
Abstract
Bcl-2-family proteins are central regulators of cell life and death. At least three major classes of Bcl-2-family proteins have been delineated, including proapoptotic proteins that contain several conserved regions of sequence similarity (termed ‘multidomain’). In mammals, the multidomain proteins (MDPs) of the Bcl-2 family include Bax, Bak, and Bok. The founding member of the MDP group of Bcl-2-family proteins was discovered by Stanley Korsmeyer and co-workers, initiating an exciting area of cell death research. The status of current knowledge about the mechanisms and functions of MDPs is reviewed here, and some areas for future research are outlined. Therapeutic opportunities emerging from a growing understanding of MDPs with respect to their three-dimensional structures, biochemical actions, and roles in disease raise hopes that the foundation of basic research laid by Korsmeyer and others will eventually be translated into clinical benefits, leaving a legacy that benefits the world for many decades.
Similar content being viewed by others

Introduction
In 1993, the first proapoptotic members of the Bcl-2-family were reported. Bax (Bcl-2 Antagonist X) was identified by the Korsmeyer group as a Bcl-2-interacting protein that opposed Bcl-2 and promoted apoptotic cell death.1 Bcl-XS (Bcl-X short) was identified by the Thompson group as a proapoptotic isoform of Bcl-X, produced by alternative mRNA splicing.2 Subsequent sequence alignment analysis led to the identification of conserved regions of amino-acid sequence similarity among the then growing family of both pro- and antiapoptotic Bcl-2 family proteins, resulting in the coining by Korsmeyer in 1994 of the term Bcl-2 Homology (BH) domain.3 Over time, four BH domains would be delineated; BH1, BH2, BH3, and BH4.
Two general classes of proapoptotic Bcl-2/Bax family proteins exist: (1) Those that share several regions of sequence homology, specifically BH1, BH2, and BH3 domain (termed ‘multidomain’ proteins by Korsmeyer in approximately 20004) and (2) Those that share little sequence similarity, save for the conserved BH3 domain (termed ‘BH3-only’ proteins by Strasser and others5). The mechanisms by which these two classes of pro-apoptotic Bcl-2/Bax-family proteins promote cell death are quite distinct. Multidomain proteins (MDPs) appear to possess intrinsic cell death-inducing activity, as evidenced for example by their ability to kill yeast when ectopically expressed in these simple eukaryotes.6, 7, 8 Mammalian MDPs also kill plant cells when ectopically expressed.9 In contrast, BH3-only proteins (BOPs) use their BH3 domains as ligands for engaging other members of the Bcl-2 family, either suppressing antiapoptotic proteins such as Bcl-2 and Bcl-XL, or activating MDPs such as Bax and Bak.10
The MDPs of mammals include Bax, Bak, and Bok, while genes encoding BOPs number at least 18 in the human genome.11 To date, the three-dimensional (3D) structure of only one MDP has been determined – namely, Bax, which consists of a bundle of α-helices resembling the pore-forming region of bacterial toxins such as Diphtheria Toxin and the Colicins.12 Amazingly, the same protein fold is found in the antiapoptotic members of the Bcl-2 family, indicating that they evolved from a common ancestor but developed opposing phenotypes from a common fold. The hypothesis that the same protein fold can accommodate opposing cell death phenotypes may explain why the genomes of invertebrate organisms such as Caenorhabditis elegans seem to be perfectly competent to regulate programmed cell death using a single Bcl-2/Bax-like gene (e.g. Ced9). Nearly the same fold is also seen in the Bcl-2/Bax-family protein, Bid, which is usually considered a BOP rather than a MDP due to the lack of sequence similarity in BH1 and BH2.13
Physiological Roles of MDPs
Gene ablation studies in mice have provided insights into the physiological roles of MDPs in mammals. For example, investigations by Korsmeyer and co-workers14, 15, 16, 17, 18 showed that homozygous disruption of bax in mice reveals roles for this MDP in neuronal cell death during development, homeostasis of lymphoid and reproductive organs, tumor suppression, and cell death responses to DNA damage, ischemia-reperfusion injury, and other forms of stress. Taking the nervous system as an example, Bax-deficient mice (‘knockouts’) have been shown to contain increased numbers of sympathetic and facial motor neurons, suggesting defective developmental neuronal cell death.19 The absence of Bax in these mice also prevents >80% of the degeneration of facial motoneurons that normally occurs after axotomy. Moreover, embryonic neurons derived from Bax knockout mice are completely resistant to cell death induced by neurotrophic factor withdrawal, demonstrating an obligatory role for Bax in the death of sympathetic neurons when deprived of NGF.19 Similarly, ablation of Bax expression using antisense oligonucleotides protects sympathetic neurons from apoptosis induced by NGF withdrawal.20 Elevations in Bax expression occur in association with cell death induced by a variety of stimuli that may be of relevance to mechanisms of neuronal cell death during ischemia, epilepsy, spinal cord injury, and certain neurodegenerative diseases such as Parkinson's and Alzheimer's. For example, the human BAX gene promoter contains typical p53-binding sites and is transcriptionally upregulated by p53.21 As elevations in p53 protein levels have been described as an early event during the neuronal cell deaths that occur in various models of forebrain ischemia and excitotoxic neurotransmitter administration, this functional connection between Bax and p53 indirectly suggests an potentially important role for Bax.22, 23, 24, 25 Elevations in Bax protein levels have been documented as an early event associated with neuronal cell death during brain ischemia. Increases in Bax are found specifically in neurons that undergo cell death in ischemia-sensitive regions of the brain such as the CA1 sector of the hippocampus, in rat, hamster, and dog models of global cerebral ischemia.26, 27, 28 Bax protein and mRNA levels also rapidly increase in neurons within the penumbra region of focal infarcts, in rodent models of middle cerebral artery occlusion.29 Moreover, evidence suggesting a cause-and-effect relation between increases in Bax expression and ischemia-associated neuronal cell death has come from experiments using bax−/− mice generated by Korsmeyer et al.30 Elevations in Bax protein and mRNA levels were described in neurons in vivo after excitotoxic lesion with the N-methyl-D-aspartate receptor agonist, quinolinic acid,31 as well as after systemic administration of kainic acid.32 Of potential relevance to neurodegenerative diseases, amyloid-beta peptide was reported to upregulate Bax and downregulate Bcl-2 in cultured human neurons,33 and systemic administration of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) has been shown to cause increases in Bax mRNA and protein in the substantia nigra.34 In addition to neuronal cell death associated with ischemia and excitotoxic neurotransmitters, Bax protein levels are markedly increased in sensory and motor neurons following sciatic nerve transection, often in association with increases in Jun protein production.35 In this regard, the bax gene promoter contains two consensus AP-1 sites, which theoretically could provide a functional connection between Jun and bax gene expression. Elevations in Jun expression have also been reported in association with neuronal cell death induced by neurotrophic factor withdrawal. Moreover, microinjection of neutralizing anti-Jun antibodies can prevent such cell deaths,36 but it has not been determined whether Bax is involved under these circumstances. Increased Bax expression also has been observed in spinal motoneurons in patients with Amyotrophic Lateral Sclerosis (ALS).37 Moreover, in transgenic mutant SOD1 mice (a model of ALS), Bax protein activation has been detected.38 Marked elevations in Bax protein levels have been reported in microglial cells and astrocytes in association with apoptosis in patients with HIV-induced encephalitis.39 Interestingly, elevations in Bax expression have also been reported in residual myocardiocytes surrounding infarcts in the heart,40 suggesting that upregulation of Bax may be a common occurrence during ischemia and is not limited to the brain. Thus, Bax has emerged as a potential drug discovery target for ischemic and degenerative diseases.
Genetic analysis of the other MDPs, namely bak and bok, has been less insightful for delineating in vivo roles for these proapoptotic proteins in vivo.41, 42 However, more should be done to investigate cell death regulation when these mice are stressed in various ways. Expression of the bok gene is limited to testis, ovary, and uterus in mice,43 though expression of the human ortholog of this gene may be more widespread, including liver and kidney.44 Genotoxic injury experiments using bok−/− mice did not reveal a requirement for bok for cell death in ovaries or other organs.41 Expression of bak is wide-spread in mammalian tissues,45 but no phenotypic abnormalities have been reported for bak−/− mice.42
The combined knockout of bax and bak in mice results in developmental defects in several tissues due to failure to eradicate cells. Cells from bax/bak double-knockout mice are also resistant to myriad stimuli that are known to kill via the mitochondria-dependent pathway for cell death, including x-irradiation, UV-irradiation, DNA-damaging drugs, kinase inhibitor staurosporine, growth factor deprivation, and agents that induce ER stress, but not killing triggered by TNF-family death receptors.42, 46 Perhaps because few bax−/−bak−/− mice survive to adulthood, these animals have not been employed widely for disease models aimed at assessing the importance of these MDPs for pathological cell death. However, in a model where intense UV-light is used to trigger photoreceptor cell death in the retina, combined deficiency of bax and bak was shown to provide protection.47 Altogether, the observations from bax/bak double-knockout mice suggest that these MDPs individually play redundant roles in control of cell death, whereby simultaneous ablation of both genes provides a clear survival benefit. However, even bax/bak double knockout cells are susceptible to apoptosis induced by the extrinsic pathway (TNF-family death receptors)42 and to autophagic cell death,48 possibly explaining why several major organs in these mice are histological normal.
Cellular Actions of MDPs
The MDPs all contain a hydrophobic stretch of amino acids at the carboxyl-terminus that allows for their insertion into intracellular membranes. The favored locations of these proteins are the outer membrane of mitochondria and membranes of the endoplasmic reticulum (ER). The actions of MDPs at mitochondrial membranes have been extensively studied, providing evidence that these proteins control mitochondrial outer membrane permeability (MOP), thus promoting release of apoptogenic proteins such as cytochrome c from these organelles.49, 50 How MDPs accomplish this feat is unclear, but their multimeric oligomerization in mitochondrial membranes (a phenomenon first demonstrated by Korsmeyer and co-workers) is correlated with MOP and release of intramitochondrial proteins. Several cell death-promoting proteins are among those released from mitochondria in response to MDPs, thus triggering a variety of downstream caspase-dependent and caspase-independent cell death mechanisms, culminating in either apoptosis or necrosis.
The proapoptotic functions of MDPs at mitochondria are suppressed by antiapoptotic Bcl-2-family members that also target to the surface of these organelles. A variety of genetic and biochemical studies argue that MDPs are the effectors of the mitochondrial cell death pathway (Figure 1), with antiapoptotic proteins such as Bcl-2 and Bcl-XL operating as upstream regulators that oppose the intrinsic death-inducing actions of MDPs such as Bax and Bak at mitochondrial membranes (see for example Wei et al.46 and Cheng et al.51). Although governing MOP perhaps through their intrinsic properties as pore-forming proteins, cross-talk between components of the inner mitochondrial membrane and MDPs in the outer membrane may also occur. For example, the adenine nucleotide translocator and F0F1-ATPase of the inner membrane can modulate the cell death activities of Bax,52, 53 though it is also clear that Bax can induce cell death independently of these proteins. Bcl-2-family proteins also may modulate activity of the mitochondrial permeability transition pore,54 a poorly understood regulator of inner membrane permeability.55 The significance of whether MDPs exclusively affect MOP versus the possibility that they also control some aspects of inner membrane permeability resides at least in part in the need for inner membrane integrity to maintain the H+ gradient that creates the driving force for ATP generation and proper flow of electrons through the respiratory chain. Disruption of inner membrane function thus leads of generation of reactive oxygen species and ATP depletion, typically culminating in necrosis.

Hierarchy of functional interactions among Bcl-2-family proteins differs at mitochondria versus endoplasmic reticulum. The apparent hierarchies of functional interactions among Bcl-2-family proteins are contrasted for mitochondria (top) and ER (bottom). BOPs can be classified as type I proteins that function as antagonists of anti-apoptotic proteins such as Bcl-2 and Bcl-XL, and type II proteins that function both as antagonists of antiapoptotic Bcl-2-family proteins and as agonists of proapoptotic MDPs. Studies using genetically engineered cells lacking Bax and Bak by the Korsmeyer group played a major role in defining the hierarchical relations among the Bcl-2-family proteins
MDPs are also implicated in remodeling of the architecture of mitochondrial cristae, perhaps poising intermembrane proteins such as cytochrome c for rapid release.56 Finally, MDPs have been implicated in pathological fission of mitochondria that often precedes cell death, where it has been reported that large aggregates of Bax and Bak accumulate at the sites of membrane constriction where pieces of these organelles are pinched off, through a process requiring Drp1.57, 58 Still other actions of MDPs may exist at downstream points in the mitochondrial cell death pathway, based on a report that the Bok (Diva) binds caspase-activator Apaf1,59 the cytochrome c-binding protein that links MOP to caspase activation.
The effects of MDPs on ER membranes are poorly understood, but a common observation is alteration in the handling of Ca2+ by this organelle. Loss of Bax and Bak causes essentially the same phenotype as over-expressing Bcl-2 or Bcl-XL with respect to ER Ca2+ – namely, it reduces internal ER Ca2+ stores, apparently by increasing the spontaneous rate of leakage of Ca2+.60, 61 The reduced Ca2+ accumulation in ER translates into a lower concentration of Ca2+ reached in the cytosol in response to stimuli that cause the ER to dump its sequestered Ca2+, and a corresponding reduction in the downstream accumulation of Ca2+ into mitochondria. Accumulation of Ca2+ in mitochondria, when excessive, can lead to depolarization and loss of osmotic homeostasis, followed by organellar swelling and subsequent membrane rupture to release apoptogenic proteins, disturb electron transport, ablate ATP production, and cause cell death.55 However, the reductions in resting ER Ca2+ seen in Bax/Bak doubly-deficient cells (or cells over-expressing Bcl-2 or Bcl-XL) may have myriad other affects on Ca2+-dependent signaling events in cells, and many of those may not be particularly germane to cell death regulation.
The hierarchy of Bcl-2/Bax-family protein actions at ER membranes is distinctly different from the situation at mitochondria. Bcl-2 and Bcl-XL remain capable of regulating ER Ca2+ even in Bax/Bak double-deficient cells that apparently express no MDPs60 (and our unpublished data). As such, it seems that the antiapoptotic proteins (Bcl-2/Bcl-XL) are the downstream effectors of ER Ca2+ levels, with Bax and Bak operating as upstream regulators that oppose the actions of proteins such as Bcl-2 and Bcl-XL (Figure 1). It is unknown whether the ability of Bcl-2 and Bcl-XL to control ER Ca2+ is a manifestation of their intrinsic function as ion-channels or an indirect consequence of their actions on Ca2+ channels in the ER. Indeed, evidence has been obtained to suggest an effect of Bcl-2 and/or Bcl-XL on Inositol Triphosphate (IP3)-gated Ca2+ channels (IP3Rs) or on the Ca2+-ATPase of the ER, but many details are lacking that would unify all the available data into a coherent model.60, 62, 63, 64, 65, 66, 67, 68
Mechanisms of MDP Activation
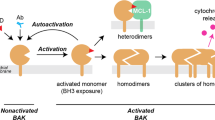
Although simply over-expressing MDPs is generally sufficient to kill cells, several factors have been identified that trigger activation of MDPs, implying that these proapoptotic proteins may reside in a latent state until provoked into action. For example, in most tissues, Bax is found in the cytosol rather than associated with membranous organelles, undergoing translocation to mitochondria in response to cell death stimuli.69 The 3D solution structure of Bax in the soluble inactive state indicates that the C-terminal membrane-anchoring domain is tucked into the same pocket that probably binds BH3 peptides.12 In response to still poorly defined signals, Bax alters its conformation, forming dimers, exposing its C-terminal membrane-anchoring domain, and inserting into mitochondrial membranes.70 The other MDPs in mammals, Bak and Bok, appear to reside constitutively within membranes. However, even though inserted in the outer mitochondrial membrane, oligomerization of Bax or Bak apparently must occur to trigger release of cytochrome c and other apoptogenic proteins. Studies first initiated by the Korsmeyer laboratory led to the demonstration that oligomerization of Bax and Bak in mitochondrial membranes occurs via a process that is promoted (perhaps even catalyzed) by agonistic BH3 domains provided by proteins such as Bid and Bim.71, 72, 73 In 2002, Korsmeyer et al.74 demonstrated that synthetic BH3 peptides that bind Bax or Bak recapitulate this activity, providing a means of triggering oligomerization experimentally. Synthetic unilammelar liposomes have been employed for attempting to identify the essential factors required for the Bax-mediated change in membrane permeability that account for release of proteins such as cytochrome c, revealing that the presence of cardiolipin in membranes and agonistic BH3 peptides (e.g. from Bid) are sufficient to trigger insertion of Bax into membranes, Bax oligomerization, and membrane permeabilization.72
Altogether, the currently available data argue that MDPs exist in two conformational states, corresponding to monomeric inactive versus oligomeric active states – at least with respect to their role at mitochondrial membranes as inducers of MOP. (The functions of these proteins at ER membranes may be different.) Further, based on differences in alkaline extraction from membranes, the oligomeric form of Bax appears to represent a membrane integrated conformation, suggesting that oligomerization is associated with conversion of MDPs to their pore-forming conformation. A critical goal of future research should include defining the structure of this membrane-integrated, oligomeric state of MDPs and the molecular basis for their induction of MOP. In this regard, studies in which various molecular weight dextrans tagged with fluorochromes have been incorporated into synthetic liposomes imply that a discrete pore may not be created by Bax,72 suggesting a more general disruption of membrane integrity where membranes rip apart, rather than a condition where a classical proteinaceous pore of discrete diameter is formed.
Since the discovery by Korsmeyer et al.75 of the first agonistic BH3-containing protein, Bid, several additional activators of MDPs have been identified. Among the nearly 20 BOPs, only Bid and Bim reportedly bind and activate MDPs.73, 76 These agonistic BOPs are often found in latent, inactive states in cells, and must be summoned into action by appropriate stimuli or conditions. In the case of Bid, one elucidated activation mechanism involves cleavage by caspases or calpains, followed by myristoylation in some cases, and insertion into mitochondrial membranes (reviewed in Strasser5). For Bim, the details depend on the isoform of the protein, as three forms of Bim have been described differing in length as a result of alternative mRNA splicing. The longest isoform of Bim, and usually the most abundant, is reportedly sequestered on microtubules in a complex with dynein light chain, and must be released to interact with membrane-associated Bcl-2-family proteins.77 However, Bim proteins can also be found constitutively associated with mitochondria even under nonapoptotic conditions.78 The phenotypes of mice in which the genes encoding Bid or Bim have been ablated are fairly normal (for details see Strasser5), suggesting that additional agonists of MDPs are available to compensate for their loss. In this regard, antiapoptotic members of the Bcl-2-family may also serve as activators of MDPs under circumstances where the phenotype of these proteins is flipped, and the proteins undergo conformational changes that expose their hidden BH3 domains. For example, cleavage of Bcl-2 and Bcl-XL by caspases removes the first α-helix from these proteins and converts them into killers, probably functioning akin to BOPs.79 Also, interaction of Bcl-2 with an orphan member of the nuclear receptor family, TR3/Nur77, induces profound conformational changes in Bcl-2, consistent with exposure of its BH3 domain, and correlating with conversion from a protector to a killer.80 Thus, these proapoptotic conformations of antiapoptotic Bcl-2-family proteins may also provide a means of triggering activation of MDPs, though definitive evidence is presently lacking that these proteins function as MDP agonists. Given that all antiapoptotic Bcl-2 family proteins possess a BH3 domain, it would be interesting to compare the BH3 peptides from the six antiapoptotic members of the mammalian Bcl-2-family for their ability to bind and activate the three mammalian MDPs, Bax, Bak, and Bok. Thus far, only Bcl-2 was tested, the BH3 domain of which failed to activate Bax/Bak.81
Additional agonists of MDPs have been reported that contain no apparent BH3 domain, including the SH3-containing protein Bif-1,82, 83 the tumor suppressor p53,84 and the caspase-activating adapter protein ASC.85 Further studies are required to delineate physiological and pathological circumstances in which Bif-1, p53, and ASC play important roles in MDP activation in vivo.
Mechanisms of Suppression of MDPs
The classical suppressors of MDPs are antiapoptotic Bcl-2-family proteins. In 1993, Korsmeyer et al.1 provided the first demonstration that proapoptotic and antiapoptotic members of the Bcl-2 family physically interact, mutually opposing each other. How physical interaction of Bcl-2 and various anti-apoptotic members of the family inhibits the pro-apoptotic functions of MDPs such as Bax, Bak and Bok remains unclear. Various mutagenesis studies have suggested that anti-apoptotic proteins Bcl-2 and Bcl-XL can suppress cell death induction by Bax or Bak even without binding to these MDPs.81, 86 In mammalian cells, this phenomenon can probably be attributed to antiapoptotic proteins sequestering the BH3 domains of MDP agonists such as Bid and Bim, thereby providing a reasonable explanation for the dissociation between binding and suppression of Bax/Bak-dependent killing.87 However, non-binding mutants of Bcl-2 and Bcl-XL can also suppress killing of yeast by Bax, a cellular setting devoid of complications caused by Bid, Bim and other Bcl-2-family proteins. One possible explanation is that assays for detecting binding of Bcl-2 and Bcl-XL to MDPs typically have been biased towards conformations of these proteins associated with the nonmembrane integrated state. Thus, it may be that mutants of Bcl-2 and Bcl-XL that apparently have lost the ability to bind Bax or Bak in solution still remain competent to bind MDPs in their membrane-integrated conformations. Given that chemical crosslinking studies have provided evidence that Bcl-2 or Bcl-XL prevent the oligomerization of Bax or Bak in mitochondrial membranes, an attractive hypothesis is that Bcl-2, Bcl-XL, and the other antiapoptotic Bcl-2-family proteins function essentially as chain-terminating molecules (Figure 2). In this speculative model, MDPs such as Bax and Bak are envisioned as forming polymers in membranes that eventually connect at their ends, analogous to clasping the ends of beaded necklace, thereby creating membrane perturbations that lead to MOP. Antiapoptotic proteins such as Bcl-2 and Bcl-XL latch onto the ends of these polymers, preventing further chain extension. This model is consistent with biophysical studies of membrane permeabilization changes produced by purified Bax in synthetic liposomes in vitro, which have failed to provide evidence of a discrete pore and instead favor a more generalized disruption of membranes.72

Proposed model for chain-termination of Bax/Bak oligomers by anti-apoptotic Bcl-2-family proteins. Bax and Bak form large oligomers, a process blocked by anti-apoptotic Bcl-2-family proteins, Bcl-2 and Bcl-XL. A speculative model is proposed in which Bax or Bak form polymers in membranes, which can loop back to form closed structures that destabilize membranes. Bcl-2 and Bcl-XL play the role of chain terminators, preventing extension of the Bax/Bak polymers. Studies of Bax/Bak oligomerization by the Korsmeyer group have provided critical insights into the mechanisms by which these proteins induce MOP
In addition to anti-apoptotic Bcl-2-family proteins, additional types of endogenous antagonists of MDPs have begun to emerge. For example, Ku70 is a noncatalytic subunit of the DNA-dependent protein kinase involved in responses to genotoxic injury. Ku70 was discovered to bind cytosolic Bax, preventing its translocation into mitochondrial membranes.88 The region of Ku70 responsible was mapped to a minimal Bax-binding domain of five amino-acids, and a synthetic peptide corresponding to this region that was reported to suppress Bax translocation to mitochondria.88, 89 It has been suggested that DNA-damage encourages Ku70 to associate with the DNA-dependent kinase and move into the nucleus, leaving Bax unattended in the cytosol. Moreover, acetylation of Ku70 may also promote its dissociation from Bax, providing a possible explanation for at least one of the many proapoptotic actions of chemical inhibitors of HDACs (reviewed in Yoo and Jones90).
Another antagonist of Bax is Humanin (HN), a small endogenous peptide of 26 amino-acids length in humans that binds to and inhibits Bax translocation to mitochondria.91 HN was originally identified as an anti-apoptotic peptide encoded in a cDNA that rescued neuronal cells from apoptosis induced by presenilin mutants associated with familial Alzheimer's disease and by amyloid-beta-protein, during a functional screen of a cDNA library prepared from the brain of an autopsy-confirmed Alzheimer's disease patient.92 Subsequently, Bax was identified among the possible cellular targets of this endogenous peptide. Mutagenesis studies of HN have so far revealed a perfect correlation between binding to Bax and ability of HN to suppress Bax-induced apoptosis. HN also suppresses Bax-mediated killing of yeast, implying that other proteins are not required. Further, direct high-affinity binding of HN to purified Bax has been demonstrated by fluorescence polarization assay. Based on the data available to date, it has been suggested that HN binds the inactive conformation of Bax and helps to keep Bax inactivate in the cytosol. In addition, another mode of action of the HN peptide has recently been revealed, whereby HN binds the MDP agonists Bid and Bim-EL, preventing them from activating Bax and Bak.93, 94 In this regard, though the 3D structure of Bim-EL is presently unknown, the structures of Bax and Bid are similar, implying that the HN peptide may recognize a conserved structural feature of these proteins. Understanding of the mechanism by which HN suppresses Bax, Bid, and BimEL would be greatly enhanced by the solving of the 3D-structure of a complex of HN with at least one of these proapoptotic proteins.
The Voltage-dependent anion channels (VDACs) are among the most abundant proteins of the outer mitochondrial membrane. Association of VDAC2 with Bak has been proposed by Korsmeyer et al.95 as a mechanism for preventing oligomerization of Bak. Apoptotic stimuli induce dissociation of VDAC2 and Bak, through an unknown mechanism.95, 96 Interestingly, VDAC1 has been reported to bind Bax, but the significance of this interaction is controversial.97, 98, 99, 100
Altogether, the available data indicate that while MDPs may be interchangeable in terms of many or most of their effector functions, the upstream afferent inputs into these proteins vary considerably thus giving each MDP its unique biology (Figure 3).

Regulators of Bax and Bak. Several of the proteins that have been reported to regulate MDP-members Bax or Bak are depicted. Upon inducing their oligomerization, Bax and Bak trigger mitochondrial outermembrane permeabilization (MOP)
Therapeutic Opportunities
Strategies for innovative therapies are beginning to emerge from the accumulating base of knowledge about MDPs and the cellular mechanisms that regulate their functions. Proof of concept experiments from the Korsmeyer group using agonistic BH3 peptides from Bid and Bim to activate MDPs imply that new therapies for inducing apoptosis of tumor cells could potentially be generated by identifying chemical compounds that mimic these BH3 peptides.74 If proven to have an acceptable therapeutic index, such a strategy could complement the ongoing efforts to use compounds mimicking BH3 peptides to antagonize antiapoptotic Bcl-2-family proteins (reviewed in101). However, somatic mutations that inactivate Bax or Bak in tumors, as well as reductions in the levels of their expression, may limit the ability of chemical agonists of MDPs to kill tumor cells.102, 103 Relieving suppression of Bax by promoting dissociation of Ku70 represents another potential strategy for promoting apoptosis of cancer cells. HDAC inhibitory compounds reportedly induce hyperacetylation of Ku70, promoting release of Bax,90 revealing at least one possible strategy. Cancer gene therapy interventions whereby Bax is delivered into tumors represents another approach to exploiting MDPs for cancer therapy.104
For diseases where cytoprotection is the goal, such as stroke, myocardial infarction, and neurodegeneration, compounds that suppress MDPs are highly desired. A series of 3,6-dibromocarbazole piperazine derivatives of 2-propanol that suppress Bax have been described, providing a potential starting point for future attempts to optimize compound potency.105 Using NMR-based strategies, compounds have also been synthesized that bind to and suppress the MDP agonist Bid,106 evidently stabilizing the inactive conformation of this MDP-activating protein. Given that bid knockout mice are protected from cell loss in several disease models, including stroke and hepatitis,107, 108 chemical inhibitors of MDP agonists could be useful for reducing tissue injury in certain disease scenarios. Another approach to MDP suppression is predicated on identification of compounds that mimick the 5′ mer peptide from Ku70 that suppresses Bax activation.89 Compounds that mimic the HN peptide could also be envisioned, and presumably would offer the advantage of simultaneously targeting three proapoptotic proteins, Bax, Bid, and BimEL.91, 93, 94 While MDPs and their endogenous agonists are attractive targets for drug discovery with respect generating novel cytoprotective agents, the requirement for some of them for normal tissue homeostasis raises concerns about chronic suppression of these death-inducing proteins. For example, bax−/− mice have an increased risk of tumorigenesis, and bid−/− mice develop leukemia after a long latent phase. Thus, clinical use of chemical antagonists of MDPs and their endogenous agonists may be limited to acute injury situations. Nevertheless, given that myocardial infraction and stroke account directly or indirectly for over one-third of all deaths in the developed world, successful development of chemical inhibitors of these proapoptotic proteins could have a huge impact, even if relegated only to acute injury scenarios.
In vitro applications of compounds or even membrane-permeable peptides that inhibit MDPs can also be envisioned for biomanufacturing processes where large-scale cell production is involved. Opportunities for ex vivo applications of chemical and peptidyl antagonists of MDPs are likely to expand as stem cell science makes strides towards cell replacement therapies for regenerative medicine.
Despite these many opportunities, it should be recognized that MDPs are challenging targets, as they lack enzymatic activity and thus do not fit the typical model of classical small-molecule drug targets. Nevertheless, with modern advances in structure-based drug optimization, it may be possible to tailor compounds that bind sites on these proteins, either stabilizing their inactive conformations for suppressing death of wanted cells or triggering MDP oligomerization for inducing demise of unwanted cells. The accomplishments in discovery research made by Stanley Korsmeyer and others working in apoptosis research have created a platform for future therapies, leaving a legacy that will benefit the world for many years.
Abbreviations
- Bax:
-
Bcl-2 Antagonist X
- MOP:
-
mitochondrial outer membrane permeability
- BH:
-
Bcl-2 homology
- MDP:
-
multi domain protein
- BOP:
-
BH3-only protein
References
Oltvai Z, Milliman C and Korsmeyer S (1993) Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell 74: 609–619.
Boise L, Gonzalez-Garcia M, Postema C, Ding L, Lindsten T, Turka L, Turka L, Mao X, Nunez G and Thompson C (1993) Bcl-x, a Bcl-2-related gene that functions as a dominant regulator of apoptotic cell death. Cell 74: 597–608.
Yin XM, Oltvai ZN and Korsmeyer SJ (1994) BH1 and BH2 domains of Bcl-2 are required for inhibition of apoptosis and heterodimerization with bax. Nature 369: 321–333.
Korsmeyer SJ, Wei MC, Saito M, Weiler S, Oh KJ and Schlesinger PH (2000) Pro-apoptotic cascade activates BID, which oligomerizes BAK or BAX into pores that result in the release of cytochrome c. Cell Death Differ. 7: 1166–1173.
Strasser A (2005) The role of BH3-only proteins in the immune system. Nat. Rev. Immunol. 5: 189–200.
Sato T, Hanada M, Bodrug S, Irie S, Iwama N, Boise LH, Thompson CB, Golemis E, Fong L, Wang H-G and Reed JC (1994) Interactions among members of the Bcl-2 protein family analyzed with a yeast two-hybrid system. Proc. Natl. Acad. Sci. USA 91: 9238–9242.
Hanada M, Aime-Sempe C, Sato T and Reed JC (1995) Structure-function analysis of Bcl-2 protein. Identification of conserved domains important for homodimerization with Bcl-2 and heterodimerization with Bax. J. Biol. Chem. 270: 11962–11969.
Jurgensmeier JM, Krajewski S, Armstrong R, Wilson GM, Oltersdorf T, Fritz LC, Reed JC and Ottilie S (1997) Bax- and Bak-induced cell death in the fission yeast Schizosaccharomyces pombe. Mol. Biol. Cell. 8: 325–339.
Kawai-Yamada M, Jin L, Yoshinaga K, Hirata A and Uchimiya H (2001) Mammalian Bax-induced plant cell death can be down-regulated by overexpression of Arabidopsis Bax Inhibitor-1 (AtBI-1). Proc. Natl. Acad. Sci. USA 98: 12295–12300.
Huang DC and Strasser A (2000) BH3-only proteins-essential initiators of apoptotic cell death. Cell 103: 839–842.
Reed JC, Doctor KS and Godzik A (2004) The domains of apoptosis: a genomics perspective. Sci. STKE 2004: RE9.
Suzuki M, Youle RJ and Tjandra N (2000) Structure of bax: coregulation of dimer formation and intracellular localization. Cell 103: 645–654.
Chou J, Li H, Salvesen G, Yuan J and Wagner G (1999) Solution structure of BID, an intracellular amplifier of apoptotic signaling. Cell 96: 615–624.
Knudson CM, Tung KSK, Tourtellotte WG, Brown GAJ and Korsmeyer SJ (1995) Bax-deficient mice with lymphoid hyperplasia and male germ cell death. Science 270: 96–99.
White F, Keller-Peck C, Knudson C, Korsmeyer S and Snider W (1998) Widespread elimination of naturally occuring neuronal death in Bax-deficient mice. J. Neurosci. 18: 1428–1439.
Shibata MA, Liu ML, Knudson MC, Shibata E, Yoshidome K, Bandey T, Korsmeyer SJ and Green JE. (1999) Haploid loss of bax leads to accelerated mammary tumor development in C3(1)/SV40-TAg transgenic mice: reduction in protective apoptotic response at the preneoplastic stage. EMBO J. 18: 2692–2701.
Knudson CM, Johnson GM, Lin Y and Korsmeyer SJ (2001) Bax accelerates tumorigenesis in p53-deficient mice. Cancer Res. 61: 659–665.
Eischen CM, Rehg JE, Korsmeyer SJ and Cleveland JL (2002) Loss of Bax alters tumor spectrum and tumor numbers in ARF-deficient mice. Cancer Res. 62: 2184–2191.
Deckwerth TL, Elliott JL, Knudson CM, Johnson JEM, Snider WD and Korsmeyer SJ (1996) BAX is required for neuronal death after trophic factor deprivation and during development. Neuron 17: 401–411.
Gillardon F, Zimmermann M, Uhlmann E, Krajewski S, Reed JC and Klimaschewski L (1996) Antisense oligodeoxynucleotides to bax mRNA promote survival of rat sympathetic neurons in culture. J. Neurosci. Res. 43: 726–734.
Miyashita T and Reed JC (1995) Tumor suppressor p53 is a direct transcriptional activator of human Bax gene. Cell 80: 293–299.
Sakhi S, Bruce A, Sun N, Tocco G, Baudry M and Schreiber SS (1994) p53 induction is associated with neuronal damage in the central nervous system. Proc. Natl. Acad. Sci. USA 91: 7525–7529.
Manev H, Kharlamov A and Armstrong DM (1994) Photochemical brain injury in rats triggers DNA fragmentation, p53 and HSP72. Neuroreport 5: 2661–2664.
Morrison RS, Wenzel HJ, Kinoshita Y, Robbins CA, Donehower LA and Schwartzkroin PA (1996) Loss of the p53 tumor suppressor gene protects neurons from kainate-induced cell death. J. Neurosci. 16: 1337–1345.
Wood KA and Youle RJ (1995) The role of free readicals and p53 in neuron apoptosis in vivo. J. Neurosci. 15: 5851–5857.
Krajewski S, Mai JK, Krajewska M, Sikorska M, Mossakowski MJ and Reed JC (1995) Upregulation of Bax protein levels in neurons following cerebral ischemia. J. Neurosci. 15: 6364–6376.
Hara A, Iwai T, Niwa M, Uematsu T, Yoshimi N, Tanaka T and Mori H (1996) Immunohistochemical detection of Bax and Bcl-2 proteins in gerbil hippocampus following transient forebrain ischemia. Brain Res. 711: 249–253.
Krajewska M, Zapata JM, Meinhold-Heerlein I, Hedayat H, Monks A, Bettendorf H and Krajewski S (2002) Expression of Bcl-2 family member Bid in normal and malignant tissues. Neoplasia 4: 129–140.
Gillardon F, Lenz C, Waschke KF, Krajewski S, Reed JC, Zimmerman M and Kuschinsky W (1996) Altered expression of Bcl-2, Bcl-X, BAX, and c-Fos colocalizes with DNA fragmentation and ischemic cell damage following middle cerebral artery occlusion in rats. Mol. Brain Res. 40: 254–260.
Gibson ME, Han BH, Choi J, Knudson CM, Korsmeyer SJ, Parsadanian M and Holtzman DM (2001) BAX contributes to apoptotic-like death following neonatal hypoxia-ischemia: evidence for distinct apoptosis pathways. Mol. Med. 7: 644–655.
Hughes PE, Alexi T, Yoshida T, Schreiber SS and Knusel B (1996) Excitotoxic lesion on rat brain with quinolinic acid induces expression of p53 messenger RNA and protein and p53-inducible genes BAX and GADD-45 in brain areas showing DNA fragmentation. Neuroscience 74: 1143–1160.
Gillardon F, Wickert H and Zimmerman M (1995) Up-regulation of bax and down-regulation of bcl-2 is associated with kainate-induced apoptosis in mouse brain. Neurosci. Lett. 192: 85–88.
Paradis E, Douillard H, Koutroumanis M, Goodyer C and LeBlanc A (1996) Amyloid β peptide of Alzheimer's disease downregulates Bcl-2 and upregulates Bax expression in human neurons. J. Neurosci. 16: 7533–7539.
Hassouna I, Wickert H, Zimmerman M and Gillardon F (1996) Increase in bax expression in substantia nigra following 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) treatment of mice. Neurosci. Lett. 204: 85–88.
Gillardon F, Klimaschewski L, Wickert H, Krajewski S, Reed JC and Zimmermann M (1996) Expression pattern of candidate cell death effector proteins Bax, Bcl-2, Bcl-X, and c-Jun in sensory and motor neurons following sciatic nerve transection in the rat. Brain Res. 739: 244–250.
Estus S, Zaks WJ, Freeman RS, Gruda M, Bravo R and Johnson Jr EM (1994) Altered gene expression in neurons during programmed cell death: identification of c-jun as necessary for neuronal apoptosis. J. Cell Biol. 127: 1717–1727.
Mu X, He J, Anderson DW, Trojanowski JQ and Springer JE (1996) Altered expression of bcl-2 and bax mRNA in amyotrophic lateral sclerosis spinal cord motor neurons. Ann. Neurol. 40: 379–386.
Vukosavic S, Dubois-Dauphin M, Romero N and Przedborski S (1999) Bax and Bcl-2 interaction in a transgenic mouse model of familial amyotrophic lateral sclerosis. J. Neurochem. 73: 2460–2468.
Krajewski S, James H, Ross J, Blumberg B, Epstein L, Gendelman H, Dewhurst S, Sharer L, Reed J and Gelbard H (1997) Expression of pro- and anti-apoptosis gene products in brains from paediatric patients with HIV-1 encephalitis. Neuropathol. Appl. Neurobiol. 23: 242–253.
Misao J, Hayakawa Y, Ohno M, Kato S, Fujiwara T and Fujiwara H (1996) Expression of bcl-2 protein, an inhibitor of apoptosis, and Bax, an accelerator of apoptosis, in ventricular myocytes of human hearts with myocardial infarction. Circulation 94: 1506–1512.
Russell Hr, Lee Y, Miller HL, Zhao J and McKinnon PJ (2002) Murine ovarian development is not affected by inactivation of the Bcl-2 family member diva. Mol. Cell. Biol. 22: 6866–6870.
Lindsten T, Ross AJ, King A, Zong W, Rathmell JC, Shiels HA, Ulrich E, Waymire KG, Mahar P, Frauwirth K, Chen Y, Wei M, Eng VM, Adelman DM, Simon MC, Ma A, Golden JA, Evan G, Korsmeyer SJ, MacGregor GR and Thompson CB (2000) The combined functions of proapoptotic Bcl-2 family members Bak and Bax are essential for normal development of multiple tissues. Mol. Cell. 6: 1389–1399.
Hsu S, Kaipa A, McGee E, Lomeli M and Hsueh A (1997) Bok is a pro-apoptotic Bcl-2 protein with restricted expression in reproductive tissues and heterodimerizes with selective anti-apoptotic Bcl-2 family members. Proc. Natl. Acad. Sci. USA 94: 12401–12406.
Lee R, Chen J, Matthews CP, McDougall JK and Neiman PE (2001) Characterization of NR13-related human cell death regulator, Boo/Diva, in normal and cancer tissues. Biochim. Biophys. Acta 1520: 187–194.
Krajewski S, Krajewska M and Reed JC (1996) Immunohistochemical analysis of in vivo patterns of Bak expression, a pro-apoptotic member of the Bcl-2 protein family. Cancer Res. 56: 2849–2855.
Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB and Korsmeyer SJ (2001) Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science 292: 727–730.
Hahn P, Lindsten T, Lyubarsky A, Ying GS, Pugh Jr EN, Thompson CB and Dunaief JL. (2004) Deficiency of Bax and Bak protects photoreceptors from light damage in vivo. Cell Death Differ. 11: 1192–1197.
Shimizu S, Kanaseki T, Mizushima N, Mizuta T, Arakawa-Kobayashi S, Thompson CB and Tsujimoto Y (2004) Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat. Cell. Biol. 6: 1221–1228.
Martinou J-C and Green DR (2001) Breaking the mitochondrial barrier. Nat. Rev. Mol. Cell. Biol. 2: 63–67.
Waterhouse NJ, Ricci JE and Green DR (2002) And all of a sudden it's over: mitochondrial outer-membrane permeabilization in apoptosis. Biochimie 84: 113–121.
Cheng EH-Y, Wei MC, Weiler S, Flavell RA, Mak TW, Lindsten T and Korsmeyer SJ (2001) BCL-2, BCL-XL sequester BH3 domain-only molecules preventing BAX- and BAK-mediated mitochondrial apoptosis. Mol. Cell. 8: 705–711.
Marzo I, Brenner C, Zamzami N, Jürgensmeier JM, Susin SA, Vieira HLA, Prevost M-C, Xie Z, Matsuyama S, Reed JC and Kroemer G (1998) Bax and adenine nucleotide translocator cooperate in the mitochondrial control of apoptosis. Science 281: 2027–2031.
Matsuyama S, Xu Q, Velours J and Reed JC (1998) Mitochondrial F0F1-ATPase proton-pump is required for function of pro-apoptotic protein bax in yeast and mammalian cells. Mol. Cell. 1: 327–336.
Milanesi E, Costantini P, Gambalunga A, Colonna R, Petronilli V, Cabrelle A, Semenzato G, Cesura AM, Pinard E and Bernardi P (2006) The mitochondrial effects of small organic ligands of BCL-2. Sensitization of BCL-2-overexpressing cells to apoptosis by a pyrimidine-2,4,6-trione derivative. J. Biol. Chem. 281: 10066–10072.
Bernardi P, Krauskopf A, Basson E, Petronilli V, Blachly-Dyson E, Di Lisa F and Forte MA (2006) The mitochondrial permeability transition from in vitro artifact to disease target. FEBS J. 273: 2077–2099.
Reed JC and Green DR (2002) Remodeling for demolition: changes in mitochondrial ultrastructure during apoptosis. Mol. Cell. 9: 1–3.
Nechushtan A, Smith CL, Lamensdorf I, Yoon S-H and Youle RJ (2001) Bax and Bak coalesce into novel mitochondria-associated clusters during apoptosis. J. Cell. Biol. 153: 1265–1276.
Szabadkai G, Simoni AM, Chami M, Wieckowski MR, Youle RJ and Rizzuto R (2004) Drp-1-dependent division of the mitochondrial network blocks intraorganellar Ca2+ waves and protects against Ca2+-mediated apoptosis. Mol. Cell. 16: 59–68.
Inohara N, Gourley TS, Carrio R, Muniz M, Merino J, Garcia I, Koseki T, Hu Y, Chen S and Nunez G (1998) Diva, a Bcl-2 homologue that binds directly to Apaf-1 and induces BH3-independent cell death. J. Biol. Chem. 273: 32479–32486.
Oakes SA, Scorrano L, Opferman JT, Bassik MC, Nishino M, Pozzan T and Korsmeyer SJ (2005) Proapoptotic BAX and BAK regulate the type 1 inositol trisphosphate receptor and calcium leak from the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA 102: 105–110.
Nutt LK, Pataer A, Pahler J, Fang B, Roth J, McConkey DJ and Swisher SG (2002) Bax and Bak promote apoptosis by modulating endoplasmic reticular and mitochondrial Ca2+ stores. J. Biol. Chem. 277: 9219–9225.
Demaurex N and Distelhorst C (2003) Apoptosis--the calcium connection. Science 300: 65–67.
Chami M, Prandini A, Campanella M, Pinton P, Szabadkai G, Reed JC and Rizzuto R (2004) Bcl-2 and Bax exert opposing effects on Ca2+ signaling, which do not depend on their putative pore-forming region. J. Biol. Chem. 279: 54581–54589.
White C, Li C, Yang J, Petrenko NB, Madesh M, Thompson CB and Foskett JK (2005) The endoplasmic reticulum gateway to apoptosis by Bcl-X(L) modulation of the InsP3R. Nat. Cell. Biol. 7: 1021–1028.
Chen R, Valencia I, Zhong F, McColl KS, Roderick HL, Bootman MD, Berridge MJ, Conway SJ, Holmes AB, Mignery GA, Velez P and Distelhorst CW (2004) Bcl-2 functionally interacts with inositol 1,4,5-trisphosphate receptors to regulate calcium release from the ER in response to inositol 1,4,5-trisphosphate. J. Cell. Biol. 166: 193–203.
Kuo TH, Kim HR, Zhu L, Yu Y, Lin HM and Tsang W (1998) Modulation of endoplasmic reticulum calcium pump by Bcl-2. Oncogene 17: 1903–1910.
Baffy G, Miyashita T, Williamson JR and Reed JC (1993) Apoptosis induced by withdrawal of interleukin-3 (IL-3) from an IL-3-dependent hematopoietic cell line is associated with repartitioning of intracellular calcium and is blocked by enforced Bcl-2 oncoprotein production. J. Biol. Chem. 268: 6511–6519.
Palmer AE, Jin C, Reed JC and Tsien RY (2004) Bcl-2-mediated alterations in endoplasmic reticulum Ca2+ analyzed with an improved genetically encoded fluorescent sensor. Proc. Natl. Acad. Sci. USA 101: 17404–17409.
Wolter KG, Hsu YT, Smith CL, Nechushtan A, Xi XG and Youle RJ (1997) Movement of bax from the cytosol to mitochondria during apoptosis. J. Cell. Biol. 139: 1281–1292.
Nechushtan A, Smith C, Hsu Y-T and Youle R (1999) Conformation of the Bax C-terminus regulates subcellular location and cell death. EMBO J. 18: 2330–2341.
Gross A, McDonnell JM and Korsmeyer SJ (1999) BCL-2 family members and the mitochondria in apoptosis. Genes Dev. 13: 1899–1911.
Kuwana T, Mackey MR, Perkins G, Ellisman MH, Latterich M, Schneiter R, Green DR and Newmeyer DD (2002) Bid, bax, and lipids cooperate to form supramolecular openings in the outer mitochondrial membrane. Cell 111: 331–342.
Wei MC, Lindsten T, Mootha VK, Weiler S, Gross A, Ashiya M, Thompson CB and Korsmeyer SJ (2000) tBID, a membrane-targeted death ligand, oligomerizes BAK to release cytochrome c. Genes Dev. 14: 2060–2071.
Letai A, Bassik MC, Walensky LD, Sorcinelli MD, Weiler S and Korsmeyer SJ (2002) Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell 2: 183–192.
Wang K, Yin W-M, Chao DT, Milliman CL and Korsmeyer SJ (1996) BID: a novel BH3 domain-only death agonist. Genes Dev. 10: 2859–2869.
Strasser A, Puthalakath H, Bouillet P, Huang DC, O'Connor L, O'Reilly LA, Cullen L, Cory S and Adams JM (2000) The role of bim, a proapoptotic BH3-only member of the Bcl-2 family in cell-death control. Ann. NY Acad Sci. 917: 541–548.
Puthalakath H, Huang D, O'Reilly L, King S and Strasser A (1999) The proapoptotic activity of the Bcl-2 family member bim is regulated by interaction with the dynein motor complex. Mol. Cell. 3: 287–296.
Zhu Y, Swanson BJ, Wang M, Hildeman DA, Schaefer BC, Liu X, Suzuki H, Mihara K, Kappler J and Marrack P (2004) Constitutive association of the proapoptotic protein Bim with Bcl-2-related proteins on mitochondria in T cells. Proc. Natl. Acad. Sci. USA 101: 7681–7686.
Cheng E, Clem R, Ravi R, Kirsch D, Kastan M, Bedi A, Ueno K and Hardwick J (1997) Conversion of Bcl-2 to a Bax-like death effector by caspases. Science 278: 1966–1968.
Lin B, Kolluri SK, Lin F, Liu W, Han Y-H, Cao X, Dawson MI, Reed JC and Zhang X-K (2004) Conversion of Bcl-2 from protector to killer by interaction with nuclear orphan receptor TR3/NGFI-B/Nur77. Cell 116: 527–540.
Zha H and Reed JC (1997) Heterodimerization-independent functions of cell death regulatory proteins Bax and Bcl-2 in yeast and mammalian cells. J. Biol. Chem. 272: 31482–31488.
Cuddeback SM, Yamaguchi H, Komatsu K, Miyashita T, Yamada M, Wu C, Singh S and Wang HG (2001) Molecular cloning and characterization of Bif-1. A novel Src homology 3 domain-containing protein that associates with Bax. J. Biol. Chem. 276: 23.
Takahashi Y, Karbowski M, Yamaguchi H, Kazi A, Wu J, Sebti SM, Youle RJ and Wang HG (2005) Loss of Bif-1 suppresses Bax/Bak conformational change and mitochondrial apoptosis. Mol. Cell. Biol. 25: 9369–9382.
Chipuk JE, Kuwana T, Bouchier-Hayes L, Droin NM, Newmeyer DD, Schuler M and Green DR (2004) Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science 303: 1010–1014.
Ohtsuka T, Ryu H, Minamishima YA, Macip S, Sagara J, Nakayama KI, Aaronson SA and Lee SW (2004) ASC is a Bax adaptor and regulates the p53-Bax mitochondrial apoptosis pathway. Nat. Cell. Biol. 6: 121–128.
Simonian PL, Grillot DAM, Merino R and Nunez G (1996) Bax can antagonize Bcl-XL during etoposide and cisplatin-induced cell death independently of its heterodimerization with Bcl-XL. J. Biol. Chem. 271: 22764–22772.
Danial NN and Korsmeyer SJ (2004) Cell death: critical control points. Cell 116: 205–219.
Sawada M, Sun W, Hayes P, Leskov K, Boothman DA and Matsuyama S (2003) Ku70 suppresses the apoptotic translocation of Bax to mitochondria. Nat. Cell. Biol. 5: 320–329.
Sawada M, Hayes P and Matsuyama S (2003) Cytoprotective membrane-permeable peptides designed from the Bax-binding domain of Ku70. Nat. Cell. Biol. 5: 352–357.
Yoo CB and Jones PA (2006) Epigenetic therapy of cancer: past, present and future. Nat. Rev. Drug Discov. 5: 37–50.
Guo B, Zhai D, Cabezas E, Welsh K, Nouraini S, Satterthwait AC and Reed JC (2003) Humanin peptide suppresses apoptosis by interfering with Bax activation. Nature 423: 456–461.
Hashimoto Y, Niikura T, Tajima H, Yasukawa T, Sudo H, Ito Y, Kita Y, Kawasumi M, Kouyama K, Doyu M, Sobue G, Koide T, Tsuji S, Lang J, Kurokawa K and Nishimoto I (2001) A resuce factor abolishing neuronal cell death by a wide spectrum of familial Alzheimer's disease genes and Aß. Proc. Natl. Acad. Sci. USA 98: 6336–6341.
Zhai D, Luciano F, Zhu X, Guo B, Satterthwait A and Reed JC (2005) Humanin binds and nullifies Bid activity by blocking its activation of Bax and Bak. J. Biol. Chem. 280: 15815–15824.
Luciano F, Zhai D, Zhu X, Bailly-Maitre B, Ricci JE, Satterthwait AC and Reed JC (2005) Cytoprotective peptide Humanin binds and inhibits pro-apoptotic Bcl-2/Bax-family protein BimEL. J. Biol. Chem. 280: 15825–15835.
Cheng EH, Sheiko TV, Fisher JK, Craigen WJ and Korsmeyer SJ (2003) VDAC2 inhibits BAK activation and mitochondrial apoptosis. Science 301: 513–517.
Chandra D, Choy G, Daniel PT and Tang DG (2005) Bax-dependent regulation of Bak by voltage-dependent anion channel 2. J. Biol. Chem. 280: 19051–19061.
Debatin KM, Poncet D and Kroemer G (2002) Chemotherapy: targeting the mitochondrial cell death pathway. Oncogene 21: 8786–8803.
Tsujimoto Y (2003) Cell death regulation by the Bcl-2 protein family in the mitochondria. J. Cell. Physiol. 195: 158–167.
Rostovtseva TK, Antonsson B, Suzuki M, Youle RJ, Colombini M and Bezrukov SM (2004) Bid, but not Bax, regulates VDAC channels. J. Biol. Chem. 279: 13575–13583.
Zalk R, Israelson A, Garty ES, Azoulay-Zohar H and Shoshan-Barmatz V (2005) Oligomeric states of the voltage-dependent anion channel and cytochrome c release from mitochondria. Biochem J. 386: 73–83.
Reed JC and Pellecchia M (2005) Apoptosis-based Therapies for Hematological Malignancies. Blood 106: 408–418.
Rampino N, Yamamoto H, Ionov Y, Li Y, Sawai H, Reed JC and Perucho M (1997) Somatic frameshift mutations in the BAX gene in colon cancers of the microsatellite mutator phenotype. Science 275: 967–969.
Kondo S, Shinomura Y, Miyazaki Y, Kiyohara T, Tsutsui S, Kitamura S, Nagasawa Y, Nakahara M, Kanayama S and Matsuzawa Y (2000) Mutations of the bak gene in human gastric and colorectal cancers. Cancer Res. 60: 4328–4330.
Xiang J, Gomez-Navarro J, Arafat W, Liu B, Barker SD, Alvarez RD, Siegal GP and Curiel DT (2000) Pro-apoptotic treatment with an adenovirus encoding Bax enhances the effect of chemotherapy in ovarian cancer. J. Gene Med. 2: 97–106.
Bombrun A, Gerber P, Casi G, Terradillos O, Antonsson B and Halazy S (2003) 3,6-dibromocarbazole piperazine derivatives of 2-propanol as first inhibitors of cytochrome c release via Bax channel modulation. J. Med. Chem. 46: 4365–4368.
Becattini B, Sareth S, Zhai D, Crowell K, Leone M, Reed JC and Pellecchia M (2004) Targeting apoptosis via chemical design: Inhibition of Bid-induced cell-death by small organic molecules. Chem. Biol. 11: 1107–1117.
Plesnila N, Zinkel S, Le DA, Amin-Hanjani S, Wu Y, Qiu J, Chiarugi A, Thomas SS, Kohane DS, Korsmeyer SJ and Moskowitz MA (2001) BID mediates neuronal cell death after oxygen/ glucose deprivation and focal cerebral ischemia. Proc. Natl. Acad. Sci. USA 98: 15318–15323.
Yin X-M, Wang K, Gross A, Klocke B, Roth KA and Korsmeyer SJ (1999) Bid-deficient mice are resistant to Fas-induced hepatocellular apoptosis. Nature 400: 886–891.
Acknowledgements
We thank Melanie Hanaii and Judie Valois for assistance with manuscript preparation, and the NIH for generous support.
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by C Borner
Rights and permissions
About this article
Cite this article
Reed, J. Proapoptotic multidomain Bcl-2/Bax-family proteins: mechanisms, physiological roles, and therapeutic opportunities. Cell Death Differ 13, 1378–1386 (2006). https://doi.org/10.1038/sj.cdd.4401975
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.cdd.4401975
Keywords
This article is cited by
Synthesis, antiproliferative activity, and molecular modeling of novel 4-methylcoumarin derivatives and/or nitric oxide donor hybrids
Molecular Diversity (2023)
Role of apoptosis and autophagy in mediating tramadol-induced neurodegeneration in the rat hippocampus
Molecular Biology Reports (2023)
Environmental hypoxia induces apoptosis in large yellow croaker Larimichthys crocea via both intrinsic and extrinsic pathways
Journal of Oceanology and Limnology (2023)
The effects of mifepristone on the structure of human decidua and chorion and Bax and Bcl-2 expression at early stage of pregnancy
BMC Pharmacology and Toxicology (2022)
Citronellol Prevents 6-OHDA-Induced Oxidative Stress, Mitochondrial Dysfunction, and Apoptosis in Parkinson Disease Model of SH-SY5Y Cells via Modulating ROS-NO, MAPK/ERK, and PI3K/Akt Signaling Pathways
Neurotoxicity Research (2022)
