


Abstract
FIP200 is a novel protein inhibitor for focal adhesion kinase (FAK), which binds to FAK directly and inhibits its kinase activity and associated cellular functions, such as cell adhesion, spreading, and motility in fibroblasts. Here we show that FIP200 inhibits G1-S phase progression, proliferation, and clonogenic survival in human breast cancer cells. Consistent with the G1 arrest induced by FIP200, we found that FIP200 increased p21 and decreased cyclin D1 protein levels in breast cancer cells. In addition, FIP200 significantly induced p21 promoter activity in MCF-7 cells and this response was abolished upon deletion of p53 binding sites within p21 promoter. Furthermore, we found that FIP200 could interact with exogenous and endogenous p53 protein and significantly increase its half-life compared with the control cells. We also found that the NH2-terminal 154 residues of FIP200 were sufficient to mediate p53 interaction and G1 arrest in cells. The increase in p53 half-life correlated with the increased phosphorylation at Ser15 and decreased proteasomal degradation via ubiquitin and Hdm2-independent mechanism. Stabilization of p53 by FIP200 could be partially reversed by NQO1 inhibitor, dicoumarol. In contrast to p53, FIP200 decreased cyclin D1 protein half-life by promoting proteasome-dependent degradation of cyclin D1. In summary, our results suggest that FIP200 increases p21 protein levels via stabilization of its upstream regulator p53 and decreases cyclin D1 protein by promoting its degradation. Both effects are critical for FIP200-induced G1 arrest and may contribute to the putative antitumor activities of FIP200 in breast cancer.
Introduction
FIP200 (focal adhesion kinase, FAK, family kinase interacting protein of 200 kDa) is a novel protein inhibitor for FAK, which directly interacts with the kinase domain of FAK and inhibits its activity and associated cellular functions, such as cell adhesion, spreading, and motility in fibroblasts (1). A potential role for FIP200 in breast cancer was suggested by its localization to the chromosome 8q11, containing several loci of putative tumor suppressor genes (2). The loss of heterozygosity for this region has been associated with breast cancer (3). Furthermore, a close correlation was found for the expression levels of FIP200 and RB1 tumor suppressor gene in several tissues and cell lines (4). In addition, 20% of primary breast cancers screened in a recent study contained deletion mutations of FIP200 gene (5). Although these data suggest that FIP200 may play an important role in breast cancer, the mechanisms of its antitumor activities are largely unknown.
Cell cycle progression is controlled by cyclins and their catalytic partners, cyclin-dependent kinases (CDK). In mammalian cells G1-S phase progression is regulated by D and E family of cyclins, which interact with CDK4/6 and CDK2, respectively, to promote pRb phosphorylation and inactivation, which is a critical step for G1 progression (6). Cyclin D1 plays an important role in mitogen-activated G1 progression and is overexpressed in about 50% of mammary carcinomas (7).
In addition, CDKs can be regulated by another family of proteins called cyclin kinase inhibitors (CKI), which could bind to the active cyclin/CDK complexes and inhibit their kinase activity (8). The most common inhibitors of the G1 CDKs are p21, p27, and p16. p21 is an important target of different signaling pathways, which induce G1 arrest, cell differentiation, and senescence (9). The expression of p21 is usually controlled at the transcriptional levels, although some posttranscriptional mechanisms of p21 regulation, such as mRNA and protein stability, have been described (10–12). p21 transcription is mainly regulated by p53-dependent and p53-independent mechanisms. p53-dependent mechanism is mediated by p53 tumor suppressor protein that binds and activates p21 promoter in response to various cellular stresses (reviewed in ref. 13). In addition, a number of agents, such as retinoic acid, phorbol 12-myristate 13-acetate, histone deacetylase inhibitors, and transforming growth factor-β, can regulate p21 transcription by p53-independent manner (13).
In the present study, we show that FIP200 inhibits G1-S phase progression, proliferation, and clonogenic survival in human breast cancer cells. We identified p21 and cyclin D1 as important mediators of FIP200-induced cell cycle arrest and investigate the molecular mechanism of their regulation by FIP200.
Materials and Methods
Cell culture and transfection. MCF-7 and MDA-MB-231 human breast cancer cells and MCF10A-immortalized nontumorigenic mammary epithelial cells were a generous gift from Dr. J. Strobl (Virginia College of Osteopathic Medicine, Blacksburg, VA). p21 null and p21+/+ mouse embryo fibroblasts (MEFs) were kindly provided by Dr. C. Deng (National Institutes of Diabetes, Digestive and Kidney Diseases, NIH). p53 null Saos-2 cells were kindly provided by Dr. Y. Shi (Department of Pathology, Harvard Medical School, Boston, MA). MCF-7, MDA-MB-231, U2OS, Saos-2, HEK293, NIH 3T3, p21 null, and p21+/+ MEF were grown in DMEM supplemented with 10% fetal bovine serum (FBS) or calf serum (for NIH3T3). MCF10A were grown in MEGM supplemented with bovine pituitary extract, hEGF, insulin, hydrocortisone (Clonetics Laboratories, Inc., San Jose, CA) and 100 ng/mL cholera toxin. Cells were maintained at 37°C and 5% CO2.
For the transfections, cells were seeded on sterile tissue culture plates at the density to give about 60% confluent monolayers 24 hours later. The transfections were carried out with LipofectAMINE and PLUS transfection reagents (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions.
Plasmids. pKH3-HA-FIP200, pKH3-HA-CT (1), pKH3-HA-MD (1), pKH3-HA-Grb7 (14), and pSG5-FLAG-FIP200 (15) plasmids were described previously. pKH3-HA-NT and pKH3-HA-N154 were constructed by PCR amplification of the first 640 and 154 NH2-terminal amino acids of FIP200 respectively with flanking SmaI and EcoRI sites and subcloning into corresponding restriction sites of the pKH3 vector containing triple HA tag. pEGFP-C3 vector was purchased from Clontech Laboratories. pCMV/Hdm2 and pCMV/p53 vectors were kindly provided by Dr. Y. Shi. The expression vector encoding human cyclin D1 (pRK5-D1) was a generous gift from Dr. T. Hunter (Salk Institute, San Diego, CA). Cyclin D1-luciferase reporter plasmids and p21-luciferase reporter plasmids were kindly provided by Dr. R. Pestell (Department of Oncology, Georgetown University, Washington, DC) and Dr. X-F. Wang (Department of Pharmacology, Duke University Medical Center, Durham, NC), respectively. pKH3-HA-p53, pHAN-Myc-p53, pHAN-Myc-p53(1-100), pHAN-Myc-p53(101-300), and pHAN-Myc-p53(301-393) plasmids were constructed by PCR amplification of the full-length human p53 cDNA or corresponding fragments from pCMV/p53 and subcloning into the vectors with the corresponding epitopes. Myc-tagged ubiquitin expression vector (pHAN-Myc-Ub) was constructed by PCR amplification and subcloning of the Ub cDNA into pHAN-Myc vector. pEGFP-FIP200 vector encoding FIP-GFP was constructed by digesting pEGFP-C3 with EcoRI, filling in with T4 polymerase, digesting with SalI, and finally ligating with the full-length FIP200 fragment excised from pSG5-FLAG-FIP200 by EcoRV and SalI digestion. pGEX-2T-N154 vector expressing GST-N154 fusion protein was constructed by PCR amplification of the first 154 NH2-terminal amino acids of FIP200 with flanking SmaI and EcoRI sites and subcloning into corresponding restriction sites of the pGEX-2T vector (Amersham Biosciences, Piscataway, NJ). A recombinant wild-type human p53 protein was obtained from BD PharMingen (San Diego, CA).
Antibodies/chemicals. Polyclonal antibodies against COOH-terminal FIP200 (clone 33) were previously described (15). Monoclonal antibodies against Mdm2 (SMP14) and Myc-probe (9E10) and polyclonal antibodies against p53 (FL-393), p21 (C-19), cyclin D1 (M-20), pRb (C-15), poly(ADP-ribose) polymerase (H-250), HA-probe (Y-11) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Phospho-p53 antibody sampler kit (9919) was purchased from Cell Signalling Technology (Beverly, MA) and monoclonal antibodies against vinculin (v-4505), bromodeoxyuridine (BrdUrd, B-2531) and anti-HA-agarose conjugates from Sigma-Aldrich Co. (St. Louis, MO). Phospho-specific antibodies against Tyr397 of FAK (44-624) was purchased from Biosource International (Camarillo, CA). MG-132 and dicoumarol were purchased from Sigma-Aldrich and dissolved in DMSO and 0.13 N NaOH water, respectively.
Preparation of the recombinant adenoviruses. The recombinant Ad-FIP200 was generated using AdEasy-1 system (Stratagene, La Jolla, CA) according to the manufacturer's instructions. Briefly, pKH3-HA-FIP200 vector was digested with EcoRI enzyme, treated with T4 polymerase to fill in the overhangs and then digested with SalI to excise the full-length human FIP200 cDNA with the NH2-terminal HA tag. This fragment was then subcloned into pAd-trackCMV shuttle vector digested with EcoRV and SalI. To get the recombinant adenovirus plasmid, the PmeI-linarized pAd-trackCMV-FIP200 plasmid was transfected into BJ5183 cells with pAdEasy-1 vector. Lastly, the PacI-linerazied pAdEasy-1-FIP200 plasmid was transfected into Ad293 cells using LipofectAMINE to generate Ad-FIP200. Ad-GFP adenovirus was generated using similar strategy. Ad-FIP200siRNA virus was generated in two steps. First, a DNA vector-based RNAi approach (16) was used to screen for the small interfering (siRNA) sequence that could efficiently (>70%) decrease endogenous FIP200 protein. The selected sequences were excised from the vector by XbaI enzyme and subcloned into pAd-track shuttle vector digested with XbaI. Ad-FIP200siRNA virus was then constructed using the same approach as described for the Ad-FIP200. The Ad-GFPsiRNA and Ad-FAKsiRNA viruses were constructed using similar approach as described for Ad-FIP200siRNA. All viruses (Ad-GFP, Ad-FIP200, Ad-GFPsiRNA, Ad-FAKsiRNA, and Ad-FIP200siRNA) were amplified using Ad 293 cells and the viral titers were determined using Adeno-X rapid titer kit (BD Biosciences, San Jose, CA). Optimal viral titer (5-50 moi) was determined for each cell line to give almost 100% infection efficiency (based on GFP expression) with no detectable cell toxicity. Cells were infected for 48 to 72 hours before the experiments.
Bromodeoxyuridine incorporation assay. Cells were seeded in 6-well plates and 24 hours later transfected with vectors encoding HA-FIP200 or irrelevant protein HA-Grb7 or mock transfected. Following transfections, cells were maintained in low serum conditions for additional 20 to 24 hours and replated on sterile glass coverslips in 10% FBS and 150 μmol/L BrdUrd. Sixteen to 20 hours later, cells were fixed in 3.7% formaldehyde, permeabilized with 0.5% Triton X-100, and cellular DNA was digested with 0.5 units/μL DNase I (New England Biolabs, Beverly, MA) for 30 minutes at 37°C. Cells were then processed for indirect immunoflourescence with rabbit anti-HA and mouse anti-BrdUrd primary antibodies (1:200) followed by FITC-conjugated anti-rabbit (green) and Texas red–conjugated anti-mouse (red) secondary antibodies (1:200). At least 100 to 400 cells from multiple fields were scored for each transfection.
Cell count and colony formation assays. MCF-7 cells infected for 48 hours with 10 moi of Ad-GFP (control) or Ad-FIP200 were replated in 10% FBS-containing medium in 6-well plates at the density of 1 × 105 and 1,000 per well for cell count and colony formation assay, respectively. On the next day, 10 moi of Ad-GFP or Ad-FIP200 were added to the cells to maintain the expression of GFP and FIP200. Cell number was determined 5 days later using hemocytometer and trypan blue (for cell viability) or cells were fixed and stained with crystal violet 14 days later to visualize colony formation.
Reporter gene assay. MCF-7 or MDA-MB-231 cells were seeded in 6-well plates at the density to give about 60% confluent monolayers on the next day. Cells were cotransfected with empty vector of FIP200 expression vector and cyclin D1-luc or p21-luc reporter plasmids. Twenty-four hours later, cells were lysed and luciferase activity was analyzed using Luciferase assay system kit (E1500) from Promega (Madison, WI) and luminometer (EG&G Berthold Lumat LB 9507).
Western blot. Whole cell lysates were collected by rinsing cell monolayer with PBS and scraping cells into the lysis buffer [1% SDS, 10 mmol/L Tris (pH 7.4)]. Cellular DNA was sheered by passing the lysates several times through the 22-gauge needle, after which the lysates were immediately boiled for 5 minutes, cooled on ice, and protease inhibitors (1 mmol/L phenylmethylsulfoxide, 10 μg/mL aprotinin, 2 μg/mL leupeptin) and 3 mmol/L Na3VO4 were added. Supernatants were collected after centrifugation and protein concentration was determined by bicinchoninic acid assay (Pierce, Rockford, IL). Twenty-five to 50 μg of proteins were resuspended in SDS-PAGE sample buffer, boiled, resolved on SDS-PAGE, and transferred into nitrocellulose membrane. Membranes were blotted with primary antibody in TBST [50 mmol/L Tris (pH 7.4), 150 mmol/L NaCl, 0.05% Tween 20] plus 5% nonfat dry milk at 4°C overnight followed by TBST washing and incubation with horseradish peroxidase–conjugated appropriate secondary antibodies. Specific signals were obtained using the Amersham enhanced chemiluminescent detection system (Arlington Heights, IL).
Immunoprecipitations. Cell monolayers were rinsed with ice-cold PBS and scraped into the ice-cold NP40 lysis buffer [20 mmol/L Tris (pH 8.0), 137 mmol/L NaCl, 1% NP40, 10% glycerol] supplemented with protease inhibitors (1 mmol/L phenylmethylsulfoxide, 10 μg/mL aprotinin, 10 μg/mL leupeptin) and 3 mmol/L Na3VO4. Lysates were incubated on ice for 20 minutes, cleared by centrifugation, and protein concentration was determined by bicinchoninic acid assay (Pierce). Four hundred to 1,000 μg of proteins were briefly precleared with protein-A beads and then used for immunoprecipitation reaction with either monoclonal anti-HA agarose conjugates (A-2095, Sigma, St. Louis, MO) or antibodies to the specific proteins at 4°C overnight followed by incubation with protein-A-Sepharose for 2 hours to collect immune complexes. Finally, beads were washed with NP40 buffer five times, resuspended in SDS-PAGE sample buffer, boiled, resolved on SDS-PAGE, and analyzed by Western blot as described above.
In vitro protein binding assay. Glutathione S-transferase (GST) alone or GST-N154 fusion protein were expressed in Escherichia coli bacteria by incubation with 0.2 mmol/L isopropyl-l-thio-B-d-galactopyranoside for 4 hours at 37 °C. After sonication of the bacterial pellet, GST and GST-N154 were purified with glutathione agarose beads. For in vitro binding assay, purified human recombinant wild-type p53 protein (0.6 μg) was precleared with GST beads for 1 hour at 4°C and incubated with 2 μg of GST or GST-N154 in 500 μL of NP40 buffer for 2 hours at 4°C. GST fusion proteins were equalized for the amount of glutathione agarose beads. After washing with NP40 buffer, bound proteins were analyzed by SDS-PAGE followed by immunoblotting with anti-p53 antibody.
Subcellular fractionation assay. Subcellular fractionation assay was done as described previously (17).
Results
FIP200 inhibits G1-S phase progression, proliferation, and clonogenic survival in MCF-7 human breast cancer cells. To investigate the potential antitumor function of FIP200 in breast cancer, we first examined the effect of FIP200 on cell cycle progression, proliferation, and clonogenic survival in MCF-7 cells. As shown in Fig. 1A, the expression of FIP200 by transient transfection significantly decreased BrdUrd incorporation compared with the mock-transfected cells or cells expressing unrelated protein (Grb7), suggesting that FIP200 inhibits G1-S phase transition in MCF-7 cells. We next examined effect of FIP200 on cell proliferation or clonogenic survival. Due to the low tranfection efficiency in MCF-7 cells, we used adenovirus to achieve almost 100% expression of FIP200 in these cells. Our results show that FIP200 dramatically decreased both cell number and clonogenic survival in MCF-7 cells compared with the cells infected with the same titer of GFP expressing virus as a control (Fig. 1B). These results suggest that ectopic expression of FIP200 can inhibit G1-S phase transition, proliferation, and clonogenic survival in MCF-7 human breast cancer cells.

Effect of FIP200 on G1-S phase progression, proliferation and cell cycle regulatory proteins in different cell lines. A, MCF-7 cells were transfected with vectors expressing an irrelevant control protein (HA-Grb7) or HA-FIP200, or mock transfected. After indirect immunofluorescence, the transfected cells (green) were analyzed for the BrdUrd incorporation (red nuclei) as described in Materials and Methods. The number of transfected cells, which incorporated BrdUrd is expressed as a percent of total number of transfected cells scored (right). B, MCF-7 cells infected for 48 hours with 10 moi of Ad-GFP (control) or Ad-FIP200 were replated in 10% FBS-containing medium in 6-well plates at the density of 1 × 105 (right) or 1,000 (left) per well. On the next day, 10 moi of Ad-GFP or Ad-FIP200 were added to the cells to maintain the expression of GFP and FIP200. Cell number was counted 5 days later for cell proliferation assay (right) or cells were stained with crystal violet 14 days later to visualize the colony formation (left). Columns, means of three to five independent experiments; bars, ±SE. C, MCF-7 or MCF10A cells plated in 60-mm plates were infected with 10 moi of Ad-GFP (G) or Ad-HA-FIP200 (F) for 48 hours. Fifty micrograms of the WCL were separated on SDS-PAGE and analyzed by Western blot with specific antibodies against HA, cyclin D1, p21, pRb, or vinculin (loading control). D, top, NIH3T3, p21−/−, or p21+/+ MEFs were transfected with vectors expressing the irrelevant control protein (HA-Grb7) or HA-FIP200. BrdUrd incorporation was analyzed as described for Fig. 1A. Percentage of BrdUrd-positive cells in FIP200 samples is expressed relative to the control samples (Grb7) set to 100%. Bottom, NIH3T3 cells were cotransfected with empty vector (V) or vector expressing FIP200 (F) ± vector expressing cyclin D1 (D1) as indicated. Vector expressing β-gal was also included in all samples. The total amount of DNA was normalized to 2 μg with the empty vector. The number of β-gal-positive cells, which incorporated BrdUrd is expressed as percentage of total number of β-gal-positive cells scored for the each sample. Columns, means of three independent experiments; bars, ±SE.
Effect of FIP200 on G1-S phase progression, proliferation and cell cycle regulatory proteins in different cell lines. A, MCF-7 cells were transfected with vectors expressing an irrelevant control protein (HA-Grb7) or HA-FIP200, or mock transfected. After indirect immunofluorescence, the transfected cells (green) were analyzed for the BrdUrd incorporation (red nuclei) as described in Materials and Methods. The number of transfected cells, which incorporated BrdUrd is expressed as a percent of total number of transfected cells scored (right). B, MCF-7 cells infected for 48 hours with 10 moi of Ad-GFP (control) or Ad-FIP200 were replated in 10% FBS-containing medium in 6-well plates at the density of 1 × 105 (right) or 1,000 (left) per well. On the next day, 10 moi of Ad-GFP or Ad-FIP200 were added to the cells to maintain the expression of GFP and FIP200. Cell number was counted 5 days later for cell proliferation assay (right) or cells were stained with crystal violet 14 days later to visualize the colony formation (left). Columns, means of three to five independent experiments; bars, ±SE. C, MCF-7 or MCF10A cells plated in 60-mm plates were infected with 10 moi of Ad-GFP (G) or Ad-HA-FIP200 (F) for 48 hours. Fifty micrograms of the WCL were separated on SDS-PAGE and analyzed by Western blot with specific antibodies against HA, cyclin D1, p21, pRb, or vinculin (loading control). D, top, NIH3T3, p21−/−, or p21+/+ MEFs were transfected with vectors expressing the irrelevant control protein (HA-Grb7) or HA-FIP200. BrdUrd incorporation was analyzed as described for Fig. 1A. Percentage of BrdUrd-positive cells in FIP200 samples is expressed relative to the control samples (Grb7) set to 100%. Bottom, NIH3T3 cells were cotransfected with empty vector (V) or vector expressing FIP200 (F) ± vector expressing cyclin D1 (D1) as indicated. Vector expressing β-gal was also included in all samples. The total amount of DNA was normalized to 2 μg with the empty vector. The number of β-gal-positive cells, which incorporated BrdUrd is expressed as percentage of total number of β-gal-positive cells scored for the each sample. Columns, means of three independent experiments; bars, ±SE.
FIP200 increases p21 and decreases cyclin D1 and hyperphosphorylated pRb protein levels in mammary epithelial cells. To understand the molecular mechanisms of FIP200-induced G1 arrest, we next examined the effect of FIP200 on the expression of important regulators of the G1-S phase transition. We found that FIP200 increased p21 and decreased cyclin D1 protein levels in MCF-7 and MCF10A (immortalized mammary epithelial) cells (Fig. 1C). The changes in p21 and cyclin D1 proteins were paralleled by the decrease in phosphorylation of their downstream target, pRb, which is a critical step for G1 arrest (Fig. 1C). In contrast, FIP200 did not affect protein levels of other important regulators of G1 progression, such as cyclin E, p27, and p16 (data not shown). Our data suggest that G1 arrest by FIP200 is associated with increase in p21 and decrease in cyclin D1 proteins leading to decreased phosphorylation of pRb.
Regulation of p21 and cyclin D1 proteins is critical for FIP200-induced cell cycle arrest. We then evaluated the potential importance of p21 and cyclin D1 regulation in FIP200-induced G1 arrest of the cell cycle. We first tested the role of p21 by using p21 null and wild-type mouse embryo fibroblasts. We found that FIP200 inhibited G1-S phase transition in both p21+/+ MEF and NIH3T3 fibroblasts, both of which have wild-type p21 (Fig. 1D , top). In contrast, FIP200 failed to induce G1 arrest in p21−/− MEF. These results strongly suggest that p21 is critical for FIP200-induced G1 arrest.
Next, we examined the role of cyclin D1 in FIP200-induced G1 arrest by testing if ectopic expression of cyclin D1 could rescue the cell cycle arrest by FIP200. NIH3T3 cells were cotranfected with empty vector or FIP200 expressing vector together with cyclin D1 and β-gal expressing vectors. The positively transfected cells (as marked by β-gal staining) were scored for BrdUrd incorporation. Figure 1D (bottom) shows that ectopic expression of cyclin D1 could rescue the cell cycle arrest by FIP200 to the levels seen in the control cells. Together, these data suggest that regulation of both p21 and cyclin D1 is important for FIP200-induced G1 arrest. Therefore, we next focused on elucidating the molecular mechanisms of p21 and cyclin D1 regulation by FIP200.
FIP200 activates p21 promoter in a p53-dependent manner. p21 expression is usually controlled at the transcriptional levels, although some posttranscriptional mechanism of p21 regulation have been described (10–12). p21 promoter is regulated by two main mechanisms: p53-dependent, mediated by p53 tumor suppressor protein, which binds to and activates p21 promoter in response to various cellular stresses and p53-independent, mediated by a number of factors that modulate p21 promoter activity by binding to the specific responsive elements within p21 promoter. To examine, whether the increase in p21 protein levels by FIP200 is due to the activation of p21 promoter, we used transient reporter gene assay. As shown in Fig. 2B, (left), FIP200 significantly (about 3.5-fold) increased p21 promoter activity in MCF-7 human breast cancer cells. The activation of p21 promoter by FIP200 was p53 dependent, because deletion of both p53-binding sites within p21 promoter abolished this response. Surprisingly, we found that FIP200 could also increase p21 promoter activity, albeit to a much lower extent, in MDA-MB-231 human breast cancer cells (Fig. 2B , right), which have nonfunctional p53 protein. Deletion of p53 binding region did not affect p21 promoter activation by FIP200 in this cell line. In summary, our data suggest that FIP200 activates p21 promoter predominantly by p53-dependent mechanism although some minor p53-independent pathway could also exist.

Functional p53 is critical for the activation of p21 promoter and cell cycle arrest by FIP200. A, a schematic diagram of the human p21 promoter-luciferase reporter constructs. Two p53-binding sites (open boxes). B, MCF-7 (left) or MDA-231 (right) cells seeded in 6-well plates were cotransfected with empty vector (control) or FIP200 expressing vector together with vector expressing human p21 promoter (with or without p53-binding sites) fused to the luciferase reporter gene. Twenty-four hours later, luciferase activity in cell lysates was analyzed as described in Materials and Methods. Luciferase activity in FIP200-transfected cells is expressed as percentage of that in control cells. Columns, means of three independent experiments; bars, ±SE. C, MCF-7, U2OS, MDA-231, or Saos-2 cells were infected with Ad-GFP (G) or Ad-HA-FIP200 (F) and analyzed by Western blot as described for Fig. 1C with specific antibodies against HA, p53, or p21. D, MCF-7, MDA-231, or Saos-2 cells were transfected with vectors expressing the irrelevant control protein (HA-Grb7) or HA-FIP200. BrdUrd incorporation was analyzed as described for Fig. 1A. Percentage of BrdUrd-positive cells in FIP200 samples is expressed relative to the control samples (Grb7) set to 100%. In coexpression experiment, Saos-2 cells were cotransfected with vector expressing HA-Grb7 or HA-FIP200 plus vector expressing p53 at the 2:1 ratio. Columns, mean of three independent experiments; bars, ±SE.
Functional p53 is critical for the activation of p21 promoter and cell cycle arrest by FIP200. A, a schematic diagram of the human p21 promoter-luciferase reporter constructs. Two p53-binding sites (open boxes). B, MCF-7 (left) or MDA-231 (right) cells seeded in 6-well plates were cotransfected with empty vector (control) or FIP200 expressing vector together with vector expressing human p21 promoter (with or without p53-binding sites) fused to the luciferase reporter gene. Twenty-four hours later, luciferase activity in cell lysates was analyzed as described in Materials and Methods. Luciferase activity in FIP200-transfected cells is expressed as percentage of that in control cells. Columns, means of three independent experiments; bars, ±SE. C, MCF-7, U2OS, MDA-231, or Saos-2 cells were infected with Ad-GFP (G) or Ad-HA-FIP200 (F) and analyzed by Western blot as described for Fig. 1C with specific antibodies against HA, p53, or p21. D, MCF-7, MDA-231, or Saos-2 cells were transfected with vectors expressing the irrelevant control protein (HA-Grb7) or HA-FIP200. BrdUrd incorporation was analyzed as described for Fig. 1A. Percentage of BrdUrd-positive cells in FIP200 samples is expressed relative to the control samples (Grb7) set to 100%. In coexpression experiment, Saos-2 cells were cotransfected with vector expressing HA-Grb7 or HA-FIP200 plus vector expressing p53 at the 2:1 ratio. Columns, mean of three independent experiments; bars, ±SE.
Functional p53 is critical for induction of p21 and G1 arrest by FIP200. To understand the mechanism of p53-dependent activation of p21 promoter by FIP200, we next examined effect of FIP200 on p53 protein. We found that FIP200 increased p53 protein levels in MCF-7 and U2OS cancer cell lines with wild-type p53 but not in MDA-MB-231 (mutant p53) and Saos-2 (p53 null) cells (Fig. 2C). High levels of mutant p53 protein in MDA-231 cells is consistent with other reports and could be due to the compensatory overexpression of the nonfunctional protein or failure to transactivate its negative regulator Hdm2 (18). As expected, the increase in p53 protein in response to FIP200 led to the significant increase in the levels of p53 transcriptional target, p21, in both MCF-7 and U2OS cells, but not p53 null Saos-2 cells. Interestingly, we found a slight increase in p21 protein levels in MDA-MB-231 cells as well, consistent with the minor activation of p21 promoter by FIP200 in MDA-MB-231 cells (Fig. 2B , right).
To test if functional p53 is critical for FIP200-induced G1 arrest, we next examined effect of FIP200 on BrdUrd incorporation in cells with different p53 status. As shown in Fig. 2D, FIP200 inhibited G1-S phase progression in MCF-7 (wild-type p53) but not Saos-2 (null p53) cells. In addition, we found that FIP200 caused G1 arrest in Saos-2 cells after ectopic reexpression of wild-type p53 in these cells (Fig. 2D). In MDA-MB-231 cells FIP200 caused only minor inhibition of cell cycle progression, which is consistent with minor p53-independent p21 induction by FIP200 in these cells (Fig. 2B , right and C). Together, these results suggest that functional p53 is critical for p21 induction and cell cycle arrest by FIP200.
Association between FIP200 and p53 proteins and its role in G1 arrest by FIP200. In attempt to elucidate the mechanisms of p53 up-regulation by FIP200, we tested whether FIP200 interacts with p53 and modulates its stability. We first looked at the interaction between exogenous p53 and FIP200 by overexpressing both proteins in HEK293 cells. Our results show that p53 coimmunoprecipitates with FIP200 in lysates from the cells expressing HA-FIP200 but not control cells expressing the empty HA vector (Fig. 3A,, left). Similarly, FIP200 can be precipitated with p53 in lysates from cells expressing HA-p53 but not empty HA-vector (Fig. 3A,, right). Furthermore, we found that endogenous FIP200 coimmunoprecipitates with endogenous p53 in MCF-7 and U2OS cell lysates using specific anti-p53 antibody but not control rabbit IgG (Fig. 3B,, left). We also tested the localization of ectopically expressed FIP-GFP and endogenous FIP200 in U2OS cells using fluorescence microscopy and subcellular fractionation, respectively. Both results show the presence of FIP200 in both cytoplasm and nucleus (Fig. 3C) and consistent with the possibility of FIP200 interaction with p53 in the nucleus.
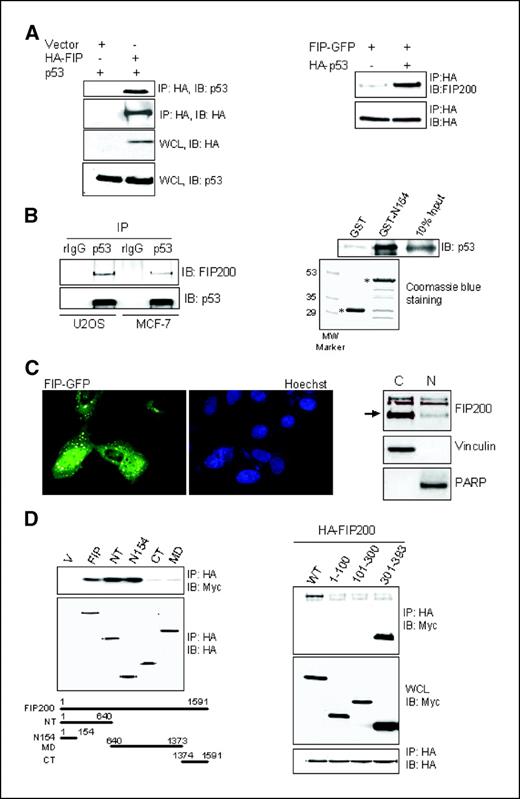
Association between FIP200 and p53. A, left, HEK293 cells were cotransfected with empty HA vector or vector expressing HA-FIP200 and Myc-p53. Twenty-four hours posttransfection, 400 μg of whole cell lysates (WCL) were incubated with HA-conjugated agarose beads as described in Materials and Methods. The immunoprecipitates (IP) were analyzed by Western blot with anti-p53 antibodies to detect p53 associated with FIP200. Equal expression of the expressed proteins in whole cell lysates is also shown. Right, 293 cells expressing FIP-GFP and HA-p53 or an empty HA vector were incubated with HA agarose beads to precipitate p53. The associated FIP200 was analyzed by Western blot with FIP200-specific antibody. B, left, 1 mg of the whole cell lysates from U2OS or MCF-7 cells were incubated with anti-p53 antibody or rabbit IgG as a control, following by incubation with protein-A-beads. The specific association between endogenous FIP200 and p53 was detected by Western blot analysis with anti-FIP200 antibody (clone 33). The amount of immunoprecipitated p53 is also shown. Right, 0.6 μg of purified human recombinant wild-type p53 protein was incubated with 2 μg of GST or GST-N154 as described in Materials and Methods for in vitro binding assay. The bound p53 was resolved on SDS-PAGE and analyzed by Western blot with anti-p53 antibody. The equal amount of GST and GST-N154 (asterisks) are shown by Coomassie blue staining of the gel with the duplicate samples. C, left, U2OS cells transiently transfected with FIP-GFP expression vector were stained with Hoechst nuclear stain (blue) and analyzed by fluorescence microscopy to detect FIP-GFP localization in cells (green). Right, U2OS cells were processed for nuclear/cytoplasmic fractionation. Fifteen micrograms of each fraction were resolved on SDS-PAGE and analyzed by Western blotting with antibodies against FIP200 (arrow on the right), vinculin (cytoplasmic protein), and poly(ADP-ribose) polymerase (PARP, nuclear protein). D, left, HEK293 cells were cotransfected with empty HA vector (V) or vector expressing HA-FIP200 or its deletion mutants and Myc-p53. Protein binding was analyzed as described for Fig. 3A (left). Right, HEK293 cells were cotransfected with HA-FIP200 and Myc-tagged full length p53 or its different fragments as indicated. Protein binding was analyzed as described for Fig. 3A (left).
Association between FIP200 and p53. A, left, HEK293 cells were cotransfected with empty HA vector or vector expressing HA-FIP200 and Myc-p53. Twenty-four hours posttransfection, 400 μg of whole cell lysates (WCL) were incubated with HA-conjugated agarose beads as described in Materials and Methods. The immunoprecipitates (IP) were analyzed by Western blot with anti-p53 antibodies to detect p53 associated with FIP200. Equal expression of the expressed proteins in whole cell lysates is also shown. Right, 293 cells expressing FIP-GFP and HA-p53 or an empty HA vector were incubated with HA agarose beads to precipitate p53. The associated FIP200 was analyzed by Western blot with FIP200-specific antibody. B, left, 1 mg of the whole cell lysates from U2OS or MCF-7 cells were incubated with anti-p53 antibody or rabbit IgG as a control, following by incubation with protein-A-beads. The specific association between endogenous FIP200 and p53 was detected by Western blot analysis with anti-FIP200 antibody (clone 33). The amount of immunoprecipitated p53 is also shown. Right, 0.6 μg of purified human recombinant wild-type p53 protein was incubated with 2 μg of GST or GST-N154 as described in Materials and Methods for in vitro binding assay. The bound p53 was resolved on SDS-PAGE and analyzed by Western blot with anti-p53 antibody. The equal amount of GST and GST-N154 (asterisks) are shown by Coomassie blue staining of the gel with the duplicate samples. C, left, U2OS cells transiently transfected with FIP-GFP expression vector were stained with Hoechst nuclear stain (blue) and analyzed by fluorescence microscopy to detect FIP-GFP localization in cells (green). Right, U2OS cells were processed for nuclear/cytoplasmic fractionation. Fifteen micrograms of each fraction were resolved on SDS-PAGE and analyzed by Western blotting with antibodies against FIP200 (arrow on the right), vinculin (cytoplasmic protein), and poly(ADP-ribose) polymerase (PARP, nuclear protein). D, left, HEK293 cells were cotransfected with empty HA vector (V) or vector expressing HA-FIP200 or its deletion mutants and Myc-p53. Protein binding was analyzed as described for Fig. 3A (left). Right, HEK293 cells were cotransfected with HA-FIP200 and Myc-tagged full length p53 or its different fragments as indicated. Protein binding was analyzed as described for Fig. 3A (left).
To provide further support for our findings, we also did mapping studies to locate the p53/FIP200-binding regions. Our results suggest that the NH2-terminal 154 residues of FIP200 are sufficient to specifically bind p53 in cells (Fig. 3D,, left). We also found that p53 interacts with FIP200 through its COOH-terminal region (Fig. 3D,, right). To check if interaction between FIP200 and p53 is direct, we did in vitro binding assay between purified recombinant wild-type human p53 and N154 fragment of FIP200. As shown in Fig. 3B, (right), N154-GST fusion protein but not GST alone could directly interact with p53. Importantly, the same small region of FIP200 (N154) could cause G1 arrest in MCF-7 cells as efficiently as full-length FIP200, whereas its COOH-terminal fragment that did not bind p53 could not cause G1 arrest (Fig. 4A , left). Together, these results suggest that FIP200 binding to p53 is critical for FIP200-mediated G1 arrest.

G1 arrest by FIP200 is associated with increase in p53 half-life and decrease in cyclin D1 half-life. A, left, MCF-7 cells were transfected with vectors expressing the irrelevant control protein (HA-Grb7), HA-FIP200, or its deletion mutants. BrdUrd incorporation was analyzed as described for Fig. 1A. Right, U2OS cells were infected for 48 hours with 50 moi of Ad-GFPsiRNA or Ad-FAKsiRNA and 10 moi of Ad-GFP or Ad-FIP200 as indicated. Twenty-five micrograms of total cell proteins were resolved on SDS-PAGE and analyzed by Western blot with specific antibodies against p53, FAK, pY397FAK, HA (for HA-FIP200), and vinculin (loading control). B, U2OS cells infected with 10 moi of Ad-GFP or Ad-FIP200 for 48 hours were treated with 10 μg/mL cycloheximide to inhibit protein synthesis. At the indicated time points, whole cell lysates were extracted, separated on SDS-PAGE, and analyzed by Western blot using specific antibodies against FIP200 (clone 33), p53, p21, cyclin D1, or vinculin as a loading control. In FIP200 blots, the lower band represents the FIP200 protein and the upper band is a nonspecific binding of antibody. C, a graphic representation of p53 (left) and cyclin D1 (right) protein levels in GFP or FIP200 infected cells shown in (A). Points, mean of three independent experiments; bars, ±SE. D, 50 μg of whole cell lysates from the U2OS cells infected with nothing (CTR), Ad-GFP, or Ad-HA-FIP200 as described for (B) were analyzed by Western blot with phospho-specific antibodies against different serine residues of p53 as indicated. All blots are representative of at least three independent experiments.
G1 arrest by FIP200 is associated with increase in p53 half-life and decrease in cyclin D1 half-life. A, left, MCF-7 cells were transfected with vectors expressing the irrelevant control protein (HA-Grb7), HA-FIP200, or its deletion mutants. BrdUrd incorporation was analyzed as described for Fig. 1A. Right, U2OS cells were infected for 48 hours with 50 moi of Ad-GFPsiRNA or Ad-FAKsiRNA and 10 moi of Ad-GFP or Ad-FIP200 as indicated. Twenty-five micrograms of total cell proteins were resolved on SDS-PAGE and analyzed by Western blot with specific antibodies against p53, FAK, pY397FAK, HA (for HA-FIP200), and vinculin (loading control). B, U2OS cells infected with 10 moi of Ad-GFP or Ad-FIP200 for 48 hours were treated with 10 μg/mL cycloheximide to inhibit protein synthesis. At the indicated time points, whole cell lysates were extracted, separated on SDS-PAGE, and analyzed by Western blot using specific antibodies against FIP200 (clone 33), p53, p21, cyclin D1, or vinculin as a loading control. In FIP200 blots, the lower band represents the FIP200 protein and the upper band is a nonspecific binding of antibody. C, a graphic representation of p53 (left) and cyclin D1 (right) protein levels in GFP or FIP200 infected cells shown in (A). Points, mean of three independent experiments; bars, ±SE. D, 50 μg of whole cell lysates from the U2OS cells infected with nothing (CTR), Ad-GFP, or Ad-HA-FIP200 as described for (B) were analyzed by Western blot with phospho-specific antibodies against different serine residues of p53 as indicated. All blots are representative of at least three independent experiments.
Because our previous data showed that FIP200 directly interacts with FAK and inhibits its phosphorylation and kinase activity, we wanted to examine a potential involvement of FAK in p53 regulation by FIP200. Surprisingly, FAK phosphorylation at Y397 was not decreased upon Ad-FIP200 infection in the U2OS cells (Fig. 4A , right; compare left two lanes). This is in contrast to our previous observations that transient transfection of FIP200 into HEK293 cells can inhibit tyrosine phosphorylation of FAK upon cell adhesion to fibronectin. The apparent discrepancy could be due to the difference in cell types used in the experiments. In any case, FIP200 was able to induce p53 with or without FAK siRNA treatment (compare p53 levels in left two lanes as well as right two lanes), suggesting that FIP200 regulation of p53 protein is independent of FAK.
FIP200 increases p53 half-life and decreases cyclin D1 half-life in U2OS cells. p53 is a short-lived protein and its levels in the cells are regulated primarily by the rate of its proteasomal degradation (19). To test if FIP200 increased p53 protein levels by affecting p53 half-life, we looked at the time course of p53 degradation in GFP (control) or FIP200 expressing U2OS cells after treatment with protein synthesis inhibitor cycloheximide. We used U2OS cells, because p53 protein regulation has been extensively studied in this cell line and it represents a good model for analyzing p53 protein degradation (20–24). As shown in Fig. 4B, and C (left), FIP200 expression dramatically increased p53 half-life compared with the control cells. As expected, stabilization of p53 correlated with the increase in p21 protein levels, suggesting that stabilized p53 is also transcriptionally active. In contrast, FIP200 decreased cyclin D1 protein half-life (Fig. 4B, and C, right) consistent with the decrease in cyclin D1 protein levels in response to FIP200 expression in mammary epithelial cells (see Fig. 1C). In summary, our results suggest that FIP200 significantly increases half-life of endogenous p53 by preventing its degradation. Stabilization of p53 by FIP200, in turn, activates p21 promoter leading to the increase in p21 protein levels.
Endogenous p53 is phosphorylated at Ser15 in response to FIP200. Stabilization of p53 protein in response to various cellular stresses is mainly achieved by posttranslational modification, such as phosphorylation or acetylation (25–27). These modifications change p53 conformation to reduce its affinity for the negative regulators. p53 phosphorylation has been extensively studied and a number of residues modified in response to p53 stabilization have been identified (28). To test if stabilization of p53 protein by FIP200 correlates with the changes in p53 phosphorylation, we examined p53 phosphorylation at the different residues using phospho-p53 antibody kit. We found that expression of FIP200 significantly increased p53 phosphorylation at Ser15 but not other residues (Fig. 4D). Phosphorylation at Ser15 has been associated with stabilization and transcriptional activation of p53 by several mechanisms (see Discussion). Consistent with this, our results show that Ser15 phosphorylation of p53 in FIP200 expressing cells correlated with more stable (Fig. 4B) and transcriptionally active p53, based on the increased promoter activity and protein levels of the p53 transcriptional target, p21 (Fig. 2B and C). These results suggest that binding of FIP200 to p53 could increase its stability and transcriptional activity by promoting p53 phosphorylation at Ser15.
FIP200 protects p53 from proteasomal degradation by ubiquitin- and Hdm2-independent mechanism. The main regulator of p53 protein stability in cells is Hdm2, which binds to p53 and acts as E3 ubiquitin ligase to promote p53 ubiquitination and subsequent proteasomal degradation (29, 30). Hence, we next examined if FIP200 stabilizes p53 by interfering with Hdm2 functions. To do so, we tested if FIP200 can inhibit Hdm2-mediated ubiquitination of p53 in HEK293 cells expressing p53, Ub, Hdm2, and FIP200 or GFP as a control. As shown in Fig. 5A, cotransfection of p53 and Ub caused appearance of ubiquitinated p53 (compare lanes 1 and 2, probably due to the endogenous Hdm2 activity), which was further increased upon cotransfection of Hdm2 (compare lanes 2 and 3). However, coexpression of FIP200 did not prevent accumulation of the ubiquitinated p53 by either endogenous (compare lanes 2 and 5) or exogenous Hdm2 (compare lanes 3 and 6). These results suggest that stabilization of p53 by FIP200 does not occur by inhibition of Hdm2-mediated ubiquitination and degradation of p53.

FIP200 protects p53 from proteasomal degradation by Hdm2-independent mechanism. A, HEK293 cells were cotransfected with vectors expressing 200 ng HA-p53, 1 μg Myc-Ub, 1 μg Hdm2, and 1 μg FIP-GFP, or GFP as indicated. Thirty-six hours posttransfection, 400 μg of whole cell lysates were incubated with HA-conjugated agarose beads to precipitate p53. Immunoprecipitates (IP) were analyzed by Western blot with specific antibodies against Myc to detect ubiquitinated forms of p53 or against HA and Hdm2 to show the levels of the expressed proteins in whole cell lysates. B, U2OS cells were infected with 10 moi of Ad-GFP or Ad-HA-FIP200 for 48 hours and treated with 10 μmol/L MG-132 or DMSO (vehicle) for 6 hours. Fifty micrograms of the whole cell lysates were resolved on SDS-PAGE and analyzed by Western blot with antibodies against HA, p53, or vinculin (loading control). C, U2OS cells were infected with 10 moi of Ad-GFP or Ad-HA-FIP200 for 48 hours and treated with 300 μmol/L dicoumarol or water (vehicle) for 5 hours. Fifty micrograms of the whole cell lysates were resolved on SDS-PAGE and analyzed by Western blot with antibodies against HA, p53, p21, or vinculin (loading control). D, U2OS cells were infected with 50 moi of Ad-GFPsiRNA or Ad-FIP200siRNA (two different clones 7 and 4) for 72 hours. Total cell proteins were resolved on SDS-PAGE and analyzed by Western blot with antibodies against FIP200, p53, p21, or vinculin (loading control).
FIP200 protects p53 from proteasomal degradation by Hdm2-independent mechanism. A, HEK293 cells were cotransfected with vectors expressing 200 ng HA-p53, 1 μg Myc-Ub, 1 μg Hdm2, and 1 μg FIP-GFP, or GFP as indicated. Thirty-six hours posttransfection, 400 μg of whole cell lysates were incubated with HA-conjugated agarose beads to precipitate p53. Immunoprecipitates (IP) were analyzed by Western blot with specific antibodies against Myc to detect ubiquitinated forms of p53 or against HA and Hdm2 to show the levels of the expressed proteins in whole cell lysates. B, U2OS cells were infected with 10 moi of Ad-GFP or Ad-HA-FIP200 for 48 hours and treated with 10 μmol/L MG-132 or DMSO (vehicle) for 6 hours. Fifty micrograms of the whole cell lysates were resolved on SDS-PAGE and analyzed by Western blot with antibodies against HA, p53, or vinculin (loading control). C, U2OS cells were infected with 10 moi of Ad-GFP or Ad-HA-FIP200 for 48 hours and treated with 300 μmol/L dicoumarol or water (vehicle) for 5 hours. Fifty micrograms of the whole cell lysates were resolved on SDS-PAGE and analyzed by Western blot with antibodies against HA, p53, p21, or vinculin (loading control). D, U2OS cells were infected with 50 moi of Ad-GFPsiRNA or Ad-FIP200siRNA (two different clones 7 and 4) for 72 hours. Total cell proteins were resolved on SDS-PAGE and analyzed by Western blot with antibodies against FIP200, p53, p21, or vinculin (loading control).
Although p53 protein levels in the cells are primarily regulated by the rate of its proteasomal degradation, it has also been shown that p53 can be cleaved by Ca2+-dependent protease, calpain (31). To confirm that FIP200 stabilizes p53 by interfering with its proteasomal degradation, we used proteasome inhibitor MG-132. Our results suggest that FIP200 inhibits p53 proteasomal degradation, because p53 protein levels were the same in control and FIP200-expressing cells after the treatment with MG-132 (Fig. 5B). The increase in the p53 levels upon MG-132 treatment is consistent with other reports (32) and suggests that in addition to FIP200, other proteasome-dependent pathways of p53 degradation (i.e., Hdm2) also exist.
Recent studies identified a distinct pathway for p53 stabilization by NAD(P)H quinone oxidoreductase 1 (NQO1) enzyme (33, 34). NQO1 has been shown to stabilize p53 by preventing its degradation by 20S proteasomes via ubiquitin and Mdm2-independent mechanism (35). Dicoumarol, which is NQO1 inhibitor, could reverse p53 stabilization by NQO1. Interestingly, we found that FIP200-mediated increase in p53 and p21 protein levels can be partially reversed by dicoumarol (Fig. 5C). In summary, our results indicate that stabilization of p53 by FIP200 is ubiquitin and Mdm2 independent and may involve NQO1 pathway.
Down-regulation of the endogenous FIP200 protein caused decrease in p53 and p21 protein levels. To test if suppression of the endogenous FIP200 protein levels will affect endogenous p53 and p21, we used siRNA strategy. U2OS cells were infected with either GFP siRNA or FIP200 siRNA expressing viruses and 72 hours later endogenous FIP200, p53, and p21 protein levels were examined by Western blotting. As shown in Fig. 5D, expression of FIP200 siRNA but not control GFP siRNA significantly reduced FIP200 protein levels, which is paralleled by a decrease in both p53 and p21 proteins. These results are consistent with our overexpression data and suggest that FIP200 is an important regulator of p53 and p21.
FIP200 regulates cyclin D1 protein levels by promoting its proteasomal degradation. Because our results suggest that cyclin D1 is another important target of FIP200 in mediating G1 arrest of the cell cycle (Fig. 1A, and D, bottom), we also examined the mechanisms of cyclin D1 regulation by FIP200. Because cyclin D1 levels can be regulated by both transcriptional and posttranscriptional mechanisms, we first examined the effect of FIP200 on cyclin D1 promoter. Using transient reporter gene assay with two different reporter constructs containing 1,745 and 962 bp of human cyclin D1 promoter fused to the luciferase reporter gene, we found that, in contrast to p21 promoter, FIP200 did not affect cyclin D1 promoter activity in two different breast cancer cell lines (Fig. 6A).

FIP200 regulates cyclin D1 protein levels by promoting its proteasomal degradation in human breast cancer cells. A, MCF-7 (left) or MDA-231 (right) cells seeded in 6-well plates were cotransfected with empty vector (control) or FIP200-expressing vector together with vector expressing human cyclin D1 promoter (1.7 or 0.96 kb) fused to the luciferase reporter gene. Twenty-four hours later, luciferase activity in cell lysates was analyzed as described in Materials and Methods. Luciferase activity in FIP200 transfected cells is expressed as percentage of that in control cells. Columns, mean of three to five independent experiments; bars, ±SE. B, MDA-231 cells were infected with 10 moi of Ad-GFP or Ad-FIP200 for 48 hours and treated with 5 μmol/L MG-132 or DMSO (vehicle) for 5 hours. Fifty micrograms of the whole cell lysates were resolved on SDS-PAGE and analyzed by Western blot with antibodies against FIP200 (clone 33), cyclin D1, or vinculin (loading control). Relative densitometric units of the cyclin D1 bands are also shown. In the FIP200 blots, the specific FIP200 band is indicated by the arrow; the top band represents a nonspecific binding of the antibody.
FIP200 regulates cyclin D1 protein levels by promoting its proteasomal degradation in human breast cancer cells. A, MCF-7 (left) or MDA-231 (right) cells seeded in 6-well plates were cotransfected with empty vector (control) or FIP200-expressing vector together with vector expressing human cyclin D1 promoter (1.7 or 0.96 kb) fused to the luciferase reporter gene. Twenty-four hours later, luciferase activity in cell lysates was analyzed as described in Materials and Methods. Luciferase activity in FIP200 transfected cells is expressed as percentage of that in control cells. Columns, mean of three to five independent experiments; bars, ±SE. B, MDA-231 cells were infected with 10 moi of Ad-GFP or Ad-FIP200 for 48 hours and treated with 5 μmol/L MG-132 or DMSO (vehicle) for 5 hours. Fifty micrograms of the whole cell lysates were resolved on SDS-PAGE and analyzed by Western blot with antibodies against FIP200 (clone 33), cyclin D1, or vinculin (loading control). Relative densitometric units of the cyclin D1 bands are also shown. In the FIP200 blots, the specific FIP200 band is indicated by the arrow; the top band represents a nonspecific binding of the antibody.
We next tested if FIP200 affects cyclin D1 protein degradation in the same experimental approach as we used for p53 (Fig. 4B). Our results suggest that FIP200 decreased cyclin D1 protein half-life (Fig. 4B, and C, right) by accelerating its degradation. To test if FIP200-mediated degradation of cyclin D1 occurs via proteasomal pathway, we used MG-132 proteasomal inhibitor. As shown in Fig. 6B, treatment with MG-132 could completely restore cyclin D1 protein levels in FIP200 expressing cells to those seen in the control cells (compare lanes 2 and 4). Together, these data suggest that FIP200 decreased cyclin D1 protein half-life by promoting its proteasomal degradation.
Discussion
Several lines of evidence have suggested a potential role for FIP200 in breast cancer, although the mechanisms of its antitumor activities are largely unknown. In this report, we showed that FIP200 inhibits G1-S phase progression, proliferation, and clonogenic survival in human breast cancer cells and explored the mechanisms of FIP200-induced cell cycle arrest.
Deregulation of the cell cycle is one of the most common alterations during tumor development (36). Therefore, it is not surprising that cancer is frequently considered a disease of the cell cycle. Cell cycle progression is tightly controlled by several families of proteins, including cyclins, their catalytic partners, CDKs, and CKIs. Here, we showed that FIP200 affects critical regulators of the cell cycle, p21 and cyclin D1, and focused on elucidating the molecular mechanisms of this regulation.
Cyclin D1 and p21 exert the opposite effects on G1 progression by modulating pRb phosphorylation, which is a critical step for G1-S phase transition (37). The major role of cyclin D1/CDK complex is to initiate pRb phosphorylation, after which cyclin E/CDK2 becomes active and completes this process by phosphorylating pRb on additional sites (6, 8, 38). In contrast, p21 regulates G1 progression by binding and inhibiting cyclin E/CDK2 complex leading to decrease in pRb phosphorylation later in G1. Interestingly, a second, noncatalytic function of cyclin D1/CDK4 complexes is to sequester p21 away from cyclin E/CDK2 to further promote pRb phosphorylation (39). We found that FIP200 decreased cyclin D1 and increased p21 protein levels, which paralleled by the decrease in phosphorylation of their downstream target, pRb, in human breast cancer cells. Interestingly, Dr. Chano's group found that FIP200 increased RB1 mRNA and protein levels in Jurkat and K562 leukima cell lines (4). However, our results show that in human breast cancer cells FIP200 affects pRb phosphorylation but not protein level (Fig. 1C). This discrepancy could be due to the cell line differences. Up-regulation of p21 by FIP200 was critical for FIP200-induced G1 arrest, because cells lacking p21 were not G1 arrested by FIP200. We also found that overexpression of cyclin D1 in cells could rescue FIP200-induced G1 arrest, perhaps by sequestering increased p21 away from the active cyclin E/CDK complex. This is supported by other reports, which showed that ectopic expression of cyclin D1 in MCF-7 human breast cancer cells is sufficient to activate cyclin E/CDK2 complex by decreasing its association with p21 (40). Together, our data suggest that balanced regulation of both cyclin D1 and p21 is important for efficient pRb hypophosphorylation and G1 arrest by FIP200.
To understand the molecular mechanisms of p21 regulation by FIP200, we first examined its effect on p21 promoter, because p21 expression in usually controlled at the transcriptional level. Our results suggest that FIP200 significantly induced p21 promoter activity in MCF-7 cells, mainly through the p53-dependent mechanism, because this response was abolished upon deletion of both p53-binding sites within p21 promoter. Furthermore, functional p53 was critical for p21 induction and cell cycle arrest by FIP200, because in p53 null cells FIP200 did not cause G1 arrest. Interestingly, we found that FIP200 interacts with both exogenous and endogenous p53 protein in different cell lines. Our mapping studies suggest that first 154 NH2-terminal residues of the FIP200 are sufficient to bind p53 in cells. Importantly, the same small region of FIP200 could cause G1 arrest in MCF-7 cells as efficiently as full-length FIP200, suggesting that interaction between FIP200 and p53 is critical for FIP200-induced G1 arrest. We also found that expression of FIP200 caused increased phosphorylation of p53 at Ser15 and stabilization by preventing its proteasomal degradation. NH2-terminal p53 phosphorylation at Ser15, Thr18, and Ser20 has been shown critical for p53 stabilization (41–43). Although all three residues have been implicated in p53 stabilization, the relative importance of each residue in regulating p53/Hdm2 interaction seems to differ among different studies (28). Our results suggest that increased phosphorylation of p53 at Ser15 in response to FIP200 correlated with stabilization of p53 proteins by Hdm2-independent mechanism, because expression of FIP200 did not prevent Hdm2-mediated ubiquitination of p53. Several other proteins, such as COP1, NQO1, c-Jun NH2-terminal kinase 1, TAFII31, Pirh2 have been shown to bind to and regulate p53 stability by both Hdm2-dependent and Hdm2-independent mechanisms (24, 33, 44–46). Recent studies identified a distinct pathway for p53 stabilization by NQO1 enzyme (33, 34). NQO1 stabilizes p53 by preventing its degradation by 20S proteasomes via ubiquitin and Mdm2-independent mechanism (35). Interestingly, we found that FIP200-mediated increase in p53 and p21 protein levels can be reversed by NQO1 inhibitor, dicoumarol, suggesting that stabilization of p53 by FIP200 may involve NQO1 pathway.
In addition to stabilization, Ser15 phosphorylation of p53 is crucial for p53-dependent transactivation by promoting p53 binding to its transcriptional coactivator p300 (47). Consistent with this, our data show that stabilization of p53 by FIP200 led to the transcriptional activation of its target gene, p21. It is possible that binding of FIP200 to p53 somehow makes it more accessible for phosphorylation at Ser15 leading to its stabilization and transcriptional activation, perhaps by modulating p53 interaction with other regulatory proteins. Clearly, more experiments should be done to further elucidate the mechanisms of p53 stabilization by FIP200, especially how this interaction affects Ser15 phosphorylation and binding to other p53-regulatory proteins, such as NQO1.
As has been mentioned above, in addition to the induction of p21, we also found that FIP200 decreased cyclin D1 protein levels, which contributed to the FIP200-induced G1 arrest. Our results suggest that regulation of cyclin D1 by FIP200 occurs at the posttranscriptional level by promoting its degradation through the proteasomal pathway. Although at present, our data regarding cyclin D1 regulation by FIP200 are preliminary and lack in-depth mechanistic explanations, it is interesting that FIP200 could simultaneously promote degradation of cyclin D1 and stabilization of p53 suggesting the complexity of FIP200 functions in regulation of G1-S phase progression.
In summary, this study showed that FIP200 increases p21 protein levels via stabilization of its upstream regulator p53 and decreases cyclin D1 protein by promoting its proteasomal degradation. Both effects are important for FIP200-induced G1 arrest and may contribute to the potential antitumor activity of FIP200 in breast cancer. Finally, our finding that FIP200 directly interacts with p53 and stabilizes it by preventing p53 proteasomal degradation is very interesting and exciting. Although the p53 gene is mutated in 50% of all human cancers, the majority of remaining tumors with wild-type p53 are still defective in p53 responses due to ineffective stabilization or increased degradation of the wild-type p53. Hence, the mechanisms of p53 stabilization by FIP200 merits further investigation and may reveal new therapeutic strategies for p53 stabilization in cancer cells.
Acknowledgments
Grant support: NIH grant GM52890 (J-L. Guan).
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
We thank Dr. J. Strobl for MCF-7, MDA-MB-231, and MCF10A cells; Dr. C. Deng for p21−/− and p21+/+ MEF; Dr. Y. Shi for Saos-2 cells, pCMV/Hdm2, and pCMV/p53 plasmids; Dr. T. Hunter for pRK5-D1 plasmid; Dr. R. Pestell for CD1-luc reporter constructs; Dr. X-F. Wang for p21-luc reporter constructs; and Richard Liang for his help with construction of Ad-FIP200.