
Abstract
Evidence is mounting that proinflammatory and proapoptotic thioredoxin-interacting protein (TXNIP) has a causative role in the development of diabetes. However, there are no studies investigating the role of TXNIP in diabetic retinopathy (DR). Here, we show that, in diabetic rats, TXNIP expression and hexosamine biosynthesis pathway (HBP) flux, which regulates TXNIP, are elevated in the retina and correlates well with the induction of inflammatory cyclooxygenase 2 (Cox-2) and sclerotic fibronectin (FN). We blocked the expression of TXNIP in diabetic rat retinas by: (i) inhibiting HBP flux; (ii) inducing post-transcriptional gene silencing (PTGS) for TXNIP mRNA; and (iii) performing an in vivo transcriptional gene silencing (TGS) approach for TXNIP knockdown by promoter-targeted small interfering RNAs and cell-penetrating peptides as RNA interference (RNAi) transducers. Each of these methods is efficient in downregulating TXNIP expression, resulting in blockade of its target genes, Cox-2 and FN, demonstrating that TXNIP has a causative role in aberrant gene induction in early DR. RNAi TGS of TXNIP abolishes diabetes-induced retinal gliosis and ganglion injury. Thus, TXNIP has a critical role in inflammation and retinal injury in early stages of DR. The successful employment of TXNIP TGS and amelioration of its pathological effects open the way for novel therapeutic strategies aimed to block disease onset and progression of DR.
Similar content being viewed by others


Main
Diabetic retinopathy (DR) is the leading cause of postnatal blindness in developed countries and one of the most severe complications of diabetes.1 DR is characterized by blood–retinal barrier breakdown, neovascularization, glial dysfunction and neuronal death. Among the pathological changes that occur early and linked causally to the development of retinopathy in diabetes are inflammation, altered extracellular matrix (ECM) gene expression, and premature demise of retinal capillary and ganglion cells.2, 3, 4, 5 However, it is not yet elucidated which components are most important for disease initiation and development of DR and which may be most useful as therapeutic targets. Several studies demonstrated a pathogenic role of proinflammatory and proapoptotic thioredoxin-interacting protein (TXNIP) in the development of both type I and II diabetes and its vascular complications.6, 7, 8, 9 TXNIP has been shown to inhibit thioredoxin activity and reduces cellular antioxidant capacity.6 Deficiency of TXNIP leads to improved glucose tolerance and insulin sensitivity in mice fed with a high-fat diet,7 protects against diabetes8 and inhibits glucose-induced pancreatic β-cell apoptosis.9 TXNIP induces caspase-1 inflammasome and innate immune response in pancreatic β-cell and macrophage.10 TXNIP is also highly induced by diabetes in renal mesangial cells,11, 12 neurons13 and retinal cells.14 We reported that TXNIP is required for RAGE-induced proinflammatory gene expression in retinal endothelial cells (ECs) under diabetic conditions in vitro and that TXNIP expression is significantly elevated in the diabetic rat retina.14 However, it is still unknown whether TXNIP is involved in the development and progression of diabetic ocular complications. Owing to the emerging relevance of TXNIP in diabetic complications and the lack of studies of TXNIP function in DR, we investigated the molecular mechanisms responsible for hyperglycemia (HG)-induced TXNIP expression in retinal EC in vitro and whether TXNIP has a causative role in early diabetic abnormalities in vivo in the retina of streptozotocin (STZ)-induced diabetic rats.
We have shown previously that the excess glucose metabolic flux through the hexosamine biosynthesis pathway (HBP) mediates cellular oxidative stress, aberrant gene expression and apoptotic demise of renal mesangial cells.15, 16 In the HBP, UDP-N-acetyl-glucosamine is the final product and is utilized as precursor for glycosylation of various cytoplasmic and nuclear proteins (Supplementary Figure S1). We reported that the HBP flux has a role in inducing TXNIP expression and ECM gene expression in renal mesangial cells and diabetic kidney.11, 16 Herein, we investigated whether HG and diabetes induce TXNIP expression via elevated HBP flux in EC in culture and in the retina of diabetic rats in vivo. We demonstrate that HG and diabetes induce TXNIP expression in retinal EC and diabetic retinas, which is mediated by HBP flux and epigenetic mechanisms involving the recruitment of p300 histone acetyltransferase (HAT) at the TXNIP promoter.
To analyze whether TXNIP is required for diabetes-induced early retinopathy, we measured the expression of two genes that are downstream of TXNIP and relevant to the development of DR: (i) cyclooxygenase 2 (Cox-2) that is involved in inflammation14, 17 and (ii) fibronectin (FN) involved in retinal angiogenesis and fibrosis.3 Moreover, we investigated whether TXNIP is associated with diabetes-induced retinal glia reactivity and neuronal injury. To elucidate the causative role of TXNIP in DR, we employed several methods to blunt TXNIP expression including a novel strategy to silence the expression of TXNIP in vivo by promoter-targeted small interfering RNA (siRNA) (RNA interference (RNAi))-mediated transcriptional gene silencing (TGS).18, 19 We show that TXNIP is required for diabetes-induced Cox-2 and FN expression, gliosis and neuronal apoptosis in the rat retina, shedding some light into a crucial role of TXNIP in disease initiation and progression of early DR.
Results
Diabetes increases retinal HBP flux, TXNIP, Cox-2 and FN expression
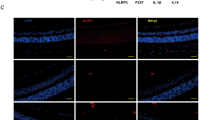
In this study, we investigated whether TXNIP upregulation in the diabetic rat retina14 has a critical role in early abnormalities of DR in vivo. First, we analyzed whether HBP flux, which activates TXNIP, and TXNIP expression are enhanced in the diabetic rat retina and whether they correlate with changes in inflammatory Cox-2 and FN expression after 4 weeks of hyperglycemic duration. Diabetes in rats was induced by STZ and their characterization is shown in Supplementary Table 1. We examined the level of HBP in retinal cell extracts from normal and diabetic rats by determining protein Ser/Thr-O-GlcNAcylation with CTD110.6 antibodies on western blots. The results show that diabetes induces retinal O-GlcNAcylation of several proteins when compared with the normal retina indicating elevated HBP flux (Figure 1A). Enhanced HBP parallels TXNIP and Cox-2 expressions both at protein (Figure 1B) and mRNA levels (Figure 1C). FN mRNA is also increased in the diabetic rat retina (Figure 1C), while protein levels appear to increase at the inner limiting membrane (ILM) and capillaries (Figure 1D).

Diabetes increases HBP, TXNIP expression, inflammation and fibrosis in the retina. (A, B) Cellular protein extracts (30 μg) were analyzed for protein Ser or Thr-O-GlcNAcylation using CTD110.6 antibodies, which specifically recognize the O-GlcNAc moieties, and TXNIP expression on western blots. In addition, Cox-2 expression was also determined. (A) Diabetes induces HBP flux as observed by enhanced CTD110.6 reactivity in diabetic retina (representative blots from n=5). At present, we do not know the identity of these proteins. (B) Similarly, diabetes also increases TXNIP protein levels (∼3.6±0.5-folds, P<0.01 versus normal retina; n=6). Furthermore, Cox-2 level is also induced in the diabetic retina (∼1.67±0.17-folds, P<0.05 versus normal retina, n=5). Actin is not changed. (C) RT-qPCR analysis reveals that TXNIP expression is significantly increased in the diabetic retina (8.06±3.18-folds) over normal retina, which correlates with increases in Cox-2 (3.18±0.49) and FN (5.99±0.78) expressions. The asterisk symbol represents significant change (P<0.01, n=7) in the diabetic retina when compared with normal retina. (D) On immunohistology, FN expression is increased in the basal membrane of blood vessels in GCL and also in the ILM as shown by the arrows. A representative of n=3 is shown. (E) Diabetes induces glial reactivity and ganglion injury in early DR. Immunohistology analysis reveals that GFAP expression is increased in the diabetic retina (a, b), which correlates with caspase-3 activation in the diabetic retina as demonstrated by positive staining for Flivo, an in vivo activated caspase-3 binding fluorescent probe (c, d, arrow). Nissl staining was used to identify neurons in flat mount retina. These results show that retinal neuronal dysregulation occur early in the retina of diabetic rats (these results are representative of three independent experiments)
Retinal inflammation and fibrosis evoke glial reactivity and neuronal injury under various diseases of the eye; therefore, we measured the expression of glial fibrillary acid protein (GFAP), a marker for gliosis, in intermediate filaments in the diabetic retina. Results show that GFAP staining increased in the diabetic retina (Figure 1E, a and b), suggesting glia activation at early DR. Activation of caspase-3 is also seen in the ganglion cell layer (GCL) in retinal flat mounts pointing to programmed cell death (Figure 1E, c and d). Furthermore, a reduction in presynaptic vesicular protein synapsin 1 expression occurs in the diabetic retina (Supplementary Figure S2). These results show that HBP and TXNIP are increased in diabetic retinas and correlate with retinal inflammation, fibrosis/gliosis and neuronal injury, the distinguishing features of early DR.
HBP is responsible for HG-induced TXNIP expression in retinal EC in culture
To ascertain that HBP is involved in diabetes-induced TXNIP and proinflammatory gene expression in the retina, we first investigated the metabolic pathway and the molecular mechanisms responsible for HG-induced TXNIP expression in retinal EC in vitro. We examined whether agents that increase or decrease the HBP alter HG-mediated TXNIP expression in a rat retinal EC, TR-iBRB2.14 We used azaserine to block HBP flux and glucosamine (GlcN) or PUGNAc to enhance the HBP. The data show that TXNIP is an early-response gene to HG and its mRNA expression is induced approximately eightfolds versus LG, within 2 h in TR-iBRB2 cells (Figure 2A, lanes 1 and 2). Pre-incubation of the cells with azaserine blocks HG-induced TXNIP mRNA expression (Figure 2A, lane 3), whereas GlcN and PUGNAc stimulate TXNIP expression (Figure 2A, lanes 4 and 5). HG also increases TXNIP protein, which is blocked by azaserine (Figures 2B and C), showing that HBP mediates TXNIP expression in EC.

HBP flux mediates HG-induced TXNIP expression by inducing histone acetylation and p300 recruitment on TXNIP promoter. (a) TXNIP mRNA expression was analyzed by RT-qPCR using β-actin as a control gene for normalization. Cells were treated for 2 h with LG (5 mM glucose), HG (25 mM glucose), PUGNAc (100 μM), GlcN (1.5 mM) and the HBP inhibitor azaserine (0.5 μM). Azaserine completely blocks the HG-induced TXNIP expression to the level of LG conditions and the decrement is statistically significant (P<0.01 HG versus HG+azaserine). GlcN and PUGNAc, which increases the HBP by inhibiting O-GlcNAcase removes O-GlcNAc, also enhance TXNIP expression significantly (P<0.01 versus LG). These data are the average of three independent experiments carried out in triplicate (n=9). (b) Western blot analysis of TXNIP expression in LG and HG conditions both in the presence and the absence of Azaserine. These data are representative of six independent experiments. (c) Quantification of the western blot shown in (b). *Significant increase versus LG (P<0.01); **significant decrease by azaserine versus HG (P<0.001). (d, e) ChIP analysis of histone H4 lysine (K) 8 acetylation on TXNIP promoter. (d) Quantitative PCR and (e) semiquantitative PCR analysis show that HG and TSA induce the acetylation of histone H4K8 on TXNIP promoter (n=6, *P<0.05 versus mock; **P<0.001 versus LG); (f, g) ChIP analysis of p300 association to TXNIP promoter. (f) Quantitative PCR and (g) semiquantitative PCR analysis show that HG and GlcN induce the binding of p300 on TXNIP promoter, whereas azaserine blocks HG effects, (n=6, *P<0.001 versus LG)
As HG alters epigenetic chromatin remodeling,20, 21 we also examined whether 5-aza-cytidine (5-AzaC), an inhibitor of DNA methyltransferase, or trichostatin A (TSA), an inhibitor histone deacetylase, has an effect on TXNIP expression. TR-iBRB2 cells were incubated with 5-AzaC for 24 h or with TSA for 2 h. 5-AzaC does not have an effect on TXNIP expression, whereas TSA increases TXNIP mRNA level (Supplementary Figure S3A, lane 3). When 5-AzaC and TSA are present together, TXNIP expression is not further enhanced as compared with TSA treatment alone (Supplementary Figure S3A, lane 4). These results indicate that TXNIP expression depends on histone acetylation and not on DNA methylation under these conditions.
We next investigated whether HG induces chromatin remodeling at the TXNIP promoter by chromatin immunoprecipitation (ChIP) assay, using an anti-acetylated lysine 8 of histone 4 (AcH4K8) antibody, an indicator of gene activation. HG and TSA increase H4K8 acetylation of the TXNIP promoter (Figures 2D and E, lanes 3 and 4). Enhanced H4K8 acetylation may be because of the recruitment of transcriptional cofactors that possess HAT activity, such as p300. We analyzed the association of p300 at the TXNIP promoter in EC by ChIP with an anti-p300 antibody followed by quantitative and semiquantitative PCR. P300 binding to the TXNIP promoter is low at LG (Figures 2E and F, lane 2); however, HG treatment induces p300 binding to the TXNIP promoter (Figures 2E and F, lane 3) and abolishes by azaserine (lane 4). GlcN also induces p300 binding to the TXNIP promoter at a level comparable to HG (Figures 2E and F, lanes 4 and 5). These results show that TXNIP mRNA induction involves p300 recruitment and histone acetylation at its promoter.
TXNIP is required for hexosamine-induced FN and Cox-2 mRNA expression in retinal EC in vitro
We have previously shown that elevated HBP flux and TXNIP upregulate FN expression in renal mesangial cells.11 Therefore, we hypothesized that TXNIP is also necessary for HBP-induced FN and Cox-2 expression in retinal EC. We generated TR-iBRB2 cell lines that stably express siRNAs targeted to TXNIP mRNA and control cells with a scramble siRNA.14 Addition of GlcN to scramble siRNA cells for 2 h induces FN and Cox-2 mRNA expression (Supplementary Figure S4A). GlcN failed to induce FN and Cox-2 mRNA expressions in siTXNIP cells (Supplementary Figure S4A), showing that TXNIP is required for GlcN induction of FN and Cox-2 in EC. Conversely, TXNIP significantly induces FN and Cox-2 mRNA expression in human TXNIP cDNA (hTXNIP)-overexpressing cells compared with LacZ control cells (Supplementary Figure S4B). Thus, the data support our hypothesis that HG-mediated FN and Cox-2 in retinal cells is mediated at least in part by the HBP and TXNIP.
TXNIP is a mediator HBP-induced inflammation and fibrosis in diabetic rat retinas
We showed above that HBP is a mediator of HG-induced TXNIP and proinflammatory gene expression in retinal EC (Figures 2A, B and Supplementary Figure S4). Therefore, we investigated further in diabetic retinas in vivo whether the observed HBP flux (Figure 1A) mediates TXNIP and proinflammatory gene expression. Diabetes enhances O-GlcNAc staining particularly in the inner plexiform layer (IPL) and around the nucleus in inner nuclear layer (INL) (Figure 3A, middle panel, arrows). Azaserine treatment blocks diabetes-induced increased O-GlcNAc modifications (Figure 3A, right panel). TXNIP staining is increased in all areas of the retina, at highest level in the GCL, IPL and INL when compared with the normal retina (Figure 3B, left and middle panels). Azaserine blocks diabetes-induced TXNIP expression (Figure 3B, right panel), demonstrating that the HBP mediates TXNIP expression in vivo in the retina. Azaserine also reduces Cox-2 and FN mRNA levels (Figure 3C). Further, azaserine also reduces GFAP immunostaining in the diabetic retina when compared with saline-treated diabetic eyes (Figure 3D).

Blockade of the HBP by azaserine prevents TXNIP expression, inflammation and gliosis in the diabetic retina. Intravitreal injection of azaserine, an inhibitor of the HBP, in the right eye of diabetic rats reduces protein O-GlcNAcylation (A), which corresponds to a decrease in TXNIP expression in the same retina (B). A representative of n=3 is shown here. The left eye of the same diabetic rats was injected with saline and used as the corresponding diabetic control retina. (C) RT-qPCR analysis for TXNIP, Cox-2 and FN mRNAs shows their elevated levels that are blunted by azaserine. *Significant change (P<0.05, n=6) for diabetic retina (left eye injected with saline) versus normal retina; #significant decrease (P<0.05, n=6) in gene expression after azaserine treatment (right eye) when compared with saline-treated left eyes of the same animal. (D). Diabetes induces glial cell reactivity in the retina as demonstrated by increased GFAP expression in intermediate filaments, which extend from epiretinal membrane to photoreceptor ONL. Azaserine prevents GFAP expression in the diabetic retina, which is more or less comparable to GFAP levels in normal rat retina. A representative of n=3 is shown here
Azaserine is an analog of glutamine; therefore, it may also influence other enzymatic pathways that require glutamine, such as glutaminase, affecting TXNIP and its downstream targets. Therefore, to demonstrate that TXNIP upregulation in the retina is indeed responsible for proinflammatory and fibrotic gene expression in early DR, we knocked down TXNIP by post-translational RNAi (post-transcriptional gene silencing (PTGS)). We injected siRNAs targeted to TXNIP mRNA twice in the right eye of diabetic rats, at days 23 and 27, and examined TXNIP, Cox-2 and FN expression at day 30. We observed a decrement of TXNIP expression upon siRNA treatment compared with the scramble siRNA-treated diabetic retina (Figure 4A). Knockdown of TXNIP in the diabetic retina leads to a significant decrement of both Cox-2 and FN mRNA (Figures 4A and B). In concert, GFAP upregulation is reduced by TXNIP siRNAs (Figure 4C). These data demonstrate that TXNIP is required for inflammatory Cox-2 and fibrotic FN expression and gliosis in early DR.

TXNIP is required for diabetes-induced Cox-2, FN and GFAP expression in the retina. Intravitreal injection of siRNA targeted to TXNIP mRNA (PTGS) on the right eye, whereas the left eye was injected with scramble siRNA as control. (a–c) RT-qPCR analysis for (a) TXNIP, (b) Cox-2 and (c) FN mRNAs shows their elevated levels that are blunted by TXNIP PTGS. *Significant change (P<0.05, n=6) for diabetic retina (left eye injected with scramble siRNA) versus normal retina; #significant decrease (P<0.05, n=3–4) in gene expression after TXNIP siRNA treatment (right eye) when compared with scramble scrRNA-treated diabetic eyes of the same animal. Some retinas in siRNA-treated diabetic rats showed too low expression (Ct, N/A) under these experiments and were excluded in the analysis. (d) TXNIP mRNA silencing also reduces GFAP expression in the diabetic retina when compared with the scr-siRNA-treated diabetic retina. A representative of n=3 is shown here
TGS for TXNIP in the diabetic retina reduces inflammation and gliosis
DR is a chronic disease and requires long-term treatment strategies. We have shown above that TXNIP is a mediator of retinal inflammation, fibrosis/gliosis and neurovascular injury in diabetes. To develop a long-term strategy for gene silencing, we tested the feasibility of a novel TGS method targeting TXNIP in vivo. We silenced TXNIP in the retina using RNAi TGS via intravitreal injection of promoter-targeted siRNA complexes with a cell-penetrating peptide (CPP) called MPG as siRNA transducers in the right eyes of diabetic rats. As control, a scramble siRNA/MPG complex was injected in the left eye of the same animal. Results show that TXNIP mRNA level is reduced after TXNIP promoter-siRNA treatment when compared with the scramble siRNA injected eye (Figure 5Aa). Furthermore, elevated Cox-2 and FN mRNA levels are reduced in diabetic retinas after TXNIP promoter-siRNA treatment (Figure 5A, b and c). To confirm the effectiveness of RNAi-mediated chromatin closing at the TXNIP promoter, we measured the binding of p300 to the TXNIP promoter by ChIP. We observed that p300 binding to the TXNIP promoter is enhanced in the diabetic retina treated with scramble siRNA when compared with the normal rat retina, while TXNIP-TGS reduces p300 recruitment to the TXNIP promoter (Figure 5B). In agreement, TXNIP protein staining is reduced in the diabetic rat retina upon TXNIP knockdown by TGS (Figure 5C).

TXNIP TGS by promoter-targeted siRNAs inhibits inflammation and fibrosis in the diabetic retina. (A) Messenger mRNA levels were analyzed by RT-qPCR after diabetic rats were treated with TXNIP promoter-siRNAs via intravitreal injection using CPPs as cargo carriers. TXNIP mRNA levels are significantly (#P<0.05, n=6) reduced in the diabetic rat retina (right eye) after TXNIP TGS as compared with the left eye of the same animal that received a scramble siRNA. Similarly, TXNIP promoter-siRNAs also significantly blocks Cox-2 and FN expressions (#P<0.05, n=6) when compared with the scr-siRNA-treated retina of the same animal. *P<0.05 represents significant increase in gene expression versus normal retina. (B) ChIP analysis of p300 binding to TXNIP promoter demonstrates that diabetes increases p300 binding to TXNIP promoter and TXNIP TGS prevents p300 binding to TXNIP promoter as revealed by qPCR of the ChIP DNA with TXNIP promoter primers (n=3 performed in duplicates). No p300 binding to TXNIP was observed in normal retina. (C) Promoter-siRNAs targeted to TXNIP gene in the diabetic retina downregulates TXNIP protein expression (A, right panel), which is comparable more or less to the level of age-matched nondiabetic rats (representative of three independent experiments)
We next investigated whether diabetes-induced gliosis and apoptosis are inhibited by TXNIP TGS. GFAP expression and caspase-3 activation are reduced after TXNIP promoter-siRNA treatment in the diabetic retina when compared with scramble siRNA treated eyes (Figures 6A–C). Together, the various approaches of TXNIP ablation in the retina demonstrate that TXNIP indeed has a critical role in inflammation, gliosis/fibrosis and apoptosis in early stages of diabetes, which will ultimately lead to disease onset and progression of blinding ocular complications.

TXNIP TGS by promoter-targeted siRNAs inhibits GFAP and caspase-3 activation in the diabetic retina. (a–b) TXNIP TGS by promoter-siRNAs also reduces GFAP mRNA (a) and protein expression (b) in the diabetic retina (right eye) when compared with scr-siRNA-treated diabetic retina (left eye) of the same animal. *Significant change (P<0.05, n=5–6) in GFAP expression for diabetic retina (left eye injected with scramble siRNA) versus normal retina; #significant decrease (P<0.05, n=5–6) in GFAP gene expression after TXNIP siRNA treatment (right eye) when compared with scramble scr-RNA-treated diabetic eyes of the same animal. A representative of n=3 is shown here of the GFAP immunohistology. (c) Further, TXNIP TGS also reduces diabetes-induced caspase-3 activation in the GCL (right eye, as shown by an arrow) when compared with left eye (diabetic control injected with scr-siRNA) in the GCL. A representative of n=3 is shown here
Discussion
This study aims to elucidate two unexplored questions that are relevant to the understanding of the molecular mechanisms that underline the development of DR: (i) analyze the metabolic pathway and the molecular network responsible for HG-induced TXNIP expression; and (ii) investigate whether TXNIP has a causative role in early abnormalities of DR. We demonstrate that increased HBP flux is a mediator of TXNIP expression in retinal EC under high ambient glucose in vitro and in vivo diabetic retinas and that transcriptional cofactor p300 is involved in TXNIP expression. Our new findings are: (i) p300 recruitment on TXNIP promoter and histone acetylation are involved in TXNIP expression in retinal EC; (ii) TXNIP mediates Cox-2 and FN expression both in vitro and in vivo; and (iii) blockade of TXNIP in vivo in the retina ameliorates diabetes-induced inflammation, gliosis/fibrosis and neuronal apoptosis (summarized in Figure 7).

A representative scheme of TXNIP-induced retinal inflammation, fibrosis/gliosis and ganglion injury in early DR. HBP is elevated early in the retina STZ-induction diabetic rats and is responsible for TXNIP expression. Recruitment of p300 on TXNIP promoter is involved in TXNIP expression. Ablation of TXNIP inhibits inflammatory gene expression for Cox-2 and FN, gliosis and neuronal injury in early diabetic retinas in vivo
We provide several evidences to demonstrate that HBP flux induces TXNIP expression in retinal EC. Inhibition of HBP by azaserine abolishes HG-induced TXNIP expression, whereas compounds that enhance HBP lead to TXNIP induction (Figures 2A–C). We show that TSA, which inhibits histone deacetylase, increases p300 recruitment to the TXNIP promoter and H4 acetylation, which leads to TXNIP expression. Similarly, RAGE activation by its endogenous ligand S100B also induces the recruitment of p300 on TXNIP promoter and H4 acetylation (Supplementary Figure S3B, C and S3D, E, respectively). We have previously shown that RAGE activates TXNIP expression in EC.14 We did not perform in this study a direct silencing of p300 to implicate TXNIP expression exclusively because p300 also mediates HG-induced expression of several genes in ECs.22 Thus, blockade of p300 expression itself may not provide a direct evidence of p300 involvement in TXNIP expression. Instead, herein we show that TGS of TXNIP abolishes p300 binding to TXNIP promoter and suppresses TXNIP mRNA and protein levels. TGS is known to lead to epigenetic chromatin closing that affect the recruitment of transcriptional factors and cofactors to target gene promoters.19 Thus, the results obtained by RNAi TGS in vivo provide further evidence of the involvement of p300 in TXNIP expression.
To investigate the functional role of TXNIP in early DR, we employ three methods to suppress TXNIP expression in the diabetic retina: (i) inhibition of HBP flux by intravitreal injection of azaserine; (ii) PTGS by siRNA targeted to TXNIP mRNA-coding region; and (iii) TGS by siRNA targeted to TXNIP promoter. With the first method we confirmed in vivo that HPB flux is responsible for diabetes-induced TXNIP expression in the retina. Further, with the PTGS knockdown of TXNIP, we demonstrate that TXNIP mediates diabetes-induced inflammatory and fibrotic gene expression in retinal EC and in vivo in the retina. Using the TXNIP TGS, we also demonstrate that TXNIP is essential for diabetes-induced inflammation and fibrosis because specific depletion of TXNIP in the retina prevents Cox-2 and FN expression while HG is still present in the animal.
Diabetes induces the formation of superoxide, which inhibits glyceraldehyde-3-phosphate dehydrogenase and accumulation of advanced glycation end products, HBP and PKC activation. These three pathways are reduced by transketolase activation via its cofactor thiamine and its analog benfotiamine.23, 24, 25 Interestingly, benfotiamine reduces HG-induced nuclear localization of FOXO1,24 a transcription factor that promotes TXNIP expression.26 Thus, benfotiamine may ameliorate DR pathogenesis via inhibition of both HBP flux and FOXO1 signaling. Our results further support that blockade of the HBP flux and TXNIP reduces inflammatory gene expression in early DR. FN is also upregulated in initial stages of diabetes (4 weeks) at the ILM and vessel basement membrane in vivo, a characteristic for early vascular abnormalities in DR. These data are consistent with findings from other laboratories that ECM proteins are increased in DR and cause blood vessel dysregulation.27 Furthermore, TXNIP mediates neuronal apoptosis (caspase-3 activation) and gliosis (GFAP upregulation) in the diabetic rat retina (this study), which is considered as an early sign of retinal injury and disease onset.2, 5 TXNIP expression has been shown to increase in the brain of diabetic animals 28 and it has a critical role in ganglion cell injury in glaucoma.29 Gliosis may be protective in acute retinal injuries to establish tissue homeostasis; however, persistent gliosis will cause inflammation and neurotoxicity in chronic diseases such as DR. Our data clearly support a crucial role of TXNIP in retinal inflammation, gliosis/fibrosis and neuronal injury/apoptosis in diabetes.
Still today, the treatment options for chronic ocular complications are limited due to the lack of safe delivery systems to target the retina. Frequent intravitreal injection of drugs increases the incidence of retinal infection and intraocular pressure leading to secondary complications. Therefore, a strategy for long-term gene and drug therapy for ocular disease treatment is important. With this view in mind, we undertook the RNAi-mediated epigenetic TGS for TXNIP silencing in vivo. The molecular mechanisms responsible for TGS have been described.19, 30, 31 RNAi TGS leads to chromatin closing at the targeted promoter via H3K9 dimethylation and subsequent CpG DNA methylation. Epigenetic gene regulation is mitotically stable and lasts long; and is therefore most desirable for long-term suppression of disease-associated genes in chronic inflammatory diseases, including DR. In our design of promoter-targeted siRNA to silence TXNIP, we selected two sites, which contain an E-box (Promoter target 1) and a FOXO1 site (Promoter target 2), which are important for TXNIP expression. We have not yet investigated whether each siRNA duplex is capable of silencing TXNIP individually. In this study, we used a combination of both promoter-siRNAs and demonstrate the feasibility of this approach in the retina in vivo. In addition, we show that CPPs are excellent agents for bioactive siRNA delivery in the retina, which in combination with RNAi TGS open the way for a new gene therapy approach to prevent diabetic ocular complications. RNAi TGS method will provide an alternative option for gene therapy in addition to viral-mediated gene delivery/silencing because CPPs do not perturb host genome. Viral vectors need to be incorporated in to host chromosome and they may introduce random mutations as viral DNA cannot be specifically targeted. In addition, viral DNAs may evoke host innate immunity and inflammation. For these considerations, we undertook the CPP-mediated RNAi TGS route to suppress TXNIP in DR. Intravitreal injection of CPPs have been used to drive siRNAs and proteins into the retina of rodents both in Muller glia and ganglion cells without affecting the retinal function.32, 33 Therefore, we expect that intravitreal injection of MPG RNAi complex used in this study targets both Muller glia and ganglion cells as gliosis and apoptosis are reduced by the TXNIP promoter-siRNA.
Several studies including our own show that TXNIP induces oxidative stress and inflammation in various cell types.34, 35 However, we previously demonstrated that TXNIP also induces inflammation without detectable ROS in cells and TXNIP-dependent Cox-2 mRNA expression is not affected by the antioxidant α-lipoic acid.14 Therefore, TXNIP-mediated inflammation itself can induce retinal oxidative and nitrosative stress in diabetes.36 TXNIP upregulation leads to epigenetic modification of the Cox-2 promoter and involves alternative histone H3K9 acetylation/methylation.14 We did not investigate here TXNIP-induced epigenetic modification of the Cox-2 and FN promoters in the diabetic rat retina in vivo, as well as the role of oxidative stress in TXNIP-mediated inflammation, fibrosis and neuronal apoptosis. Herein, we focus our attention to demonstrate first that TXNIP has a causative role in the development of early DR. We extensively show that a blockade of TXNIP expression in the diabetic rat retina inhibit the early abnormalities of DR.
In conclusion, we provide a critical function of TXNIP in inflammation and neurovascular injury in DR. TXNIP provides an excellent target for gene and drug therapies to prevent ocular complications of diabetes. Further long-term studies are warranted to establish the role of TXNIP in pathoetiology of DR and therapeutic potentials of RNAi TGS.
Materials and Methods
Materials
Antibody for TXNIP was purchased from MBL Biotechnology (Woburn, MA, USA), while CTD110.6 and Cox-2 antibodies were from Abcam (Cambridge, MA, USA). Antibodies for modified histones were also obtained from Abcam. α-Tubulin and actin were also obtained from Cell Signaling Solutions (Lake Placid, NY, USA). Fluorescent-labeled mouse anti-GFAP antibodies conjugated with Alexa Fluor 488, Nissl stain, and fluorescent secondary antibodies for mouse and rabbit were purchased from Molecular Probes (Invitrogen, Carlsbad, CA, USA). The enhanced chemiluminescence (ECL) system was purchased from Amersham (Arlington Heights, IL, USA). DMEM and F-12 nutrient mixture (Ham's) were from GIBCO (Grand Island, NY, USA).
Cell culture
In this study, we used a conditionally immortalized temperature-sensitive rat retinal EC line, TR-iBRB2, as recently described.14 Briefly, these cells maintain spindle fiber shapes, multicellular nodules and exhibit bipolar morphology like the primary retinal ECs. TR-iBRB2 cells express endothelial markers, such as von Willebrand factor, acetylated low-density lipoprotein receptors, VEGF receptor 2 (KDF/Flk-1), glucose transporter Glut-1 and maintain other endothelial properties.14 The TR-iBRB2 cells were maintained at 33°C in DMEM and F-12 Nutrient Mixture (Ham's) (4 : 1 ratio) containing a normal D-glucose concentration of 5.0 mM, 10% FCS, 100 μg/ml treptomycin, 100 U/ml penicillin and 2 mM glutamine. After the cells reach 70–80% confluence, they were differentiated at 37°C in a humidified incubator of 5% CO2 and then incubated in the starvation medium with the desired concentrations of glucose (5 mM, LG or 25 mM, HG) or GlcN (1.5 mM+LG) for different time periods. When azaserine (an inhibitor of HBP pathway, 0.5 μM) (from Sigma, St. Louis, MO, USA) was added to the culture, they were introduced 1 h before adding glucose and were present throughout the period of incubation. Inhibitors for p38 MAPK and NF-κB were purchased from Calbiochem (San Diego, CA, USA).
Generation of stably transfectant TR-iBRB2 cell lines
The CMV-human TXNIP and CMV-LacZ in pcDNA3.1 plasmid were used to generate stably overexpressing TXNIP and corresponding control LacZ cell lines as previously described.11 To knock down TXNIP expression, we performed four independent transfections with four different plasmids carrying a double-stranded oligonucleotide targeted to different regions of the rat TXNIP mRNA (Super Array Bioscience Corporation, Frederick, MD, USA), as recently described.14 As control, a plasmid encoding a scramble RNA was transfected. The clone that strongly silenced TXNIP expression was identified by real-time quantitative PCR (qPCR) and western blot analysis.
Reverse transcriptase qPCR
mRNA expression were analyzed by reverse transcriptase (RT)-qPCR using the BioRad Chromo 4 detection system (BioRad, Hercules, CA, USA) and SYBR Green PCR Master Mix from Applied Biosystems (Foster City, CA, USA) as previously described.11, 14 Primers were synthesized by Invitrogen.
Primer sequences of genes for the RT-qPCR are the following:
- 1. Rat TXNIP::
-
Forward: 5′-GTGAAGTTACCCGAGTCAAAGC-3′
Reverse: 5′-CTCACCTGTAGGCTGGTCTTCT-3
- 2. mFibronectin::
-
Forward: 5′-CTGGGGTCA CGTACCTCTTCA-3′
Reverse: 5′-AGTCGGTAGCCTGCTATACGG-3′
- 3. Rat Cox-2::
-
Forward: 5′- TACCCGGACTGGATTCTACG- 3′
Reverse: 5′- AAGTTGGTGGGCTGTCAATC-3′
- 4. Rat GFAP::
-
Forward: 5′-GAGGAGATCCAGTTCTTGAGG-3′
Reverse: 5′-CGTACTGAGTGCGAATCTCTC-3′
- 5. mActin::
-
Forward: 5′- ATTATCACCAACTGGGATGACATGG -3′
Reverse: 5′- CCAGCAGATTCCATACCAATGAAAG -3′
Cycle threshold (Ct) values were used to calculate the relative expression level of the various mRNAs that were normalized to actin mRNA. Primers for FN and actin are for mouse and also react with rat genes. As negative controls, the same reaction was performed on RNA samples without the reverse transcriptase reaction, and no PCR products were detected.
ChIP from retinal ECs
ChIP is a widely utilized method to detect transcription factor binding to specific DNA sequences or gene promoters in a chromatic complex. We used EZ-ZymeTM Enzymatic Chromatin Prep Kit from Upstate (now Millipore Corp., Billerica, MA, USA), which produces enzymatic cleavage of DNA to 180–360 bp, as described recently.14 Cells were fixed in 1% final parafolmaldeide for 10 min. Chromatin was obtained according to the manufacturer instructions. Antibodies to AcH4K8 (Abcam), acetylated H3K9 and phosphorylated in Ser10, trimethylated H3K914 and p300 (Santa Cruz Biotechnology, Santa Cruz, CA, USA) were used at 1 : 100 dilution. The proximal promoter regions for TXNIP, Cox-2 and FN were analyzed by qPCR and semiquantitative PCR using the following primers:
- 1. TXNIPC1F::
-
5′-CCCGAACAACAACCATTTTCC-3′
- TXNIPC1R::
-
5′-TTATATAGCCGCCTGGCTTG-3′
- 2. Cox-2 promF::
-
5′-CAGCAGCCCTCTCATTTCATT-3′
- Cox-2 promR::
-
5′-TCTTTGAGGTCTCGGGTTTCC-3′
- 3. FNpromF::
-
5′-CGGACTCCGGCCAATCGGC-3′
- FNpromR::
-
5′-GTGCAGTGCAGCGGGTGCG-3′
ChIP from the rat retina
We optimize a ChIP method for TXNIP promoter analysis from the frozen rat retina. Briefly, one frozen retina was fixed for 30 min in 1% paraformaldehyde (PFA) in PBS on ice. Tissue was dispersed with a hand-held homogenizer and further fixation of retinal tissue was continued for an additional 30 min on ice. Chromatin was purified using the Nuclear Extraction Kit (Active Motif, Carlsbad, CA, USA; catalog number 40010). Immunoprecipitation was carried out using the Immunoprecipitation Kit-Dynabeads-Protein A (Invitrogen, catalog number 100.07D); the rabbit anti-p300 polyclonal antibody was used at 1 : 100 dilution for 16 h. Dynabeads were added for further 2 h. Beads were resuspended in 0.1 ml TE buffer (10 mM Tris/Cl pH 8; 1 mM EDTA) and treated for 30 min with RNase 50 μg/ml at 37°C. Reversion of the crosslinking was carried out by adding proteinase K 500 μg/ml and SDS 0.5% for 3 h at 44°C and further at 65°C overnight.14 DNA was extracted using the ChargeSwitch gDNA Micro Tissue kit (Invitrogen, catalog number CS11203) and eluted from the magnetic beads in 60 μl of elution buffer. The promoter region for TXNIP (regions −478 and −297) was analyzed by qPCR using the following primer pair as in ECs:
- TXNIPC1F::
-
5′-CCCGAACAACAACCATTTTCC-3′
- TXNIPC1R::
-
5′-TTATATAGCCGCCTGGCTTG-3′
SDS-PAGE and western blotting
SDS-PAGE and western blot analysis of proteins were performed as described.14 The primary antibodies were used at a 1 : 1000 dilution (unless otherwise mentioned) in Tris-buffered saline (pH 7.4) containing 5% nonfat dry milk or 3% BSA. The dilution for HRP-conjugated secondary antibodies was in the proportion of 1 : 3000 (v/v). ECL was used to detect the immunoreactive bands.
Diabetes induction of rats
Diabetes of adult male Sprague–Dawley rats (∼275 g) was induced by injection of a single dose of STZ, (65 mg/kg i.v., Sigma) dissolved in 0.01 M citrate buffer, pH 4.5. Normal rats received a similar volume of vehicle alone. The rats were treated in accordance with the principles of NIH guidelines for the Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee. Before starting the experiments, the rats were acclimatized for 1 week. Diabetes was established after 48 h of STZ-injection and considered as day 1 of diabetes in rats. The treatment conditions and characterization of the diabetic rats are shown in Supplementary Table 1. At 4 weeks, diabetes rats have a blood glucose level of ∼500 mg per 100 ml compared with ∼102 mg per ml in control rats. Diabetic rats were given 1–2 U of insulin (Humulin-N, Lilly, Indianapolis, IN, USA) to maintain body weight in alternate days or daily, depending on the blood glucose levels as monitored by a glucometer. Intravitreal injection of azaserine (2.5 μl of 10 μM azaserine) was performed in anesthetized rats with 40 mg/kg body weight on the right eyes of diabetic rats (treatment) and a similar volume of saline on the left eye (control). The blood level of glucose stays elevated after this treatment (Supplementary Table 1). The injections were performed twice at days 23 and 27 and they were killed at day 30. An overdose of pentobarbital (200 mg/kg weight) was given to euthanize the rats. The retina were removed, processed for immunohistological analysis or frozen immediately in liquid N2 and stored at −80°C until used.
TGS for TXNIP by promoter-targeted siRNAs in the diabetic rat retina
As epigenetic TGS by chromatin reprogramming in vivo is a new technique for gene silencing in mammals, we provide a brief background of the process below.
(I) TXNIP promoter-siRNA targets: Short duplex small inhibitory RNAs (siRNAs) can target mRNAs and inhibit translation by mRNA degradation, which is known as PTGS. This method for blocking mRNA translation has become the commonly employed method for gene silencing. On the other hand, TGS caused by antigene siRNAs has been known for several years in the plant and lower animals. However, until recently it remained unknown whether siRNA-mediated TGS occurs in mammals. It has been demonstrated that the antisense strand of double-stranded RNAi targeted toward the promoter leads to an assembly of repressor complexes containing argonaute 1, histone deacetylase HDAC-1, H3K9 dimethyltransferase G2a and DNA methyltransferase DNMT3a without cleaving the DNA.30, 37, 38 This assembly leads to H3K9 dimethylation, chromatin condensation and induces gene silencing. The advantage of TGS over PTGS is that it tackles the root cause of gene expression and has the potential to correct aberrant gene expression directly while PTGS is a secondary remedy. Therefore, we developed this novel technique for epigenetic gene silencing for TXNIP in the retina of diabetic rats using the following promoter targets.
- (i). TXNIP Promoter Target 1::
-
5′-AATGGTCACGTCGAAATGAAT-3′
- siRNAs: sense::
-
5′-UGGUCACGUCGAAAUGAAUTT-3′
- antisense::
-
5′-TTACCAGUGCAGCUUUACUUA-3′
- (ii). TXNIP promoter target 2.:
-
5′-AACTGTGCACGAGGGATGCAC-3′
- siRNAs: sense::
-
5′-CUGUGCACGAGGGAUGCACTT-3′
- antisense::
-
5′-TTGACACGUGCUCCCUACGUG-3′
These double-stranded siRNAs targeting the rat TXNIP promoter (UniGene: Rn.2758, NCBI Entrez) atg (i) −413 to −434 (Promoter Target 1) and (ii) −8 to −29 (Promoter Target 2) from the transcription start site (TSS +1) were custom synthesized by Applied Biosystems (Ambion).
(II) CPPs as promoter-siRNA transducting agents: In order to drive these TXNIP promoter-siRNAs to cell nucleus in the retina of diabetic rats, we chose recently developed CPPs as cargo carriers. Recent advances have been developed in the employment of CPPs for siRNA delivery to mammalian cells, particularly to cell nucleus, which mediates epigenetic gene silencing by chromatin reprogramming.19 This is a very attractive approach for ocular disease treatment in diabetes. The potential advantage of this delivery system is that they penetrate the cell membranes carrying the cargos (nucleic acids or protein) into the nucleus in nondividing fully differentiated cells. These peptides are usually <40 amino acids and have three components: (i) an N-terminus hydrophobic amino acid sequence; (ii) a C-terminus nuclear localization signal; and (iii) a linker domain between the two regions. In this study, we used an siRNA-transducing CPP known as MPG.30, 39 The amino-acid sequence of MPG is as follows:
- MPG (27 residues)::
-
Acetyl-GALFLGFLGAAGSTMGAWSQP KKKRKV-cysteamide
MPG was custom synthesized by Anaspec (San Jose, CA, USA). The linker portion is shown in bold and the nuclear localization sequence in italics. The peptide is acetylated at N-terminus and carries a cysteamide group at the C-terminus, which enhance peptide stability and efficiency in transduction. The N-terminus of MPG is derived from HIV glycoprotein 41 (gp41) for membrane penetration and the C-terminus is the NLS of SV40 large T-antigen.39
(III) Intravitreal injection of MPG and promoter-siRNA complex in the rat retina
All the procedures are performed in a sterile BSL2 facility under a dissecting microscope. Briefly, under anesthesia (40 mg/kg pentobarbiatal), a small puncture is made in the eye with a single-use monojet 250 needle. To analyze the efficacy of this method, we tested several concentration ranges for TXNIP silencing using two promoter-targeted siRNAs together in rat retinal ECs in culture. MPG:prom-siRNA molar ratio of 10 : 1 at 40–100 nM siRNA concentrations causes a significant silencing of TXNIP mRNA (∼70%) at 24–72 h (data not shown). Then, 2 μl of a preformed MPG/siRNA complex at 10 : 1 molar ratio containing 2 μM siRNA (30 min incubation at room temperature before the injections) was injected in the vitreous through the limbus using a 10 μl nanofil syringe and a fine 35-gauge needle (WPI, Sarasota, FL, USA). The TXNIP Prom-siRNA injections were performed on the right eye (treatment) and scramble siRNAs on the left eye (RNAi Negative Control Duplex; Invitrogen, catalog number 1295112 Medium GC) of the same animal. Injections were performed twice at days 16 and 20 on each retina in order to establish a stable chromatin remodeling.
PTGS for TXNIP in the diabetic rat retina
Rat TXNIP mRNA siRNAs (#1330001, RSS332043) and Negative Control Duplexes (#12935-300) were purchased from Invitrogen. MPG-δNSL (27 residues): We used a C-terminus modified MPG at NSL called MPG-δNSL – (27 residue): acetyl-GALFLGFLGAAGSTMGAWSQP K S KRKV-cysteamide. MPG-δNSL was also custom-synthesized by Anaspec. Intravitreal injection of TXNIP siRNA (∼100 nM) and MPG-δNSL complexes (10 : 1 ratio of MPG-δNSL:siRNA) was performed in anesthetized rats with 40 mg/kg body weight on the right eyes of diabetic rats (treatment) and a similar volume of MPG-δNSL in saline on the left eye (control). The injections were performed twice at days 23 and 27 and they were killed at day 30 similar to azaserine treatment. An overdose of pentobarbital (200 mg/kg weight) was given to kill the rats. The retina were removed, processed for immunohistological analysis or frozen immediately in liquid N2 and stored at −80°C until used.
Immunohistochemistry
Immunohistological methods were similar those described recently by Drs. Ivanova and Pan40 as described in Supplementary methods. The retinas were fixed in the eyecups with 4% PFA in 0.1 M phosphate buffer (PB) for 20 min. The retinas were cryoprotected in a sucrose gradient (10, 20 and 30% w/v in PB, respectively). Cryostat sections were cut at 20 ìm in Tissue Tek (VWR, Batavia, IL, USA) (OCT mounting medium). Retinal sections were blocked for 1 h in a PB solution that contained 5% Chemiblocker (Chemicon, Temecula, CA, USA), 0.5% Triton X-100 and 0.05% sodium azide. The primary antibodies were diluted in the same solution (1 : 200 to 1 : 400 dilution depending on the antibody) and applied overnight followed by appropriate secondary antibodies (1 : 600 dilutions) conjugated with Alexa Fluor 488 or Alexa Fluor 594 for 2 h. The images were captured by an OLYMPUS BX 51 fluorescence microscope (Center Valley, PA, USA), which is fitted with a triple DAPI/FITC/TRITC cube, a DP70 digital camera and image acquisition software. Some images were also captured with a Zeiss Apotome microscope with Z-section (Zeiss, Oberkochen, Germany). Similar magnification ( × 400) and exposure time were maintained throughout for comparing images unless otherwise mentioned.
In vivo caspase-3 activation assay using FAM-FLIVO reagent
We used green Flivo as an in vivo reagent to detect activated caspase-3 in intact animals according to manufacturer's instruction (Immunohistochemistry Technologies, Bloomington, MN, USA). Flivo is cell and blood–retinal barrier permeable and binds covalently to active caspase-3 giving green fluorescence undergoing apoptosis. Under deep anesthesia, 150 μl of the Flivo reagent was injected in the tail vein 30 min before euthanization. At the end of 30 min, additional anesthesia was applied and the chest cavity in cells was opened; a small cut was made in the atrium to drain blood. A small cut was then made at the left ventricle and 20 ml of saline was injected to flush blood and retina was removed quickly. Retinal was fixed in 4% PFA for 20 min and processed for retinal flat mount or cryosectioning in OCT. This is the first demonstration of in vivo caspase-3 activation assay in the retina. Furthermore, FAM-Flivo method for detecting activated caspase-3 in vivo was confirmed by using NMDA-induced excitotoxicity and ganglion cell death in rat retinas (Dr D Goebel, Department of Anatomy and Cell Biology, Wayne State University, Detroit, MI, USA (personal communication)).
Statistical analysis
Results are expressed as means±S.E. of indicated number of experiments. Comparison between two sets of experiments was analyzed by Student's t-test while one-way ANOVA was used to determine differences among means in multiple sets of experiments followed by Bonferroni post hoc test. In both cases, a preset P-value of <0.05 was considered statistically significant.
Abbreviations
- 5-AzaC:
-
5-aza-cytidine
- AcH4K8:
-
acetylated lysine 8 of histone 4
- Cox-2:
-
cyclooxygenase 2
- CPP:
-
cell-penetrating peptide
- ChIP:
-
chromatin immunoprecipitation
- DR:
-
diabetic retinopathy
- ECM:
-
extracellular matrix
- FN:
-
fibronectin
- GFAP:
-
glial fibrillary acid protein
- HBP:
-
hexosamine biosynthesis pathway
- HAT:
-
histone acetyltransferase
- PTGS:
-
post-transcriptional gene silencing
- TXNIP:
-
thioredoxin-interacting protein
- TGS:
-
transcriptional gene silencing
- TSA:
-
trichostatin A
References
Congdon N, O’Colmain B, Klaver CC, Klein R, Muñoz B, Friedman DS et al. Causes and prevalence of visual impairment among adults in the United States. Arch Ophthalmol 2004; 122: 477–485.
Antonetti DA, Barber AJ, Bronson SK, Freeman WM, Gardner TW, Jefferson LS et al. Diabetic retinopathy: seeing beyond glucose-induced microvascular disease. Diabetes 2006; 55: 2401–2411.
Khan ZA, Chan BM, Uniyal S, Barbin YP, Farhangkhoee H, Chen S et al. EDB fibronectin and angiogenesis – a novel mechanistic pathway. Angiogenesis 2005; 8: 183–196.
Pambianco G, Costacou T, Ellis D, Becker DJ, Klein R, Orchard TJ . The 30-year natural history of type 1 diabetes complications: the Pittsburgh Epidemiology of Diabetes Complications Study experience. Diabetes 2006; 55: 1463–1469.
Kern TS . Contributions of inflammatory processes to the development of the early stages of diabetic retinopathy. Exp Diabetes Res 2007; 2007: 95103.
Junn E, Han S, Im J, Yang Y, Cho E, Um H et al. Vitamin D3 up-regulated protein 1 mediates oxidative stress via suppressing the thioredoxin function. J Immunol 2000; 164: 6287–6295.
Hui ST, Andres AM, Miller AK, Spann NJ, Potter DW, Post NM et al. Txnip balances metabolic and growth signaling via PTEN disulfide reduction. Proc Natl Acad Sci USA 2008; 105: 3921–3926.
Chen J, Hui ST, Couto FM, Mungrue IN, Davis DB, Attie AD et al. Thioredoxin-interacting protein deficiency induces Akt/Bcl-xL signaling and pancreatic beta-cell mass and protects against diabetes. FASEB J 2008; 22: 3581–3594.
Chen J, Saxena G, Mungrue IN, Lusis AJ, Shalev A . Thioredoxin-interacting protein: a critical link between glucose toxicity and beta-cell apoptosis. Diabetes 2008; 57: 938–1044.
Zhou R, Tardivel A, Thorens B, Choi I, Tschopp J . Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol 2010; 11: 136–140.
Cheng DW, Jiang Y, Shalev A, Kowluru R, Crook ED, Singh LP . An analysis of high glucose and glucosamine-induced gene expression and oxidative stress in renal mesangial cells. Arch Physiol Biochem 2006; 112: 189–218.
Hamada Y, Fukagawa M . A possible role of thioredoxin interacting protein in the pathogenesis of streptozotocin-induced diabetic nephropathy. Kobe J Med Sci 2007; 53: 53–61.
Price SA, Gardiner NJ, Duran-Jimenez B, Zeef LA, Obrosova IG, Tomlinson DR . Thioredoxin interacting protein is increased in sensory neurons in experimental diabetes. Brain Res 2006; 1116: 206–214.
Perrone L, Devi TS, Hosoya KI, Terasaki T, Singh LP . Thioredoxin interacting protein (TXNIP) induces inflammation through chromatin modification in retinal capillary endothelial cells under diabetic conditions. J Cell Physiol 2009; 221: 262–272.
Singh L, Andy J, Anyamale V, Greene K, Alexander M, Crook ED . Hexosamine-induced fibronectin protein synthesis in mesangial cells is associated with increases in cAMP responsive element binding (CREB) phosphorylation and nuclear CREB: the involvement of protein kinases A and C. Diabetes 2001; 50: 2355–2362.
Singh LP, Cheng D, Kowluru RA, Levi E, Jiang Y . Hexosamine induction of oxidative stress, hypertrophy and laminin expression in renal mesangial cells: effect of the anti-oxidant alpha-lipoic acid. Cell Biochem Funct 2007; 25: 537–550.
Cherney DZ, Miller JA, Scholey JW, Bradley TJ, Slorach C, Curtis JR et al. The effect of cyclooxygenase-2 inhibition on renal hemodynamic function in humans with type 1 diabetes. Diabetes 2008; 5: 688–695.
Mette MF, Aufsatz W, van der Winden J, Matzke MA, Matzke AJ . Transcriptional silencing and promoter methylation triggered by double-stranded RNA. EMBO J 2000; 19: 5194–5201.
Morris KV, Chan SW, Jacobsen SE, Looney DJ . Small interfering RNA-induced transcriptional gene silencing in human cells. Science 2004; 305: 1289–1292.
Miao F, Wu X, Zhang L, Yuan YC, Riggs AD, Natarajan R . Genome-wide analysis of histone lysine methylation variations caused by diabetic conditions in human monocytes. J Biol Chem 2007; 282: 13854–13863.
Mönkemann H, De Vriese AS, Blom HJ, Kluijtmans LA, Heil SG, Schild HH et al. Early molecular events in the development of the diabetic cardiomyopathy. Amino Acids 2002; 23: 331–336.
Chen S, Feng B, George B, Chakrabart R, Chen M, Chakrabarti S . Transcriptional co-activator p300 regulates glucose induced gene expression in the endothelial cells. Am J Physiol Endocrinol Metab 2010; 298: E127–E137.
Hammes HP, Du X, Edelstein D, Taguchi T, Matsumura T, Ju Q et al. Benfotiamine blocks three major pathways of hyperglycemic damage and prevents experimental diabetic retinopathy. Nat Med 2003; 9: 294–299.
Marchetti V, Menghini R, Rizza S, Vivanti A, Feccia T, Lauro D et al. Benfotiamine counteracts glucose toxicity effects on endothelial progenitor cell differentiation via Akt/FoxO signaling. Diabetes 2006; 55: 2231–2237.
Du X, Edelstein D, Brownlee M . Oral benfotiamine plus alpha-lipoic acid normalises complication-causing pathways in type 1 diabetes. Diabetologia 2008; 51: 1930–1932.
Al-Mubarak B, Soriano FX, Hardingham GE . Synaptic NMDAR activity suppresses FOXO1 expression via a cis-acting FOXO binding site: FOXO1 is a FOXO target gene. Channels (Austin) 2009; 3: 233–238.
George B, Chen S, Chaudhary V, Gonder J, Chakrabarti S . Extracellular matrix proteins in epiretinal membranes and in diabetic retinopathy. Curr Eye Res 2009; 34: 134–144.
Lappalainen Z, Lappalainen J, Oksala NK, Laaksonen DE, Khanna S, Sen CK et al. Diabetes impairs exercise training-associated thioredoxin response and glutathione status in rat brain. J Appl Physiol 2009; 106: 461–467.
Munemasa Y, Ahn JH, Kwong JM, Caprioli J, Piri N . Redox proteins thioredoxin 1 and thioredoxin 2 support retinal ganglion cell survival in experimental glaucoma. Gene Ther 2009; 16: 17–25.
Morris KV . The antisense strand of small interfering RNAs directs histone methylation and transcriptional gene silencing in human cells. RNA 2006; 12: 256–262.
Morris KV . Long antisense non-coding RNAs function to direct epigenetic complexes that regulate transcription in human cells. Epigenetics 2009; 4: 296–301.
Johnson LN, Cashman SM, Kumar-Singh R . Cell-penetrating peptide for enhanced delivery of nucleic acids and drugs to ocular tissues including retina and cornea. Mol Ther 2008; 6: 107–114.
Wang MH, Frishman LJ, Otteson DC . Intracellular delivery of proteins into mouse Müller glial cells in vitro and in vivo using Pep-1 transfection reagent. J Neurosci Methods 2009; 177: 403–419.
Kaimul AM, Nakamura H, Masutani H, Yodoi J . Thioredoxin and thioredoxin-binding protein-2 in cancer and metabolic syndrome. Free Radic Biol Med 2007; 43: 861–868.
Schroder K, Zhou R, Tschopp J . The NLRP3 inflammasome: a sensor for metabolic danger? Science 2010; 327: 296–300.
Chan PS, Kanwar M, Kowluru RA . Resistance of retinal inflammatory mediators to suppress after reinstitution of good glycemic control: novel mechanism for metabolic memory. J Diabetes Complicat 2010; 24: 55–63.
Lim HG, Suzuki K, Cooper DA, Kelleher AD . Promoter-targeted siRNAs induce gene silencing of simian immunodeficiency virus (SIV) infection in vitro. Mol Ther 2008; 16: 565–570.
Suzuki K, Juelich T, Lim H, Ishida T, Watanebe T, Cooper DA et al. Closed chromatin architecture is induced by an RNA duplex targeting the HIV-1 promoter region. J Biol Chem 2008; 283: 23353–23363.
Morris MC, Deshayes S, Heitz F, Divita G . Cell-penetrating peptides: from molecular mechanisms to therapeutics. Biol Cell 2008; 100: 201–217.
Ivanova E, Pan ZH . Evaluation of the adeno-associated virus mediated long-term expression of channelrhodopsin-2 in the mouse retina. Mol Vis 2009; 15: 1680–1689.
Acknowledgements
This study was supported by Research grants from Mid-West Eye Bank, Michigan, and the Juvenile Diabetes Research Foundation International to Dr Singh; Marie Curie International Reintegration Grant from European Community (number 224892) to L Perrone; and the Research to Prevent Blindness to the Department of Ophthalmology, WSU School of Medicine, Detroit, to LP Singh. Research funding to the Department of Anatomy and Cell Biology for Core facility by Grant number P30 EY04068 from the National Eye Institute, the National Institutes of Health, is also acknowledged. We also thank Drs Pan and Ivanova and Ms. Lu Qui in the Department of Anatomy and Cell Biology, WSU, Detroit, for helping us initially in performing the intravitreal injection and immunohistochemistry methods, and Dr Dennis Goebel for reading the paper critically and for his valuable suggestions.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by M Federici
Parts of the studies were presented at 2009 ARVO Summer Meeting at Bethesda, MD, and ARVO 2010 at Fort Lauderdale, FL.
Supplementary Information accompanies the paper on Cell Death and Disease website
Supplementary information
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Perrone, L., Devi, T., Hosoya, KI. et al. Inhibition of TXNIP expression in vivo blocks early pathologies of diabetic retinopathy. Cell Death Dis 1, e65 (2010). https://doi.org/10.1038/cddis.2010.42
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2010.42
Keywords
This article is cited by
TXNIP: A key protein in the cellular stress response pathway and a potential therapeutic target
Experimental & Molecular Medicine (2023)
Thioredoxin-Interacting Protein Inhibited Vascular Endothelial Cell–Induced HREC Angiogenesis Treatment of Diabetic Retinopathy
Applied Biochemistry and Biotechnology (2023)
Role of ASM/Cer/TXNIP signaling module in the NLRP3 inflammasome activation
Lipids in Health and Disease (2021)
Expression of TXNIP is associated with angiogenesis and postoperative relapse of conventional renal cell carcinoma
Scientific Reports (2021)
Verapamil as an Adjunct Therapy to Reduce tPA Toxicity in Hyperglycemic Stroke: Implication of TXNIP/NLRP3 Inflammasome
Molecular Neurobiology (2021)
