manuscript.
Conflicts of Interest
O.C. is an employee of AbbVie and owns AbbVie stock. The authors L.M., P.M.C.S., F.S., and A.M.L.S. declare they have no conflict of interest.
References
- Li, Q.; Wang, Z.; Xie, Y.; Hu, H. Antitumor activity and mechanism of costunolide and dehydrocostus lactone: Two natural sesquiterpene lactones from the Asteraceae family. Biomed. Pharmacother. 2020, 125, 109955. [Google Scholar] [CrossRef] [PubMed]
- Stavrianidi, A. A classification of liquid chromatography mass spectrometry techniques for evaluation of chemical composition and quality control of traditional medicines. J. Chromatog. A 2020, 1609, 460501. [Google Scholar] [CrossRef] [PubMed]
- Gou, J.; Hao, F.; Huang, C.; Kwon, M.; Chen, F.; Li, C.; Liu, C.; Ro, D.-K.; Tang, H.; Zhang, Y. Discovery of a non-stereoselective cytochrome P450 catalyzing either 8α- or 8β-hydroxylation of germacrene A acid from the Chinese medicinal plant, Inula hupehensis. Plant J. 2018, 93, 92–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perassolo, M.; Cardillo, A.B.; Busto, V.D.; Giulietti, A.M.; Talou, J.R. Biosynthesis of sesquiterpene lactones in plants and metabolic engineering for their biotechnological production. In Sesquiterpene Lactones: Advances in Their Chemistry and Biological Aspects; Sülsen, V.P., Martino, V.S., Eds.; Springer International Publishing: Cham, Switzerland, 2018; pp. 47–91. [Google Scholar] [CrossRef]
- Sülsen, V.P.; Martino, V.S. Overview. In Sesquiterpene Lactones: Advances in Their Chemistry and Biological Aspects; Sülsen, V.P., Martino, V.S., Eds.; Springer International Publishing: Cham, Switzerland, 2018; pp. 3–17. [Google Scholar] [CrossRef]
- Choodej, S.; Pudhom, K.; Yamauchi, K.; Mitsunaga, T. Inhibition of melanin production by sesquiterpene lactones from Saussurea lappa and their analogues. Med. Chem. Res. 2019, 28, 857–862. [Google Scholar] [CrossRef]
- Seca, A.M.L.; Grigore, A.; Pinto, D.C.G.A.; Silva, A.M.S. The genus Inula and their metabolites: From ethnopharmacological to medicinal uses. J. Ethnopharmacol. 2014, 154, 286–310. [Google Scholar] [CrossRef] [Green Version]
- Bosco, A.; Golsteyn, R.M. Emerging anti-mitotic activities and other bioactivities of sesquiterpene compounds upon human cells. Molecules 2017, 22, 459. [Google Scholar] [CrossRef] [Green Version]
- Beer, M.F.; Bivona, A.E.; Sánchez Alberti, A.; Cerny, N.; Reta, G.F.; Martín, V.S.; Padrón, J.M.; Malchiodi, E.L.; Sülsen, V.P.; Donadel, O.J. Preparation of sesquiterpene lactone derivatives: Cytotoxic activity and selectivity of action. Molecules 2019, 24, 1113. [Google Scholar] [CrossRef] [Green Version]
- Sokovic, M.; Ciric, A.; Glamoclija, J.; Skaltsa, H. Biological activities of sesquiterpene lactones isolated from the genus Centaurea L. (Asteraceae). Curr. Pharm. Des. 2017, 23, 2767–2786. [Google Scholar] [CrossRef]
- Ma, C.; Meng, C.-W.; Zhou, Q.-M.; Peng, C.; Liu, F.; Zhang, J.-W.; Zhou, F.; Xiong, L. New sesquiterpenoids from the stems of Dendrobium nobile and their neuroprotective activities. Fitoterapia 2019, 138, 104351. [Google Scholar] [CrossRef]
- Ghantous, A.; Gali-Muhtasib, H.; Vuorela, H.; Saliba, N.A.; Darwiche, N. What made sesquiterpene lactones reach cancer clinical trials? Drug Discov. Today 2010, 15, 668–678. [Google Scholar] [CrossRef]
- Cheriti, A.; Belboukhari, N. Terpenoids of the Saharan medicinal plants Launaea Cass. genus (Asteraceae) and their biological activities. In Terpenoids and Squalene: Biosynthesis, Functions and Health Implications; Bates, A.R., Ed.; Nova Science Publishers, Inc.: New York, NY, USA, 2015; pp. 51–70. ISBN 978-1-63463-656-8. [Google Scholar]
- Wang, J.; Su, S.; Zhang, S.; Zhai, S.; Sheng, R.; Wu, W.; Guo, R. Structure-activity relationship and synthetic methodologies of α-santonin derivatives with diverse bioactivities: A mini-review. Eur. J. Med. Chem. 2019, 175, 215–233. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, T.J. Structure-activity and activity-activity relationships of sesquiterpene lactones. In Sesquiterpene Lactones: Advances in Their Chemistry and Biological Aspects; Sülsen, V., Martino, V., Eds.; Springer International Publishing AG: Cham, Switzerland, 2018; pp. 349–371. [Google Scholar] [CrossRef]
- Wang, G.-W.; Qin, J.-J.; Cheng, X.-R.; Shen, Y.-H.; Shan, L.; Jin, H.-Z.; Zhang, W.-D. Inula sesquiterpenoids: Structural diversity, cytotoxicity and anti-tumor activity. Expert Opin. Investig. Drugs 2014, 23, 317–345. [Google Scholar] [CrossRef] [PubMed]
- Reddy, D.M.; Qazi, N.A.; Sawant, S.D.; Bandey, A.H.; Srinivas, J.; Shankar, M.; Singh, S.K.; Verma, M.; Chashoo, G.; Saxena, A.; et al. Design and synthesis of spiro derivatives of parthenin as novel anti-cancer agents. Eur. J. Med. Chem. 2011, 46, 3210–3217. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Cao, J.; Huang, G.; Zhao, Q.; Shen, J. Biological activities of artemisinin derivatives beyond malaria. Curr. Top. Med. Chem. 2019, 19, 205–222. [Google Scholar] [CrossRef]
- Zhang, C.; Zhu, Y.; Yin, X.-P.; Wei, Q.-H.; Zhang, N.-N.; Li, C.-X.; Xie, T.; Chen, R. [Advances in synthesis of artemisinin based on plant genetic engineering]. Zhongguo Zhong Yao Za Zhi 2019, 44, 4285–4292. [Google Scholar] [CrossRef]
- Gao, F.; Sun, Z.; Kong, F.; Xiao, J. Artemisinin-derived hybrids and their anticancer activity. Eur. J. Med. Chem. 2020, 188, 112044. [Google Scholar] [CrossRef]
- Xu, R.; Peng, Y.; Wang, M.; Li, X. Intestinal absorption of isoalantolactone and alantolactone, two sesquiterpene lactones from radix inulae, using Caco-2 cells. Eur. J. Drug Metab. Pharmacokinet. 2019, 44, 295–303. [Google Scholar] [CrossRef]
- Circioban, D.; Pavel, I.Z.; Ledeti, A.; Ledeti, I.; Danciu, C.; Dehelean, C. Cytotoxic activity evaluation on breast cells of guest-host complexes containing artemisinin. Rev. Chim. 2019, 70, 2843–2846. [Google Scholar] [CrossRef]
- Shakeri, A.; Amini, E.; Asili, J.; Masullo, M.; Piacente, S.; Iranshahi, M. Screening of several biological activities induced by different sesquiterpene lactones isolated from Centaurea behen L. and Rhaponticum repens (L.) Hidalgo. Nat. Prod. Res. 2018, 32, 1436–1440. [Google Scholar] [CrossRef]
- Seca, A.M.L.; Pinto, D.C.G.A.; Silva, A.M.S. Metabolomic profile of the genus Inula. Chem. Biodivers. 2015, 12, 859–906. [Google Scholar] [CrossRef]
- Lim, H.S.; Jin, S.E.; Kim, O.S.; Shin, H.K.; Jeong, S.J. Alantolactone from Saussurea lappa exerts antiinflammatory effects by inhibiting chemokine production and STAT1 phosphorylation in TNF-α and IFN-γ-induced in HaCaT cells. Phytother. Res. 2015, 29, 1088–1096. [Google Scholar] [CrossRef] [PubMed]
- Chun, J.; Choi, R.J.; Khan, S.; Lee, D.-S.; Kim, Y.-C.; Nam, Y.-J.; Lee, D.-U.; Kim, Y.S. Alantolactone suppresses inducible nitric oxide synthase and cyclooxygenase-2 expression by down-regulating NF-κB, MAPK and AP-1 via the MyD88 signaling pathway in LPS-activated RAW 264.7 cells. Int. Immunopharmacol. 2012, 14, 375–383. [Google Scholar] [CrossRef] [PubMed]
- Marshall, J.A.; Cohen, N. The stereoselective total synthesis of alantolactone. J. Am. Chem. Soc. 1965, 87, 2773–2774. [Google Scholar] [CrossRef]
- Schultz, A.G.; Godfrey, J.D. An annelation approach to the synthesis of eudesmane and elemane sesquiterpene lactones. Total synthesis of dl-dihydrocallitrisin, dl-7,8-epialantolactone, dl-7,8-epiisoalantolactone, and dl-atractylon. J. Am. Chem. Soc. 1980, 102, 2414–2428. [Google Scholar] [CrossRef]
- Trendafilova, A.; Chanev, C.; Todorova, M. Ultrasound-assisted extraction of alantolactone and isoalantolactone from Inula helenium roots. Pharmacogn. Mag. 2010, 6, 234–237. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.M.; Wang, J.; Liu, H.B.; Guo, C.Y.; Zhang, W.M. Microwave-assisted extraction of alantolactone and isoalantolactone from Inula helenium. Indian J. Pharm. Sci. 2015, 77, 116–120. [Google Scholar] [CrossRef] [Green Version]
- Chi, X.-F.; Yue, H.-L.; Zhao, X.-H.; Hu, F.-Z. Obtaining alantolactone and isoalantolactone from Inula racemose Hook.f. by optimized supercritical fluid extraction. Ind. Crops Prod. 2016, 79, 63–69. [Google Scholar] [CrossRef]
- Nikonova, L.P. Lactones of Inula magnifica. Chem. Nat. Comp. 1973, 9, 528. [Google Scholar] [CrossRef] [Green Version]
- Stampf, J.L.; Benezra, C.; Klecak, G.; Geleick, H.; Schulz, K.H.; Hausen, B. The sensitizing capacity of helenin and of two of its main constituents, the sesquiterpene lactones alantolactone and isoalantolactone: A comparison of epicutaneous and intradermal sensitizing methods in different strains of guinea pig. Contact Derm. 1982, 8, 16–24. [Google Scholar] [CrossRef]
- Kuno, Y.; Kawabe, Y.; Sakakibara, S. Allergic contact dermatitis associated with photosensitivity, from alantolactone in a chrysanthemum farmer. Contact Derm. 1999, 40, 224–225. [Google Scholar] [CrossRef]
- Marc, E.B.; Nelly, A.; Annick, D.-D.; Frederic, D. Plants used as remedies antirheumatic and antineuralgic in the traditional medicine of Lebanon. J. Ethnopharmacol. 2008, 120, 315–334. [Google Scholar] [CrossRef] [PubMed]
- Sekera, A.; Rahm, J. Natural antihelminthics. III. Helenin. Cesk. Farm. 1953, 2, 22–24. [Google Scholar] [PubMed]
- Kang, X.; Wang, H.; Li, Y.; Xiao, Y.; Zhao, L.; Zhang, T.; Zhou, S.; Zhou, X.; Li, Y.; Shou, Z.; et al. Alantolactone induces apoptosis through ROS-mediated AKT pathway and inhibition of PINK1-mediated mitophagy in human HepG2 cells. Artif. Cells Nanomed. Biotechnol. 2019, 47, 1961–1970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Y.; Cao, X.; Kong, Y.; Wang, S.; Xia, Y.; Bi, R.; Liu, J. Apoptosis-promoting and migration-suppressing effect of alantolactone on gastric cancer cell lines BGC-823 and SGC-7901 via regulating p38 MAPK and NF-κB pathways. Hum. Exp. Toxicol. 2019, 38, 1132–1144. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Yang, Z.; Kong, Y.; He, Y.; Xu, Y.; Cao, X. Antitumor activity of alantolactone in lung cancer cell lines NCI-H1299 and Anip973. J. Food Biochem. 2019, 43, 12972. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, H.-M. Alantolactone induces gastric cancer BGC-823 cell apoptosis by regulating reactive oxygen species generation and the AKT signaling pathway. Oncol. Lett. 2019, 17, 4795–4802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Lan, Y.L.; Xing, J.S.; Lan, X.Q.; Wang, L.T.; Zhang, B. Alantolactone plays neuroprotective roles in traumatic brain injury in rats via anti-inflammatory, anti-oxidative and anti-apoptosis pathways. Am. J. Transl. Res. 2018, 10, 368–380. [Google Scholar]
- Tavares, W.R.; Seca, A.M.L. Inula L. secondary metabolites against oxidative stress-related human diseases. Antioxidants 2019, 8, 122. [Google Scholar] [CrossRef] [Green Version]
- Da Silva Castro, E.; Azeredo Alves Antunes, L.; Felipe Revoredo Lobo, J.; Arthur Ratcliffe, N.; Moreira Borges, R.; Rocha, L.; Burth, P.; Maria Fonte Amorim, L. Antileukemic properties of sesquiterpene lactones: A systematic review. Anti-Cancer Agents Med. Chem. 2018, 18, 323–334. [Google Scholar] [CrossRef]
- Xu, X.; Huang, L.; Zhang, Z.; Tong, J.; Mi, J.; Wu, Y.; Zhang, C.; Yan, H. Targeting non-oncogene ROS pathway by alantolactone in B cell acute lymphoblastic leukemia cells. Life Sci. 2019, 227, 153–165. [Google Scholar] [CrossRef]
- Chun, J.; Li, R.-J.; Cheng, M.-S.; Kim, Y.S. Alantolactone selectively suppresses STAT3 activation and exhibits potent anticancer activity in MDA-MB-231 cells. Cancer Lett. 2015, 357, 393–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, W.; Cao, P.; Xia, Y.; Hong, L.; Zhang, T.; Shen, X.; Zheng, P.; Shen, H.; Zhao, Y.; Zou, P. Potent inhibition of gastric cancer cells by a natural compound via inhibiting TrxR1 activity and activating ROS-mediated p38 MAPK pathway. Free Radic. Res. 2019, 53, 104–114. [Google Scholar] [CrossRef] [PubMed]
- Cao, P.; Xia, Y.; He, W.; Zhang, T.; Hong, L.; Zheng, P.; Shen, X.; Liang, G.; Cui, R.; Zou, P. Enhancement of oxaliplatin-induced colon cancer cell apoptosis by alantolactone, a natural product inducer of ROS. Int. J. Biol. Sci. 2019, 15, 1676–1684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, R.; Shi, X.; Zhou, M.; Zhao, Y.; Pan, S.; Zhao, C.; Guo, X.; Wang, M.; Li, X.; Qin, R. Alantolactone induces apoptosis and improves chemosensitivity of pancreatic cancer cells by impairment of autophagy-lysosome pathway via targeting TFEB. Toxicol. Appl. Pharmacol. 2018, 356, 159–171. [Google Scholar] [CrossRef]
- Rasul, A.; Khan, M.; Ali, M.; Li, J.; Li, X. Targeting apoptosis pathways in cancer with alantolactone and isoalantolactone. Sci. World J. 2013, 2013, 248532. [Google Scholar] [CrossRef] [Green Version]
- Quintana, J.; Estévez, F. Recent advances on cytotoxic sesquiterpene lactones. Curr. Pharm. Des. 2018, 24, 4355–4361. [Google Scholar] [CrossRef]
- Ren, Y.; Yue, B.; Ren, G.; Yu, Z.; Luo, X.; Sun, A.; Zhang, J.; Han, M.; Wang, Z.; Dou, W. Activation of PXR by alantolactone ameliorates DSS-induced experimental colitis via suppressing NF-κB signaling pathway. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef]
- Seo, J.Y.; Lim, S.S.; Kim, J.; Lee, K.W.; Kim, J.-S. Alantolactone and isoalantolactone prevent amyloid β25-35 -induced toxicity in mouse cortical neurons and scopolamine-induced cognitive impairment in mice. Phytother. Res. 2017, 31, 801–811. [Google Scholar] [CrossRef]
- Kumar, C.; Kumar, A.; Nalli, Y.; Lone, W.I.; Satti, N.K.; Verma, M.K.; Ahmed, Z.; Ali, A. Design, synthesis and biological evaluation of alantolactone derivatives as potential anti-inflammatory agents. Med. Chem. Res. 2019, 28, 849–856. [Google Scholar] [CrossRef]
- Li, X.; Lu, C.; Liu, S.; Liu, S.; Liu, S.; Su, C.; Xiao, T.; Bi, Z.; Sheng, P.; Huang, M.; et al. Synthesis and discovery of a drug candidate for treatment of idiopathic pulmonary fibrosis through inhibition of TGF-β1 pathway. Eur. J. Med. Chem. 2018, 157, 229–247. [Google Scholar] [CrossRef]
- Khan, M.; Yi, F.; Rasul, A.; Li, T.; Wang, N.; Gao, H.; Gao, R.; Ma, T. Alantolactone induces apoptosis in glioblastoma cells via GSH depletion, ROS generation, and mitochondrial dysfunction. IUBMB Life 2012, 64, 783–794. [Google Scholar] [CrossRef] [PubMed]
- Markowiak, T.; Kerner, N.; Neu, R.; Potzger, T.; Großer, C.; Zeman, F.; Hofmann, H.-S.; Ried, M. Adequate nephroprotection reduces renal complications after hyperthermic intrathoracic chemotherapy. J. Surg. Oncol. 2019, 120, 1220–1226. [Google Scholar] [CrossRef] [PubMed]
- Leach, C. Complications of systemic anti-cancer treatment. Medicine 2020, 48, 48–51. [Google Scholar] [CrossRef]
- Jain, K.K. An overview of drug delivery systems. Meth. Mol. Biol. 2020, 2059, 1–54. [Google Scholar] [CrossRef]
- Guo, C.; Zhang, S.; Teng, S.; Niu, K. Simultaneous determination of sesquiterpene lactones isoalantolactone and alantolactone isomers in rat plasma by liquid chromatography with tandem mass spectrometry: Application to a pharmacokinetic study. J. Sep. Sci. 2014, 37, 950–956. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Zhou, G.; Peng, Y.; Wang, M.; Li, X. Pharmacokinetics, tissue distribution and excretion of isoalantolactone and alantolactone in rats after oral administration of radix inulae extract. Molecules 2015, 20, 7719–7736. [Google Scholar] [CrossRef] [Green Version]
- Zhou, B.; Ye, J.; Yang, N.; Chen, L.; Zhuo, Z.; Mao, L.; Liu, Q.; Lan, G.; Ning, J.; Ge, G.; et al. Metabolism and pharmacokinetics of alantolactone and isoalantolactone in rats: Thiol conjugation as a potential metabolic pathway. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2018, 1072, 370–378. [Google Scholar] [CrossRef]
- Bottex-Gauthier, C.; Vidal, D.; Picot, F.; Potier, P.; Menichini, F.; Appendino, G. In vitro biological activities of arglabin, a sesquiterpene lactone from the Chinese herb Artemisia myriantha Wall. (Asteraceae). Biotechnol. Ther. 1993, 4, 77–98. [Google Scholar]
- Zan, K.; Chen, X.Q.; Tu, P.F. Guaianolides from aerial parts of Artemisia myriantha. Zhongguo Zhongyao Zazhi. 2018, 43, 2295–2299. [Google Scholar] [CrossRef]
- Dylenova, E.P.; Randalova, T.E.; Tykheev, Z.A.; Zhigzhitzhapova, S.V.; Radnaeva, L.D. Artemisia jacutica Drob. as the source of terpenoids. IOP Conf. Ser. Earth Environ. Sci. 2019, 320, 012054. [Google Scholar] [CrossRef]
- Shaikenov, T.E.; Adekenov, S.M.; Williams, R.M.; Prashad, N.; Baker, F.L.; Madden, T.L.; Newman, R. Arglabin-DMA, a plant derived sesquiterpene, inhibits farnesyltransferase. Oncol. Rep. 2001, 8, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Lone, S.H.; Bhat, K.A.; Khuroo, M.A. Arglabin: From isolation to antitumor evaluation. Chem.-Biol. Interact. 2015, 240, 180–198. [Google Scholar] [CrossRef] [PubMed]
- Seitz, M.; Reiser, O. Synthetic approaches towards structurally diverse gamma-butyrolactone natural-product-like compounds. Curr. Opin. Chem. Biol. 2005, 9, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Kalidindi, S.; Jeong, S.W.B.; Schall, A.; Bandichhor, R.; Nosse, B.; Reiser, O. Enantioselective synthesis of arglabin. Angew. Chem. Int. Ed. 2007, 46, 6361–6363. [Google Scholar] [CrossRef] [PubMed]
- Gharpure, S.J.; Nanda, L.N. Application of oxygen/nitrogen substituted donor-acceptor cyclopropanes in the total synthesis of natural products. Tetrahedron Lett. 2017, 58, 711–720. [Google Scholar] [CrossRef]
- Lone, S.H.; Bhat, K.A. Hemisynthesis of a naturally occurring clinically significant antitumor arglabin from ludartin. Tetrahedron Lett. 2015, 56, 1908–1910. [Google Scholar] [CrossRef]
- Ganesan, A. The impact of natural products upon modern drug discovery. Curr. Opin. Chem. Biol. 2008, 12, 306–317. [Google Scholar] [CrossRef]
- Adekenov, S.M. Chemical modification of arglabin and biological activity of its new derivatives. Fitoterapia 2016, 110, 196–205. [Google Scholar] [CrossRef]
- Musulmanbekov, K.Z. Clinical use of the antitumor drug arglabin. In Proceedings of the International Scientific-Practical Confcrcnce, Karaganda, Kazakhstan, 16–18 October 2002; p. 46. [Google Scholar]
- Zhangabylov, N.S.; Dederer, L.Y.; Gorbacheva, L.B.; Vasil’eva, S.V.; Terekhov, A.S.; Adekenov, S.M. Sesquiterpene lactone arglabin influences DNA synthesis in P388 leukemia cells in vivo. Pharm. Chem. J. 2004, 38, 651–653. [Google Scholar] [CrossRef]
- Sirota, V.B.; Bochkova, N.V.; Kostrova, E.V.; Tselikova, N.L.; Kabildina, N.A. Application of the phytopreparate arglabin in combined treatment of patients with cancer. In Proceedings of the 1st Russian Phytotherapeutic Congress, Moscow, Russia, 14–16 March 2008; p. 148. [Google Scholar]
- Schepetkin, I.A.; Kirpotina, L.N.; Mitchell, P.T.; Kishkentaeva, A.C.; Shaimerdenova, Z.R.; Atazhanova, G.A.; Adekenov, S.M.; Quinn, M.T. The natural sesquiterpene lactones arglabin, grosheimin, agracin, parthenolide, and estafiatin inhibit T cell receptor (TCR) activation. Phytochemistry 2018, 146, 36–46. [Google Scholar] [CrossRef]
- He, W.; Lai, R.F.; Lin, Q.; Huang, Y.M.; Wang, L. Arglabin is a plant sesquiterpene lactone that exerts potent anticancer effects on human oral squamous cancer cells via mitochondrial apoptosis and downregulation of the mTOR/PI3K/Akt signaling pathway to inhibit tumor growth in vivo. J. Buon 2018, 23, 1679–1685. [Google Scholar] [PubMed]
- Tewari, D.; Rawat, P.; Singh, P.K. Adverse drug reactions of anticancer drugs derived from natural sources. Food Chem. Toxicol. 2019, 123, 522–535. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Jia, H.; Xia, H. Arglabin as a potential drug in the treatment of Freund’s complete adjuvant-induced arthritis in rats. Trop. J. Pharm. Res. 2018, 17, 1585–1590. [Google Scholar] [CrossRef]
- Abderrazak, A.; Couchie, D.; Mahmood, D.F.; Elhage, R.; Vindis, C.; Laffargue, M.; Mateo, V.; Buchele, B.; Ayala, M.R.; El Gaafary, M.; et al. Anti-inflammatory and antiatherogenic effects of the NLRP3 inflammasome inhibitor arglabin in ApoE2.Ki mice fed a high-fat diet. Circulation 2015, 131, 1061–1070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldrighi, M.; Mallat, Z.; Li, X. NLRP3 inflammasome pathways in atherosclerosis. Atherosclerosis 2017, 267, 127–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, A.S.; Kelkar, G.R.; Bhattacharyya, S.C. Terpenoids—XXI: The structure of costunolide, a new sesquiterpene lactone from costus root oil. Tetrahedron 1960, 9, 275–283. [Google Scholar] [CrossRef]
- Ikezawa, N.; Göpfert, J.C.; Nguyen, D.T.; Kim, S.-U.; O’Maille, P.E.; Spring, O.; Ro, D.-K. Lettuce costunolide synthase (CYP71BL2) and its homolog (CYP71BL1) from sunflower catalyze distinct regio- and stereoselective hydroxylations in sesquiterpene lactone metabolism. J. Biol. Chem. 2011, 286, 21601–21611. [Google Scholar] [CrossRef] [Green Version]
- Kassuya, C.A.L.; Cremoneze, A.; Barros, L.F.L.; Simas, A.S.; Lapa, F.d.R.; Mello-Silva, R.; Stefanello, M.E.A.; Zampronio, A.R. Antipyretic and anti-inflammatory properties of the ethanolic extract, dichloromethane fraction and costunolide from Magnolia ovata (Magnoliaceae). J. Ethnopharmacol. 2009, 124, 369–376. [Google Scholar] [CrossRef]
- De Kraker, J.-W.; Franssen, M.C.R.; Joerink, M.; de Groot, A.; Bouwmeester, H.J. Biosynthesis of costunolide, dihydrocostunolide, and leucodin. Demonstration of cytochrome p450-catalyzed formation of the lactone ring present in sesquiterpene lactones of chicory. Plant Physiol. 2002, 129, 257–268. [Google Scholar] [CrossRef] [Green Version]
- Abdelwahab, S.I.; Taha, M.M.E.; Alhazmi, H.A.; Ahsan, W.; Rehman, Z.U.; Bratty, M.A.; Makeen, H. Phytochemical profiling of costus (Saussurea lappa Clarke) root essential oil, and its antimicrobial and toxicological effects. Trop. J. Pharm. Res. 2019, 18, 2155–2160. [Google Scholar] [CrossRef]
- Yang, Z.-J.; Ge, W.-Z.; Li, Q.-Y.; Lu, Y.; Gong, J.-M.; Kuang, B.-J.; Xi, X.; Wu, H.; Zhang, Q.; Chen, Y. Synthesis and biological evaluation of costunolide, parthenolide, and their fluorinated analogues. J. Med. Chem. 2015, 58, 7007–7020. [Google Scholar] [CrossRef] [PubMed]
- Pavan Kumar, C.; Devi, A.; Ashok Yadav, P.; Rao Vadaparthi, R.; Shankaraiah, G.; Sowjanya, P.; Jain, N.; Suresh Babu, K. “Click” reaction mediated synthesis of costunolide and dehydrocostuslactone derivatives and evaluation of their cytotoxic activity. Asian Nat. Prod. Res. 2016, 18, 1063–1078. [Google Scholar] [CrossRef] [PubMed]
- Poon, P.S.; Banerjee, A.K.; Vera, W.J.; Bedoya, L. Reagents for bromination; Application in the synthesis of diterpenes, sesquiterpenes and bioactive compounds. Curr. Org. Chem. 2017, 21, 889–907. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.Y.; Choi, B.Y. Costunolide - A bioactive sesquiterpene lactone with diverse therapeutic potential. Int. J. Mol. Sci. 2019, 20, 2926. [Google Scholar] [CrossRef] [Green Version]
- Lin, X.; Peng, Z.; Su, C. Potential anti-cancer activities and mechanisms of costunolide and dehydrocostuslactone. Int. J. Mol. Sci. 2015, 16, 10888–10906. [Google Scholar] [CrossRef] [Green Version]
- Jin, X.; Wang, C.; Wang, L. Costunolide inhibits osteosarcoma growth and metastasis via suppressing STAT3 signal pathway. Biomed. Pharmacother. 2020, 121, 109659. [Google Scholar] [CrossRef]
- Kolosenko, I.; Yu, Y.; Busker, S.; Dyczynski, M.; Liu, J.; Haraldsson, M.; Palm Apergi, C.; Helleday, T.; Tamm, K.P.; Page, B.D.G.; et al. Identification of novel small molecules that inhibit STAT3-dependent transcription and function. PLoS ONE 2017, 12, 0178844. [Google Scholar] [CrossRef]
- Yan, Z.; Xu, T.; An, Z.; Hu, Y.; Chen, W.; Ma, J.; Shao, C.; Zhu, F. Costunolide induces mitochondria-mediated apoptosis in human gastric adenocarcinoma BGC-823 cells. BMC Complement. Altern. Med. 2019, 19, 151. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, F.; Dixit, D.; Sharma, V.; Kumar, A.; Joshi, S.D.; Sarkar, C.; Sen, E. Nrf2-driven TERT regulates pentose phosphate pathway in glioblastoma. Cell Death Dis. 2016, 7, 2213. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, F.; Patrick, S.; Sheikh, T.; Sharma, V.; Pathak, P.; Malgulwar, P.B.; Kumar, A.; Joshi, S.D.; Sarkar, C.; Sen, E. Telomerase reverse transcriptase (TERT) - enhancer of zeste homolog 2 (EZH2) network regulates lipid metabolism and DNA damage responses in glioblastoma. J. Neurochem. 2017, 143, 671–683. [Google Scholar] [CrossRef] [Green Version]
- Menendez, J.A.; Lupu, R. Fatty acid synthase (FASN) as a therapeutic target in breast cancer. Expert Opin. Ther. Targets 2017, 21, 1001–1016. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Bell, E.H.; Mischel, P.; Chakravarti, A. Targeting SREBP-1-driven lipid metabolism to treat cancer. Curr. Pharm. Des. 2014, 20, 2619–2626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bost, F.; Kaminski, L. The metabolic modulator PGC-1α in cancer. Am. J. Cancer Res. 2019, 9, 198–211. [Google Scholar] [PubMed]
- Park, E.; Song, J.H.; Kim, M.S.; Park, S.-H.; Kim, T.S. Costunolide, a sesquiterpene lactone, inhibits the differentiation of pro-inflammatory CD4+ T cells through the modulation of mitogen-activated protein kinases. Int. Immunopharmacol. 2016, 40, 508–516. [Google Scholar] [CrossRef] [PubMed]
- Turk, A.; Ahn, J.H.; Jo, Y.H.; Song, J.Y.; Khalife, H.K.; Gali-Muhtasib, H.; Kim, Y.; Hwang, B.Y.; Lee, M.K. NF-κB inhibitory sesquiterpene lactones from Lebanese Laurus nobilis. Phytochem. Lett. 2019, 30, 120–123. [Google Scholar] [CrossRef]
- Saraswati, S.; Alhaider, A.A.; Abdelgadir, A.M. Costunolide suppresses an inflammatory angiogenic response in a subcutaneous murine sponge model. APMIS 2018, 126, 257–266. [Google Scholar] [CrossRef]
- Xie, Y.; Li, Q.-J.; Wang, Z.-G.; Hu, H.-L. Effects of active components from vladimiriae radix and their combinations on ethanol-induced acute gastric ulcer in mice. Chin. J. New Drugs 2019, 28, 2754–2760. [Google Scholar]
- Chen, Z.; Zhang, D.; Li, M.; Wang, B. Costunolide ameliorates lipoteichoic acid-induced acute lung injury via attenuating MAPK signaling pathway. Int. Immunopharm. 2018, 61, 283–289. [Google Scholar] [CrossRef]
- Chen, Y.-T.; Du, Y.; Zhao, B.; Gan, L.-X.; Yu, K.-K.; Sun, L.; Wang, J.; Qian, F. Costunolide alleviates HKSA-induced acute lung injury via inhibition of macrophage activation. Acta Pharmacol. Sin. 2019, 40, 1040–1048. [Google Scholar] [CrossRef]
- Liu, B.; Rong, Y.; Sun, D.; Li, W.; Chen, H.; Cao, B.; Wang, T. Costunolide inhibits pulmonary fibrosis via regulating NF-κB and TGF-β1/Smad2/Nrf2-NOX4 signaling pathways. Biochem. Biophys. Res. Commun. 2019, 510, 329–333. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, X.; Zhao, L.; Shi, M.; Wei, Z.; Yang, Z.; Guo, C.; Fu, Y. Costunolide protects lipopolysaccharide/D-galactosamine–induced acute liver injury in mice by inhibiting NF-κB signaling pathway. J. Surg. Res. 2017, 220, 40–45. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Moqbel, S.A.A.; Xu, L.; Ran, J.; Ma, C.; Xu, K.; Bao, J.; Jiang, L.; Chen, W.; Xiong, Y.; et al. Costunolide inhibits matrix metalloproteinases expression and osteoarthritis via the NF-κB and Wnt/β-catenin signaling pathways. Mol. Med. Rep. 2019, 20, 312–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, B.-K.; Park, S.-J.; Nam, S.-Y.; Kang, S.; Hwang, J.; Lee, S.-J.; Im, D.-S. Anti-allergic effects of sesquiterpene lactones from Saussurea costus (Falc.) Lipsch. determined using in vivo and in vitro experiments. J. Ethnopharmacol. 2018, 213, 256–261. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.E.; Choi, H.C.; Nam, G.; Choi, B.Y. Costunolide promotes the proliferation of human hair follicle dermal papilla cells and induces hair growth in C57 BL/6 mice. J. Cosmet. Dermatol. 2019, 18, 414–421. [Google Scholar] [CrossRef] [Green Version]
- Cala, A.; Zorrilla, J.G.; Rial, C.; Molinillo, J.M.G.; Varela, R.M.; Macías, F.A. Easy access to alkoxy, amino, carbamoyl, hydroxy, and thiol derivatives of sesquiterpene lactones and evaluation of their bioactivity on parasitic weeds. J. Agric. Food Chem. 2019, 67, 10764–10773. [Google Scholar] [CrossRef]
- Elsebai, M.F.; Mocan, A.; Atanasov, A.G. Cynaropicrin: A comprehensive research review and therapeutic potential as an anti-hepatitis C virus agent. Front. Pharmacol. 2016, 7, 472. [Google Scholar] [CrossRef] [Green Version]
- Suchy, M.; Herout, V.; Šorm, F. On terpenes. CXVI. Structure of cynaropicrin. Collect. Czech. Chem. Commun. 1960, 25, 2777–2782. [Google Scholar] [CrossRef]
- Eljounaidi, K.; Comino, C.; Moglia, A.; Cankar, K.; Genre, A.; Hehn, A.; Bourgaud, F.; Beekwilder, J.; Lanteri, S. Accumulation of cynaropicrin in globe artichoke and localization of enzymes involved in its biosynthesis. Plant Sci. 2015, 239, 128–136. [Google Scholar] [CrossRef]
- Colantuono, A.; Ferracane, R.; Vitaglione, P. Potential bioaccessibility and functionality of polyphenols and cynaropicrin from breads enriched with artichoke stem. Food Chem. 2018, 245, 838–844. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, J.; Han, X.; Xu, J.; Wu, Y.; Fang, J. Promotion of HeLa cells apoptosis by cynaropicrin involving inhibition of thioredoxin reductase and induction of oxidative stress. Free Rad. Biol. Med. 2019, 135, 216–226. [Google Scholar] [CrossRef]
- Formisano, C.; Sirignano, C.; Rigano, D.; Chianese, G.; Zengin, G.; Seo, E.J.; Efferth, T.; Taglialatela-Scafati, O. Antiproliferative activity against leukemia cells of sesquiterpene lactones from the Turkish endemic plant Centaurea drabifolia subsp. detonsa. Fitoterapia 2017, 120, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Nawrot, J.; Budzianowski, J.; Nowak, G. Phytochemical profiles of the leaves of Stizolophus balsamita and Psephellus sibiricus and their chemotaxonomic implications. Phytochemistry 2019, 159, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Cis, J.; Nowak, G.; Kisiel, W. Antifeedant properties and chemotaxonomic implications of sesquiterpene lactones and syringin from Rhaponticum pulchrum. Biochem. Syst. Ecol. 2006, 34, 862–867. [Google Scholar] [CrossRef]
- Schinor, E.C.; Salvador, M.J.; Ito, I.Y.; de Albuquerque, S.; Dias, D.A. Trypanocidal and antimicrobial activities of Moquinia kingii. Phytomedicine 2004, 11, 224–229. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.Z.; Choi, S.U.; Lee, K.R. Cytotoxic sesquiterpene lactones from Saussurea calcicola. Arch. Pharmacol. Res. 2005, 28, 1142–1146. [Google Scholar] [CrossRef]
- Cho, J.Y.; Kim, A.R.; Jung, J.H.; Chun, T.; Rhee, M.H.; Yoo, E.S. Cytotoxic and pro-apoptotic activities of cynaropicrin, a sesquiterpene lactone, on the viability of leukocyte cancer cell lines. Eur. J. Pharmacol. 2004, 492, 85–94. [Google Scholar] [CrossRef]
- Pandey, M.M.; Rastogi, S.; Rawat, A.K.S. Saussurea costus: Botanical, chemical and pharmacological review of an ayurvedic medicinal plant. J. Ethnopharmacol. 2007, 110, 379–390. [Google Scholar] [CrossRef]
- Bhattacharyya, P.R.; Barua, N.C.; Ghosh, A.C. Cynaropicrin from Tricholepis glaberrima: A potential insect feeding deterrent compound. Ind. Crops Prod. 1995, 4, 291–294. [Google Scholar] [CrossRef]
- Brás, T.; Neves, L.A.; Crespo, J.G.; Duarte, M.F. Effect of extraction methodologies and solvent selection upon cynaropicrin extraction from Cynara cardunculus leaves. Sep. Purif. Technol. 2019, 236, 116283. [Google Scholar] [CrossRef]
- Zimmermann, S.; Kaiser, M.; Brun, R.; Hamburger, M.; Adams, M. Cynaropicrin: The first plant natural product with in vivo activity against Trypanosoma brucei. Planta Med. 2012, 78, 553–556. [Google Scholar] [CrossRef]
- Zimmermann, S.; Oufir, M.; Leroux, A.; Krauth-Siegel, R.L.; Becker, K.; Kaiser, M.; Brun, R.; Hamburger, M.; Adams, M. Cynaropicrin targets the trypanothione redox system in Trypanosoma brucei. Bioorg. Med. Chem. 2013, 21, 7202–7209. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, C.F.; Batista, D.d.G.; De Araújo, J.S.; Batista, M.M.; Lionel, J.; de Souza, E.M.; Hammera, E.R.; Silva, P.B.; De Mieri, M.; Adams, M.; et al. Activities of psilostachyin A and cynaropicrin against Trypanosoma cruzi in vitro and in vivo. Antimicrob. Agents Chemother. 2013, 57, 5307–5314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimmermann, S.; Fouché, G.; De Mieri, M.; Yoshimoto, Y.; Usuki, T.; Nthambeleni, R.; Parkinson, C.J.; van der Westhuyzen, C.; Kaiser, M.; Hamburger, M.; et al. Structure-activity relationship study of sesquiterpene lactones and their semi-synthetic amino derivatives as potential antitrypanosomal products. Molecules 2014, 19, 3523–3538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Usuki, T.; Sato, M.; Hara, S.; Yoshimoto, Y.; Kondo, R.; Zimmermann, S.; Kaiser, M.; Brun, R.; Hamburger, M.; Adams, M. Antitrypanosomal structure-activity-relationship study of synthetic cynaropicrin derivatives. Bioorg. Med. Chem. Lett. 2014, 24, 794–798. [Google Scholar] [CrossRef]
- Tanaka, Y.T.; Tanaka, K.; Kojima, H.; Hamada, T.; Masutani, T.; Tsuboi, M.; Akao, Y. Cynaropicrin from Cynara scolymus L. suppresses photoaging of skin by inhibiting the transcription activity of nuclear factor-kappa B. Bioorg. Med. Chem. Lett. 2013, 23, 518–523. [Google Scholar] [CrossRef]
- Willuhn, G. Arnica flowers: Pharmacology, toxicolgy, and analysis of the sesquiterpene lactones—Their main active substances. ACS Symp. Ser. 1998, 691, 118–132. [Google Scholar]
- Iannitti, T.; Morales-Medina, J.C.; Bellavite, P.; Rottigni, V.; Palmieri, B. Effectiveness and safety of Arnica montana in post-surgical setting, pain and inflammation. Am. J. Ther. 2016, 23, 184–197. [Google Scholar] [CrossRef]
- Todorova, M.; Trendafilova, A.; Vitkova, A.; Petrova, M.; Zayova, E.; Antonova, D. Developmental and environmental effects on sesquiterpene lactones in cultivated Arnica montana L. Chem. Biodiv. 2016, 13, 976–981. [Google Scholar] [CrossRef]
- Widen, J.C.; Kempema, A.M.; Villalta, P.W.; Harki, D.A. Targeting NF-κB p65 with a helenalin inspired bis-electrophile. Chem. Biol. 2017, 12, 102–113. [Google Scholar] [CrossRef]
- Li, Y.; Zeng, Y.; Huang, Q.; Wen, S.; Wei, Y.; Chen, Y.; Zhang, X.; Bai, F.; Lu, Z.; Wei, J.; et al. Helenalin from Centipeda minima ameliorates acute hepatic injury by protecting mitochondria function, activating Nrf2 pathway and inhibiting NF-κB activation. Biomed. Pharmacother. 2019, 119, 109435. [Google Scholar] [CrossRef]
- Kriplani, P.; Guarve, K.; Baghael, U.S. Arnica montana L.—A plant of healing: Review. J. Pharm. Pharmacol. 2017, 69, 925–945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chadwick, M.; Trewin, H.; Gawthrop, F.; Wagstaff, C. Sesquiterpenoids lactones: Benefits to plants and people. Int. J. Mol. Sci. 2013, 14, 12780–12805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, P.R.; Yeh, Y.M.; Wang, T.C. Potent inhibition of human telomerase by helenalin. Cancer Lett. 2005, 227, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Berges, C.; Fuchs, D.; Opelz, G.; Daniel, V.; Naujokat, C. Helenalin suppresses essential immune functions of activated CD4+ T cells by multiple mechanisms Mol. Immunol. 2009, 46, 2892–2901. [Google Scholar] [CrossRef]
- Lyss, G.; Knorre, A.; Schmidt, T.J.; Pahl, H.L.; Merfort, I. The anti-inflammatory sesquiterpene lactone helenalin inhibits the transcription factor NF-κB by directly targeting p65. J. Biol. Chem. 1998, 273, 33508–33516. [Google Scholar] [CrossRef] [Green Version]
- Zwicker, P.; Schultze, N.; Niehs, S.; Albrecht, D.; Methling, K.; Wurster, M.; Wachlin, G.; Lalk, M.; Lindequist, U.; Haertel, B. Differential effects of helenalin, an anti-inflammatory sesquiterpene lactone, on the proteome, metabolome and the oxidative stress response in several immune cell types. Toxicol. In Vitro 2017, 40, 45–54. [Google Scholar] [CrossRef]
- Büchele, B.; Zugmaier, W.; Lunov, O.; Syrovets, T.; Merfort, I.; Simmet, T. Surface plasmon resonance analysis of nuclear factor-κB protein interactions with the sesquiterpene lactone helenalin. Anal. Biochem. 2010, 401, 30–37. [Google Scholar] [CrossRef]
- Tornhamre, S.; Schmidt, T.J.; Nasman-Glaser, B. Inhibitory effects of helenalin and related compounds on 5-lipoxygenase and leukotriene C(4) synthase in human blood cells. Biochem Pharmacol. 2001, 62, 903–911. [Google Scholar] [CrossRef]
- Schröder, H.; Lösche, W.; Strobach, H.; Leven, W.; Willuhn, G.; Till, U.; Schrör, K. Helenalin and 11α,13-dihydrohelenalin, two constituents from Arnica montana L., inhibit human platelet function via thiol-dependent pathway. Thromb. Res. 1990, 57, 839–845. [Google Scholar] [CrossRef]
- Macêdo, S.B.; Ferreira, L.R.; Perazzo, F.F.; Tavares Carvalho, J.C. Anti-inflammatory activity of Arnica montana 6cH: Preclinical study in animals. Homeopathy 2004, 93, 84–87. [Google Scholar] [CrossRef]
- Castro, F.C.; Magre, A.; Cherpinski, R.; Zelante, P.M.; Neves, L.M.; Esquisatto, M.A.; Mendonça, F.A.; Santos, G.M. Effects of microcurrent application alone or in combination with topical Hypericum perforatum L. and Arnica montana L. on surgically induced wound healing in Wistar rats. Homeopathy 2012, 101, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Widrig, R.; Suter, A.; Saller, R.; Melzer, J. Choosing between NSAID and Arnica for topical treatment of hand osteoarthritis in a randomised, double-blind study. Rheumatol. Int. 2007, 27, 585–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boulanger, D.; Brouillette, E.; Jaspar, F.; Malouin, F.; Mainil, J.; Bureau, F.; Lekeux, P. Helenalin reduces Staphylococcus aureus infection in vitro and in vivo. Vet. Microbiol. 2007, 119, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Valan, M.F.; Britto, J.B.; Venkataraman, R. Phytoconstituents with hepatoprotective activity. Int. J. Chem. Sci. 2010, 8, 1421–1432. [Google Scholar]
- Usui, K.; Ikeda, T.; Horibe, Y.; Nakao, M.; Hoshino, T.; Mizushima, T. Identification of HSP70- inducing activity in Arnica montana extract and purification and characterization of HSP70-inducers. J. Dermatol. Sci. 2015, 78, 67–75. [Google Scholar] [CrossRef] [Green Version]
- Lin, X.; Huang, Q.; Bai, F.; Wei, J.; Huang, R.; Wen, S.; Wei, Y.; Zhang, X. Preparation Method of Traditional Chinese Medicine Centipeda minima Sesquiterpene Monomer Helenalin, and Its Application in Preparing Drug for Treating Hepatic Fibrosis and. Inflammation. Patent No. CN 110283151, 27 September 2019. [Google Scholar]
- Lin, X.; Huang, Q.; Bai, F.; Wei, J.; Huang, R.; Wen, S.; Wei, Y.; Zhang, X. Application of Helenalin for Inhibiting Hepatic Stellate Cell Activation from Centipeda. minima. Patent No. CN 110179790, 30 August 2019. [Google Scholar]
- Messaoudi, M.; Chahmi, N.; El-Mzibri, M.; Gmouh, S.; Amzazi, S.; Benbacer, L.; El-Hassouni, M. Cytotoxic effect and chemical composition of Inula viscosa from three different regions of Morocco. Eur. J. Med. Plants 2016, 16, 1–9. [Google Scholar] [CrossRef]
- Hernández, V.; Recio, M.D.C.; Máñz, S.; Prieto, J.M.; Giner, R.M.; Ríos, J.L. A mechanistic approach to the in vivo anti-inflammatory activity of sesquiterpenoid compounds isolated from Inula viscosa. Planta Med. 2001, 67, 726–731. [Google Scholar] [CrossRef]
- Dolejš, L.; Souček, M.; Horák, M.; Herout, V.; Šorm, F. On terpenes. XCIV. The structure of lactucin. Collect. Czech. Chem. Commun. 1958, 23, 2195–2200. [Google Scholar] [CrossRef]
- Ruban, G.; Zabel, V.; Gensch, K.H.; Smalla, H. The crystal structure and absolute configuration of lactucin. Acta Cryst. 1978, 34, 1163–1167. [Google Scholar] [CrossRef] [Green Version]
- Besharat, S.; Besharat, M.; Jabbari, A. Wild lettuce (Lactuca virosa) toxicity. BMJ Case Rep. 2009, 2009, 10–13. [Google Scholar] [CrossRef] [Green Version]
- De Kraker, J.W.; Franssen, M.C.R.; Dalm, M.C.F.; De Groot, A.; Bouwmeester, H.J. Biosynthesis of germacrene a carboxylic acid in chicory roots. Demonstration of a cytochrome p450 (+)-germacrene a hydroxylase and NADP+-dependent sesquiterpenoid dehydrogenase(s) involved in sesquiterpene lactone biosynthesis. Plant Physiol. 2001, 125, 1930–1940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sessa, R.A.; Bennett, M.H.; Lewis, M.J.; Mansfield, J.W.; Beale, M.H. Metabolite profiling of sesquiterpene lactones from Lactuca Species. J. Biol. Chem. 2000, 275, 26877–26884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Testone, G.; Mele, G.; di Giacomo, E.; Tenore, G.C.; Gonnella, M.; Nicolodi, C.; Frugis, G.; Iannelli, M.A.; Arnesi, G.; Schiappa, A.; et al. Transcriptome driven characterization of curly- and smooth-leafed endives reveals molecular differences in the sesquiterpenoid pathway. Hortic. Res. 2019, 6, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Seo, M.W.; Yang, D.S.; Kays, S.J.; Lee, G.P.; Park, K.W. Sesquiterpene lactones and bitterness in korean leaf lettuce cultivars. HortScience 2009, 44, 246–249. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.D.; Hong, K.B.; Noh, D.O.; Suh, H.J. Sleep-inducing effect of lettuce (Lactuca sativa) varieties on pentobarbital-induced sleep. Food Sci. Biotechnol. 2017, 26, 807–814. [Google Scholar] [CrossRef]
- Bahmani, M.; Shahinfard, N.; Rafieian-Kopaei, M.; Saki, K.; Shahsavari, S.; Taherikalani, M.; Ghafourian, S.; Baharvand-Ahmadi, B. Chicory: A review on ethnobotanical effects of Cichorium intybus L. J. Chem. Pharm. Sci. 2015, 8, 672–682. [Google Scholar]
- Ludwig, H. Ueber die Bestandtheile des Lactucariums. Arch. Pharm. Pharm. Med. Chem. 1847, 100, 1–19. [Google Scholar] [CrossRef]
- Christison, R. A Dispensatory, or Commentary on the Pharmacopoeias of Great Britain; Adam and Charles Black Ed.: Edinburgh, UK, 1842. [Google Scholar]
- Thomson, A.T. A Conspectus of the Pharmacopoeias of the London, Edinburgh and Dublin Collegues of Physicians and of the United States Pharmacopoeia Being a Practical Compendium of Materia Medica and Pharmacy; Lee, C.A., Ed.; Henry, G. Langley, 8 Astor House: New York, NY, USA, 1844. [Google Scholar]
- Wesołowska, A.; Nikiforuk, A.; Michalska, K.; Kisiel, W.; Chojnacka-Wójcik, E. Analgesic and sedative activities of lactucin and some lactucin-like guaianolides in mice. J. Ethnopharmacol. 2006, 107, 254–258. [Google Scholar] [CrossRef]
- Yakoot, M.; Helmy; Fawal, K. Pilot study of the efficacy and safety of lettuce seed oil in patients with sleep disorders. Int. J. Gen. Med. 2011, 4, 451–456. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, B.; Ilic, N.; Poulev, A.; Raskin, I. Toxicological evaluation of chicory extract (humans). Food Chem. Toxicol. 2007, 45, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Ghantous, A.; Sinjab, A.; Herceg, Z.; Darwiche, N. Parthenolide: From plant shoots to cancer roots. Drug Discov. Today 2013, 18, 894–905. [Google Scholar] [CrossRef] [PubMed]
- Kreuger, M.R.O.; Grootjans, S.; Biavatti, M.; Vandanabeele, P.; Dherde, K. Sesquiterpene lactones as drugs with multiple targets in cancer treatment: Focus on parthenolide. Anticancer Drugs 2011, 23, 883–896. [Google Scholar] [CrossRef]
- Jafari, N.; Nazeri, S.; Enferadi, S.T. Parthenolide reduces metastasis by inhibition of vimentin expression and induces apoptosis by suppression elongation factor α−1 expression. Phytomedicine 2018, 41, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Freund, R.R.A.; Gobrecht, P.; Fischer, D.; Arndt, H.-D. Advances in chemistry and bioactivity of parthenolide. Nat. Prod. Rep 2020. [Google Scholar] [CrossRef] [PubMed]
- Seca, A.M.L.; Silva, A.M.S.; Pinto, D.C.G.A. Parthenolide and parthenolide-like sesquiterpene lactones as multiple targets drugs: Current knowledges and new developments. In Studies in Natural Products Chemistry; Atta-Ur-Rahman, Ed.; Elsevier Science Publishers: Amsterdam, The Netherlands, 2017; Volume 52, pp. 337–372. [Google Scholar] [CrossRef]
- Ren, Y.; Yu, J.; Kinghorn, A.D. Development of anticancer agents from plant-derived sesquiterpene lactones. Curr. Med. Chem. 2016, 23, 2397–2420. [Google Scholar] [CrossRef]
- Babaei, G.; Aliarab, A.; Abroon, S.; Rasmi, Y.; Aziz, S.G. Application of sesquiterpene lactone: A new promising way for cancer therapy based on anticancer activity. Biomed. Pharmacother. 2018, 106, 239–246. [Google Scholar] [CrossRef]
- Dey, S.; Sarkar, M.; Giri, B. Anti-inflammatory and anti-tumor activities of parthenolide: An update. Chem. Biol. Ther. 2016, 1, 107. [Google Scholar] [CrossRef] [Green Version]
- Saadane, A.; Eastman, J.; Berger, M.; Bonfield, T.M. Parthenolide inhibits ERK and AP-1 which are dysregulated and contribute to excessive IL-8 expression and secretion in cystic fibrosis cells. J. Inflamm. 2011, 8, 26. [Google Scholar] [CrossRef] [Green Version]
- Carlisi, D.; D’Anneo, A.; Angileri, L.; Lauricella, M.; Emanuele, S.; Santulli, A.; Vento, R.; Tesoriere, G. Parthenolide sensitizes hepatocellular carcinoma cells to TRAIL by inducing the expression of death receptors through inhibition of STAT3 activation. J. Cell Physiol. 2011, 226, 1632–1641. [Google Scholar] [CrossRef]
- Nakshatri, P.; Rice, S.E.; Bhat-Nakshatri, P. Antitumor agent parthenolide reverses resistance of breast cancer cells to tumor necrosis factor-related apoptosis-inducing ligand through sustained activation of c-Jun N-terminal kinase. Oncogene 2004, 23, 7330–7344. [Google Scholar] [CrossRef] [Green Version]
- Rüngeler, P.; Castro, V.; Mora, G.; Gören, N.; Vichnewski, W.; Pah, H.L.; Merfort, I.; Schmidt, T.J. Inhibition of transcription factor NFkappaB by sesquiterpene lactones: A proposed molecular mechanism of action. Bioorg. Med. Chem. 1999, 7, 2343–2352. [Google Scholar] [CrossRef]
- Steele, A.J.; Jones, D.T.; Ganeshaguru, K.; Duke, V.; Yogashangary, B.C.; North, J.M.; Lowdell, M.W.; Kottaridis, P.D.; Mehta, A.; Prentice, A.G.; et al. The sesquiterpene lactone parthenolide induces selective apoptosis of B-chronic lymphocytic leukemia cells in vitro. Leukemia 2006, 20, 1073–1079. [Google Scholar] [CrossRef] [PubMed]
- D’Anneo, A.; Carlisi, D.; Lauricella, M.; Puleio, R.; Martinez, R.; Di Bella, S.; Di Marco, P.; Emanuele, S.; Di Fiore, R.; Guercio, A.; et al. Parthenolide generates reactive oxygen species and autophagy inMDA-MB231 cells. A soluble parthenolide analogue inhibits tumour growth and metastasis in a xenograft model of breast cancer. Cell Death Dis. 2013, 4, 89. [Google Scholar] [CrossRef] [Green Version]
- Guzman, M.L.; Rossi, R.M.; Karnischky, L.; Li, X.; Peterson, D.R.; Howard, D.S.; Jordan, C.T. The sesquiterpene lactone parthenolide induces apoptosis of human acute myelogenous leukemia stem and progenitor cells. Blood 2005, 105, 4163–4169. [Google Scholar] [CrossRef] [PubMed]
- Zunino, S.J.; Ducore, J.M.; Storms, D.H. Parthenolide induces significant apoptosis and production of reactive oxygen species in high-risk pre-B leukemia cells. Cancer Lett. 2007, 254, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Gopal, Y.N.; Arora, T.S.; Van Dyke, M.W. Parthenolide specifically depletes histone deacetylase 1 protein and induces cell death through ataxia telangiectasia mutated. Chem. Biol. 2007, 14, 813–823. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Liu, S.; Xie, Z.; Pavlovicz, R.E.; Wu, J.; Chen, P.; Aimiuwu, J.; Pang, J.; Bhasin, D.; Neviani, P.; et al. Modulation of DNA methylation by a sesquiterpene lactone parthenolide. J. Pharmacol. Exp. Ther. 2009, 329, 505–514. [Google Scholar] [CrossRef]
- Fonrose, X.; Ausseil, F.; Soleilhac, E.; Masson, V.; David, B.; Pouny, I.; Cintrat, J.C.; Rousseau, B.; Barette, C.; Massiot, G.; et al. Parthenolide inhibits tubulin carboxypeptidase activity. Cancer Res. 2007, 67, 3371–3378. [Google Scholar] [CrossRef] [Green Version]
- Mathema, V.B.; Koh, Y.S.; Thakuri, B.C.; Sillanpa, M. Parthenolide, a sesquiterpene lactone, expresses multiple anti-cancer and anti-inflammatory activities. Inflammation 2012, 35, 560–565. [Google Scholar] [CrossRef]
- Pei, S.; Jordan, C.T. How close are we to targeting the leukemia stem cell? Best Pract. Res. Clin. Haematol. 2012, 25, 415–418. [Google Scholar] [CrossRef]
- Gach, K.; Długosz, A.; Janecka, A. The role of oxidative stress in anticancer activity of sesquiterpene lactones. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2015, 388, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Liu, L.; Lee, S.O.; Kim, Y.T.; You, K.R.; Kim, D.G. Susceptibility of cholangiocarcinoma cells to parthenolide-induced apoptosis. Cancer Res. 2005, 65, 6312–6320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.L.; Trang, K.T.; Kim, A.H.; Kim, I.H.; Lee, S.O.; Lee, S.T.; Kim, D.G.; Kim, S.W. Parthenolide suppresses tumor growth in a xenograft model of colorectal cancer cells by inducing mitochondrial dysfunction and apoptosis. Int. J. Oncol. 2012, 41, 1547–1553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yun, B.R.; Lee, M.J.; Kim, J.H.; Kim, I.H.; Yu, G.R.; Kim, D.G. Enhancement of parthenolide-induced apoptosis by a PKC-alpha inhibition through heme oxygenase-1 blockage in cholangiocarcinoma cells. Exp. Mol. Med. 2010, 42, 787–797. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.L.; Liu, Y.C.; Seo, S.Y.; Kim, S.H.; Kim, I.H.; Lee, S.O.; Lee, S.T.; Kim, D.; Kim, S.W. Parthenolide induces apoptosis in colitis-associated colon cancer, inhibiting NF-κB signaling. Oncol. Lett. 2015, 9, 2315–2342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baskaran, N.; Selvam, G.S.; Yuvaraj, S.; Abhishek, A. Parthenolide attenuates 7,12-dimethylbenz[a]anthracene induced hamster buccal pouch carcinogenesis. Mol. Cell. Biochem. 2018, 440, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Nakabayashi, H.; Shimizu, K. Involvement of Akt/NF-κB pathway in antitumor effects of parthenolide on glioblastoma cells in vitro and in vivo. BMC Cancer. 2012, 12, 453. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Liu, R.T.; Zhang, P.; Zhang, N.; Yang, C.L.; Yue, L.T.; Li, X.L.; Liu, Y.; Li, H.; Du, J.; et al. Parthenolide inhibits the initiation of experimental autoimmune neuritis. J. Neuroimmunol. 2017, 15, 154–161. [Google Scholar] [CrossRef]
- Fiebich, B.L.; Lieb, K.; Engels, S.; Heinrich, M. Inhibition of LPS-induced p42/44 MAP kinase activation and iNOS/NO synthesis by parthenolide in rat primary microglial cells. J. Neuroimmunol. 2002, 132, 18–24. [Google Scholar] [CrossRef]
- Gobrecht, P.; Andreadaki, A.; Diekmann, H.; Heskamp, A.; Leibinger, M.; Fischer, D. Promotion of functional nerve regeneration by inhibition of microtubule detyrosination. J. Neurosci. 2016, 36, 3890–3902. [Google Scholar] [CrossRef]
- Diekmann, H.; Fischer, D. Parthenolide: A novel pharmacological approach to promote nerve regeneration. Neural. Regen. Res. 2016, 11, 1566–1567. [Google Scholar] [CrossRef] [PubMed]
- Li, X.H.; Xiao, T.; Yang, J.H.; Qin, Y.; Gao, J.J.; Liu, H.J.; Zhou, H.G. Parthenolide attenuated bleomycin-induced pulmonary fibrosis via the NF-κB/Snail signaling pathway. Respir. Res. 2018, 19, 111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Heiden, K.; Cuhlmann, S.; Luong, A.; Zakkar, M.; Evans, P.C. Role of nuclear factor kappaB in cardiovascular health and disease. Clin. Sci. (Lond.). 2010, 23, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Latanich, C.A.; Toledo-Pereyra, L.H. Searching for NF-kappaB-based treatments of ischemia reperfusion injury. J. Investig. Surg. 2009, 22, 301–315. [Google Scholar] [CrossRef] [PubMed]
- Zingarelli, B.; Hake, P.W.; Denenberg, A.; Wong, H.R. Sesquiterpene lactone parthenolide, an inhibitor of IkappaB kinase complex and nuclear factor-kappaB, exerts beneficial effects in myocardial reperfusion injury. Shock 2002, 17, 127–134. [Google Scholar] [CrossRef]
- Tsai, T.Y.; Lou, S.L.; Cheng, K.S.; Wong, K.L.; Wang, M.L.; Su, T.H.; Chan, P.; Leu, Y.M. Repressed Ca2+ clearance in parthenolide-treated murine brain bEND.3 endothelial cells. Eur. J. Pharmacol. 2015, 769, 280–286. [Google Scholar] [CrossRef]
- Nasim, S.; Crooks, P.A. Antileukemic activity of aminoparthenolide analogs. Bioorg. Med. Chem. Lett. 2008, 18, 3870–3873. [Google Scholar] [CrossRef]
- Neelakantan, S.; Nasim, S.; Guzman, M.L.; Jordan, C.T.; Crooks, P.A. Aminoparthenolides as novel anti-leukemic agents: Discovery of the NF-kappaB inhibitor, DMAPT (LC-1). Bioorg. Med. Chem. Lett. 2009, 19, 4346–4349. [Google Scholar] [CrossRef]
- Guzman, M.L.; Rossi, R.M.; Neelakantan, S.; Li, X.; Corbett, C.A.; Hassane, D.C.; Becker, M.W.; Bennett, J.M.; Sullivan, E.; Lachowicz, J.L.; et al. An orally bioavailable parthenolide analog selectively eradicates acute myelogenous leukemia stem and progenitor cells. Blood 2007, 110, 4427–4431. [Google Scholar] [CrossRef]
- Carlisi, D.G.; Di Fiore, B.R.; Scerri, C.; Drago-Ferrante, R.; Tesoriere, V.G. Parthenolide and DMAPT exert cytotoxic effects on breast cancer stem-like cells by inducing oxidative stress, mitochondrial dysfunction and necrosis. Cell Death Dis. 2016, 7, 2194. [Google Scholar] [CrossRef] [Green Version]
- Song, J.M.; Qian, X.; Upadhyayya, P.; Hong, K.H.M.; Kassie, F. Dimethylaminoparthenolide, a water soluble parthenolide, suppresses lung tumorigenesis through down-regulating the STAT3 signaling pathway. Curr. Cancer Drug Targets 2014, 14, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Nakshatri, H.; Appaiah, H.N.; Anjanappa, M.; Gilley, D.; Tanaka, H.; Badve, S.; Crooks, P.A.; Mathews, W.; Sweeney, C.; Bhat-Nakshatri, P. NF-κB-dependent and -independent epigenetic modulation using the novel anti-cancer agent DMAPT. Cell Death Dis. 2015, 22, 1608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adis Insight. Available online: http://adisinsight.springer.com/drugs/800029612 (accessed on 15 January 2020).
- Morel, K.L.; Ormsby, R.J.; Klebe, S.; Sweeney, C.J.; Sykes, P.J. Parthenolide selectively sensitizes prostate tumor tissue to radiotherapy while protecting healthy tissues in vivo. Radiat. Res. 2017, 187, 501–512. [Google Scholar] [CrossRef] [PubMed]
- Morel, K.L.; Ormsby, R.J.; Klebe, S.; Sweeney, C.J.; Sykes, P.J. DMAPT is an effective radioprotector from long-term radiation-induced damage to normal mouse tissues in vivo. Radiat. Res. 2019, 192, 231–239. [Google Scholar] [CrossRef]
- Mendonca, M.; Turchan, W.; Alpucha, M.; Watson, C.N.; Estabrook, N.; Chin-Sinex, H.; Shapiro, J.B.; Imasuen-Williams, I.E.; Rangela, G.; Gilley, D.; et al. DMAPT inhibits NF-κB activity and increases sensitivity of prostate cancer cells to X-rays in vitro and in tumor xenografts in vivo. Free Radic. Biol. Med. 2017, 112, 318–326. [Google Scholar] [CrossRef]
- Li, X.; Payne, D.T.; Ampolu, B.; Bland, N.; Brown, J.T.; Dutton, M.J.; Fitton, C.A.; Gulliver, A.; Hale, L.; Hamza, D.; et al. Derivatisation of parthenolide to address chemoresistant chronic lymphocytic leukaemia. Med. Chem. Commun. 2019, 10, 1379. [Google Scholar] [CrossRef] [Green Version]
- Baranello, M.P.; Bauer, L.; Danielle, S.W.; Benoit, D. Poly (styrene-alt-maleic anhydride)-based diblock copolymer micelles exhibit versatile hydrophobic drug loading, drug-dependent release, and internalization by multidrug resistant ovarian cancer cells. Biomacromolecules 2014, 15, 2629–2641. [Google Scholar] [CrossRef]
- Karmakar, A.; Xu, Y.; Mustafa, T.; Kannarpady, G.; Bratton, S.M.; Radominska-Pandya, A.; Crooks, P.A.; Biris, A.S. Nanodelivery of parthenolide using functionalized nanographene enhances its anticancer activity. RSC Adv. 2015, 5, 2411. [Google Scholar] [CrossRef] [Green Version]
- Jin, X.; Zhou, J.; Zhang, Z.; Lv, H. The combined administration of parthenolide and ginsenoside CK in long circulation liposomes with targeted tLyp-1 ligand induce mitochondria-mediated lung cancer apoptosis. artificial cells. Artif. Cells Nanomed. Biotechnol. 2018, 46, S931–S942. [Google Scholar] [CrossRef] [Green Version]
- Darwish, N.H.E.; Sudha, T.; Godugu, K.; Bharali, D.J.; Elbaz, O.; El-Ghaffar, H.A.A.; Azmy, E.; Anber, N.; Mousa, S.A. Novel targeted nano-parthenolide molecule against NF-kB in acute myeloid leukemia. Molecules 2019, 24, 2103. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.L.; Liu, Y.C.; Park, Y.R.; Seo, S.Y.; Kim, S.H.; Kim, I.H.; Lee, S.O.; Lee, S.; Kim, D.G.; Kim, S.W. Parthenolide enhances sensitivity of colorectal cancer cells to TRAIL by inducing death receptor 5 and promotes TRAIL-induced apoptosis. Int. J. Oncol. 2015, 46, 1121–1130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holcomb, B.K.; Yip-Schneider, M.T.; Waters, J.A.; Beane, J.D.; Crooks, P.A.; Schmidt, C.M. Dimethylamino parthenolide enhances the inhibitory effects of gemcitabine in human pancreatic cancer cells. J. Gastrointest. Surg. 2012, 16, 1333–1340. [Google Scholar] [CrossRef] [PubMed]
- Yip-Schneider, M.T.; Wu, H.; Njoku, V.; Ralstin, M.; Holcomb, B.; Crooks, P.A.; Neelakantan, S.; Sweeney, C.J.; Schmidt, C.M. Effect of celecoxib and the novel anti-cancer agent, dimethylamino-parthenolide, in a developmental model of pancreatic cancer. Pancreas 2008, 37, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Yip-Schneider, M.T.; Wu, H.; Hruban, R.H.; Lowy, A.M.; Crooks, P.A.; Schmidt, C.M. Efficacy of dimethylaminoparthenolide and sulindac in combination with gemcitabine in a genetically engineered mouse model of pancreatic cancer. Pancreas 2013, 42, 160–167. [Google Scholar] [CrossRef]
- Yip-Schneider, M.T.; Wu, H.; Stantz, K.; Agaram, N.; Crooks, P.A.; Schmidt, C.M. Dimethylaminoparthenolide and gemcitabine: A survival study using a genetically engineered mouse model of pancreatic cancer. BMC Cancer 2013, 13, 194. [Google Scholar] [CrossRef] [Green Version]
- Carlisi, D.; Lauricella, M.; D’Anneo, A.; Buttitta, G.; Emanuele, S.; di Fiore, R.; Martinez, R.; Rolfo, C.; Vento, R.; Tesoriere, G. The synergistic effect of SAHA and parthenolide in MDA-MB231 breast cancer cells. J. Cell Physiol. 2015, 230, 1276–1289. [Google Scholar] [CrossRef] [Green Version]
- Lamture, G.; Crooks, P.A.; Borrelli, M.J. Actinomycin-D and dimethylamino-parthenolide synergism in treating human pancreatic cancer cells. Drug Dev. Res. 2018, 79, 287–294. [Google Scholar] [CrossRef]
- Rasmussen, U.; Broogger, C.S.; Sandberg, F. Thapsigargine and thapsigargicine, two new histamine liberators from Thapsia garganica L. Acta Pharm. Suec. 1978, 15, 133–140. [Google Scholar]
- Andersen, T.B.; López, C.Q.; Manczak, T.; Martinez, K.; Simonsen, H.T. Thapsigargin - from Thapsia L. to mipsagargin. Molecules 2015, 20, 6113–6127. [Google Scholar] [CrossRef] [Green Version]
- Doan, N.T.Q.; Paulsen, E.S.; Sehgal, P.; Møller, J.V.; Nissen, P.; Denmeade, S.R.; Isaacs, J.T.; Dionne, C.A.; Christensen, S.B. Targeting thapsigargin towards tumors. Steroids 2015, 97, 2–7. [Google Scholar] [CrossRef]
- Mohamed Ibrahim, A.M.; Martinez-Swatson, K.A.; Benkaci-Ali, F.; Cozzi, F.; Zoulikha, F.; Simonsen, H.T. Effects of gamma irradiation and comparison of different extraction methods on sesquiterpene lactone yields from the medicinal plant Thapsia garganica L. (Apiaceae). J. Appl. Res. Med. Aromat. Plants 2018, 8, 26–32. [Google Scholar] [CrossRef] [Green Version]
- Crestey, F.; Toma, M.; Christensen, S.B. Concise synthesis of thapsigargin from nortrilobolide. Tetrahedron Lett. 2015, 56, 5896–5898. [Google Scholar] [CrossRef]
- Chen, D.; Evans, P.A. A concise, efficient and scalable total synthesis of thapsigargin and nortrilobolide from (R)-(-)-carvone. J. Am. Chem. Soc. 2017, 139, 6046–6049. [Google Scholar] [CrossRef] [PubMed]
- Denmeade, S.R.; Isaacs, J.T. The SERCA pump as a therapeutic target: Making a “smart bomb” for prostate cancer. Cancer Biol. Ther. 2005, 4, 14–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, Y.; Meng, M.; Tian, Z.; Xie, F.; Yin, Q.; Dai, C.; Wang, J.; Zhang, Q.; Liu, Y.; Liu, C. Pharmacological preconditioning with the cellular stress inducer thapsigargin protects against experimental sepsis. Pharmacol. Res. 2019, 141, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Michelangeli, F.; East, J.M. A diversity of SERCA Ca2+ pump inhibitors. Biochem. Soc. Transact. 2011, 39, 789–797. [Google Scholar] [CrossRef]
- Wootton, L.L.; Michelangeli, F. The effects of the phenylalanine to valine mutation on the sensitivity of sarcoplasmic/endoplasmic reticulum Ca2+ ATPase (SERCA) Ca2+ pump isoforms 1, 2, and 3 to thapsigargin and other inhibitors. J. Biol. Chem. 2006, 281, 6970–6976. [Google Scholar] [CrossRef] [Green Version]
- Skytte, D.M.; Moller, J.V.; Liu, H.; Nielsen, H.O.; Svenningsen, L.E.; Jensen, C.M.; Olsen, C.E.; Christensen, S.B. Elucidation of the topography of the thapsigargin binding site in the sarco-endoplasmic calcium ATPase. Bioorg. Med. Chem. 2010, 18, 5634–5646. [Google Scholar] [CrossRef]
- Shiraishi, M.; Hirasawa, N.; Kobayashi, Y.; Oikawa, S.; Murakami, A.; Ohuchi, K. Participation of mitogen-activated protein kinase in thapsigargin- and TPA-induced histamine production in murine macrophage RAW 264.7 cells. Brit. J. Pharmacol. 2000, 129, 515–524. [Google Scholar] [CrossRef] [Green Version]
- Muramatsu, Y.; Maemoto, T.; Iwashita, A.; Matsuoka, N. Novel neuroprotective compound SCH-20148 rescues thymocytes and SH-SY5Y cells from thapsigargin-induced mitochondrial membrane potential reduction and cell death. Eur. J. Pharmacol. 2007, 563, 40–48. [Google Scholar] [CrossRef]
- Wang, C.; Li, T.; Tang, S.; Zhao, D.; Zhang, C.; Zhang, S.; Deng, S.; Zhou, Y.; Xiao, X. Thapsigargin induces apoptosis when autophagy is inhibited in HepG2 cells and both processes are regulated by ROS-dependent pathway. Environ. Toxicol. Pharmacol. 2016, 41, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Silva, Z.; Verissimo, T.; Videira, P.A.; Novo, C. Protein disulfide isomerases: Impact of thapsigargin treatment on their expression in melanoma cell lines. Int. J. Biol. Macromol. 2015, 79, 44–48. [Google Scholar] [CrossRef] [PubMed]
- Meldrum, D.R.; Cleveland, J.C.J.; Mitchell, M.B.; Rowland, R.T.; Banerjee, A.; Harken, A.H. Constractive priming of myocardium against ishemia-reperfusion injury. Shock 1996, 6, 238–242. [Google Scholar] [CrossRef] [PubMed]
- Karunakaran, U.; Lee, J.E.; Elumalai, S.; Moon, J.S.; Won, K.C. Myricetin prevents thapsigargin-induced CDK5-P66Shc signalosome mediated pancreatic β-cell dysfunction. Free Rad. Biol. Med. 2019, 141, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Janyou, A.; Changtam, C.; Suksamrarn, A.; Tocharus, C.; Tocharus, J. Suppression effects of O-demethyldemethoxycurcumin on thapsigargin triggered on endoplasmic reticulum stress in SK-N-SH cells. Neurotoxicology 2015, 50, 92–100. [Google Scholar] [CrossRef]
- Einbond, L.S.; Wu, H.A.; Sandu, C.; Ford, M.; Mighty, J.; Antonetti, V.; Redenti, S.; Ma, H. Digitoxin enhances the growth inhibitory effects of thapsigargin and simvastatin on ER negative human breast cancer cells. Fitoterapia 2016, 109, 146–154. [Google Scholar] [CrossRef]
- Denmeade, S.R.; Jakobsen, C.M.; Janssen, S.; Khan, S.R.; Garrett, E.S.; Lilja, H.; Christensen, S.B.; Isaacs, J.T. Prostate-specific antigen-activated thapsigargin prodrug as targeted therapy for prostate cancer. J. Nat. Cancer Inst. 2003, 95, 990–1000. [Google Scholar] [CrossRef] [Green Version]
- Zhong, W.; Chebolu, S.; Darmani, N.A. Thapsigargin-induced activation of Ca2+-CaMKII-ERK in brainstem contributes to substance P release and induction of emesis in the least shrew. Neuropharmacology 2016, 103, 195–210. [Google Scholar] [CrossRef]
- Kmonickova, E.; Harmatha, J.; Vokac, K.; Kostecka, P.; Farghali, H.; Zidek, Z. Sesquiterpene lactone trilobolide activates production of interferon-gamma and nitric oxide. Fitoterapia 2010, 81, 1213–1219. [Google Scholar] [CrossRef]
- Mahalingam, D.; Wilding, G.; Denmeade, S.; Sarantopoulas, J.; Cosgrove, D.; Cetnar, J.; Azad, N.; Bruce, J.; Kurman, M.; Allgood, V.E.; et al. Mipsagargin, a novel thapsigargin-based PSMA-activated prodrug: Results of a first-in-man phase I clinical trial in patients with refractory, advanced or metastatic solid tumours. Br. J. Cancer 2016, 114, 986–994. [Google Scholar] [CrossRef]
- Mahalingam, D.; Peguero, J.; Cen, P.; Arora, S.P.; Sarantopoulos, J.; Rowe, J.; Allgood, V.; Tubb, B.; Campos, L. A phase II, multicenter, single-arm study of mipsagargin (G-202) as a second-line therapy following sorafenib for adult patients with progressive advanced hepatocellular carcinoma. Cancers 2019, 11, 833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, H.H.; Kim, S.G.; Kim, M.J.; Lee, J.; Choi, B.K.; Jin, M.H.; Lee, E. Suppressive effect of tomentosin on the production of inflammatory mediators in RAW264.7 cells. Biol. Pharm. Bull. 2014, 37, 1177–1183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merghoub, N.; El Btaouri, H.; Benbacer, L.; Gmouh, S.; Trentesaux, C.; Brassart, B.; Attaleb, M.; Madoulet, C.; Wenner, T.; Amzazi, S.; et al. Tomentosin induces telomere shortening and caspase-dependant apoptosis in cervical cancer cells. J. Cell. Biochem. 2017, 118, 1689–1698. [Google Scholar] [CrossRef]
- Göpfert, J.C.; Heil, N.; Conrad, J.; Spring, O. Cytological development and sesquiterpene lactone secretion in capitate glandular trichomes of sunflower. Plant Biol. 2005, 7, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.M.; Lee, J.; Nam, M.J.; Choi, Y.S.; Park, S.H. Tomentosin displays anti-carcinogenic effect in human osteosarcoma MG-63 cells via the induction of intracellular reactive oxygen species. Int. J. Mol. Sci. 2019, 20, 1508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Laurentis, N.; Losacco, V.; Milillo, M.A.; Lai, O. Chemical investigations of volatile constituents of Inula viscosa (L.) Aiton (Asteraceae) from different areas of Apulia, Southern Italy. Delpinoa 2002, 44, 115–119. [Google Scholar]
- Bar-Shalom, R.; Bergman, M.; Grossman, S.; Azzam, N.; Sharvit, L.; Fares, F. Inula Viscosa extract inhibits growth of colorectal cancer cells in vitro and in vivo through induction of apoptosis. Front. Oncol. 2019, 9, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Cohen, Y.; Wang, W.; Ben-Daniel, B.H.; Ben-Daniel, Y. Extracts of Inula viscosa control downy mildew of grapes caused by Plasmopara viticola. Phytopathology 2006, 96, 417–424. [Google Scholar] [CrossRef] [Green Version]
- Ahern, J.R.; Whitney, K.D. Stereochemistry affects sesquiterpene lactone bioactivity against an herbivorous grasshopper. Chemoecology 2014, 24, 35–39. [Google Scholar] [CrossRef]
Figure 1. Basic chemical structure of each of the sesquiterpene lactone subclasses: eudesmanolide, guaianolide, pseudoguaianolide, germacranolide, and xanthanolide.
Figure 1. Basic chemical structure of each of the sesquiterpene lactone subclasses: eudesmanolide, guaianolide, pseudoguaianolide, germacranolide, and xanthanolide.

Figure 2. Structure of alantolactone (1).

Figure 3. Structure of arglabin (2).

Figure 4. Structure of costunolide (3).

Figure 5. Structure of cynaropicrin (4).

Figure 6. Structure of helenalin (5).

Figure 7. Structure of inuviscolide (6).

Figure 8. Structures of lactucin (7) and its derivatives lactupicrin (8) and lactucopicrin (9).

Figure 9. Structure of parthenolide (10) and its derivatives DMAPT (11) and HMPPPT (12).

Figure 10. Structure of thapsigargin (13) and its derivative mipsagargin (14).

Figure 11. Structure of tomentosin (15).

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Moujir, L.; Callies, O.; Sousa, P.M.C.; Sharopov, F.; Seca, A.M.L. Applications of Sesquiterpene Lactones: A Review of Some Potential Success Cases. Appl. Sci. 2020, 10, 3001. https://doi.org/10.3390/app10093001
AMA Style
Moujir L, Callies O, Sousa PMC, Sharopov F, Seca AML. Applications of Sesquiterpene Lactones: A Review of Some Potential Success Cases. Applied Sciences. 2020; 10(9):3001. https://doi.org/10.3390/app10093001
Chicago/Turabian StyleMoujir, Laila, Oliver Callies, Pedro M. C. Sousa, Farukh Sharopov, and Ana M. L. Seca. 2020. "Applications of Sesquiterpene Lactones: A Review of Some Potential Success Cases" Applied Sciences 10, no. 9: 3001. https://doi.org/10.3390/app10093001
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.