
Abstract
Cell death is an essential biological process for physiological growth and development. Three classical forms of cell death—apoptosis, autophagy, and necrosis—display distinct morphological features by activating specific signaling pathways. With recent research advances, we have started to appreciate that these cell death processes can cross-talk through interconnecting, even overlapping, signaling pathways, and the final cell fate is the result of the interplay of different cell death programs. This review provides an insight into the independence of and associations among these three types of cell death and explores the significance of cell death under the specific conditions of human diseases, particularly neurodegenerative diseases and cancer.
Similar content being viewed by others


Introduction
For unicellular organisms, cell death is the end of life. However, for multicellular organisms, cell death is an essential biological process for physiological growth and development. Deregulation of cell death is involved in the pathogenesis of a wide range of human diseases, such as neurodegenerative diseases and cancer1. Three classical forms of cell death—apoptosis, autophagy, and necrosis—display distinct morphological features by activating specific signaling pathways2. In brief, apoptosis is a caspase-mediated programmed cell death3,4 that is characterized by chromosome condensation, nuclear fragmentation, and membrane blebbing5. In contrast to apoptosis, necrosis is considered to be an unregulated, accidental cell death caused by nonspecific, or non-physiological stress inducers and is characterized by the expansion of cellular organelles, plasma membrane rupture, and subsequent inflammatory responses caused by release of the intracellular contents6,7. The third form of cell death, autophagy, is accompanied by the formation of the autophagosome, which is a bilayer vesicle containing damaged organelles, proteins, and other cytoplasmic components. The autophagosomes fuse with the lysosomes, degrading cellular macromolecules and organelles and producing renewable energy and metabolites for cells8. Autophagy acts as a pro-survival mechanism but can also induce autophagic cell death, which is currently an active research area in cell death9,10. Our understanding has been rapidly expanded in the last decades owing to the great advances in cell death research. Identification of the programmed forms of necrosis11 has changed our perception about necrosis. More importantly, we have started to appreciate that the molecular mechanisms of various types of cell death are distinct but also overlapping. There are multiple signaling pathways independently controlling different types of cell death. However, they are interconnected, can be activated simultaneously, and can operate in parallel in cells in response to stress.
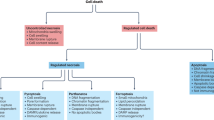
Necrosis and apoptosis are two types of cell death with different mechanisms5,12. Autophagy can be described as a degradation mechanism rather than as a form of cell death, although it can also induce cell death9. Of the cell death types, autophagy has the highest survival superiority, followed by apoptosis, with necrosis having the lowest survival superiority. Autophagy is instinctively induced prior to apoptosis when cells are stimulated by stress, and apoptosis rather than necrosis is induced if autophagy is inhibited or ineffective8,13,14,15,16. Thus, two or three types of cell death may be induced simultaneously or successively when cells are exposed to certain stimuli. If the three types of cell death are placed on an axis according to their survival superiority, autophagy, and necrosis would be placed at opposing ends, whereas apoptosis would be placed in the middle; furthermore, programmed necrosis would be placed between necrosis and apoptosis (Fig. 1).

Survival superiority among the different types of cell death
Herein, we review the different types of cell death, discuss the specific mechanisms involved in each type of cell death and connections among them, and explore the impact of different types of cell death on disease treatment.
Distinct characteristics of apoptosis, necrosis, and autophagy
Apoptosis
Apoptosis is generally considered a caspase-mediated programmed cell death3,4,17,18. Apoptotic cells display distinct morphological characteristics, including cell shrinkage, chromosome condensation, nuclear fragmentation (late stage), plasma membrane blebbing and the formation of apoptotic bodies, and exhibit biochemical changes, such as the exposure of phosphatidyl-l-serine on the outer plasma membrane (early stage)19,20,21. Apoptosis can be activated via either the death receptor-mediated apoptosis pathway, the mitochondria-dependent apoptosis pathway or endoplasmic reticulum (ER) stress-induced apoptosis pathways (Fig. 2)22.

Mechanisms of apoptosis. In the exogenous pathway, the binding of FASL, TNF-α, or TRAIL to their corresponding receptors can transform procaspase-8 to caspase-8 through autohydrolysis. In type I cells, activated caspase-8 can activate caspase-3, followed by apoptosis. In type II cells, activated caspase-8 can hydrolyze Bid to tBid, and then tBid interacts with Bax/Bak, which is located on mitochondria, to induce apoptosis. In the intrinsic apoptosis pathway, DNA damage, growth factor withdrawal, oxidative stress, or toxic damage can destroy the homeostasis of the mitochondria, typically controlled by the Bcl-2 family members, and can lead to increased mitochondrial membrane permeability to induce cytochrome c release from the intermembrane space of the mitochondria. In addition, the released cytochrome c can interact with Apaf-1 and caspase-9 to activate caspase-3 and induce apoptosis. In the endoplasmic reticulum stress-induced apoptosis pathway, the disturbance in calcium homeostasis and excessive accumulation of unwanted proteins in the endoplasmic reticulum induce caspase-12-mediated apoptosis, in which activated caspase-12 translocates from the ER into the cytosol to directly cleave caspase-9 and then activate caspase-3
The death receptor-mediated apoptosis pathway is activated upon the binding of the Fas ligand, TNF-α (tumor necrosis factor α), or TRAIL to the corresponding death receptors23,24. The adaptor protein FADD23,25 and the procaspase-8 protein form a complex, namely, death-inducing signaling complex (DISC). In DISC, procaspase-8 is activated by autohydrolysis26. The activated caspase-8 transduces the apoptosis signal through either the activation of caspase-3 or cleavage of Bid to truncated Bid (tBid). tBid translocates to the mitochondria, resulting in conformational changes in Bax and Bak and their oligomerization for pore formation in the outer mitochondrial membrane26,27.
The mitochondrial-dependent pathway can be activated by various stress inducers such as DNA damage, growth factor withdrawal, and oxidative stress28,29. The Bcl-2 family of proteins controls this intrinsic pathway by regulating the permeability of the mitochondrial outer membrane30,31,32. Upon release from the mitochondria into the cytoplasm, cytochrome c combines with Apaf-1 to promote caspase-9 activation, which, in turn, activates effector caspases33,34 to trigger a cascade of proteolytic events.
In addition, ER stresses, such as calcium homeostasis disturbance, excessive unfolded, or misfolded protein accumulation in the ER, nutrient deprivation, and hypoxia, can induce apoptosis. This apoptosis is mediated by caspase-12, an ER-resistant caspase22. Activated caspase-12 directly cleaves caspase-9 after translocation from the ER into the cytosol, followed by caspase-3 activation35. The molecular mechanisms of activation of caspase-12 during ER stress include forming a complex with the inositol-requiring enzyme-1α-TNF receptor-associated factor 2 (TRAF2) complex36, or by calpains, a family of Ca2+-dependent intracellular cysteine proteases37.
Autophagy
Autophagy is a self-degradative process in response to various stresses, including nutrient deficiency, organelle damage, hypoxia, reactive oxygen species (ROS), ER stress, and drug treatment. The process of autophagy involves four key steps—initiation, nucleation, fusion of autophagosome and lysosome, and hydrolyzation. Our understanding of the molecular mechanisms of autophagy starts from research in yeast. A set of autophagy regulatory molecules was identified by genetic screening in yeast, particularly autophagy (Atg)-related proteins, which are the main players in autophagy. The assembly and aggregation of the Atg1 complex, which includes Atg1, Atg13, Atg17, Atg29, and Atg3138,39, are required for the formation of the phagophore at the initiation step40. However, in mammals, the UNC-51-like kinase 1 (ULK)-mAtg13-FIP200 complex, comprising the homologous analogs to yeast Atg1, Atg13, and Atg17, is formed41. At the step of nucleation, phagophore formation at the ER and other membranes is controlled by a complex of the class III PI-3 kinase VPS34, Atg6 (known as Beclin1 in mammals), Atg14, and Vps15. Atg9 and vesicle membrane protein VMP1, which circulate in the Golgi complex, autophagosomes, and endosomes, may be involved in the transport of lipids to the isolation membrane42,43. The expansion and closure of the autophagosome require two ubiquitin-like protein-conjugated systems, namely, Atg12 and Atg8 (Atg8 is also known as LC3 in mammals)44. The Atg12 system includes five Atg proteins, Atg5, Atg7, Atg10, Atg12, and Atg1645,46,47. Atg12 is activated by Atg7, which is an E1-like enzyme45, and is then transferred to the E2-like enzyme Atg1047. Finally, the C-terminal glycine of Atg12 covalently binds to the Lys149 side chain of Atg5 before binding to the dimer protein Atg16 to form the E3-like complex45. The Atg8 system including four Atg proteins, Atg3, Atg4, Atg7, and Atg8, represents another ubiquitin-like protein-conjugated system48. Atg8 is cleaved by Atg4, a cysteine protease, and exposes its C-terminal glycine residue (LC3 I in mammals)49. Atg8 is further activated by Atg7, an E1-like enzyme and is then transferred to Atg3, an E2-like enzyme48, before covalently binding to the amidogen of PE through the E3-like Atg12-Atg5-Atg16 complex48,50. The Atg8-PE covalent structure (LC3 II in mammals) confers Atg8 membrane tethering and hemifusion ability and plays a critical role in autophagosome formation. LC3 II is associated with both the outer and inner membranes of the autophagosome and is a typical marker of autophagy formation. The Atg8-PE covalent structure can be reversibly cleaved to Atg8 by Atg4 for the recycling of Atg8. Subsequently, the fusion of autophagosome and lysosome is mediated by SNARE (soluble N-ethylmaleimide-sensitive factor attachment protein receptor)-like proteins51,52,53,54. Finally, at a low pH, various lysosomal enzymes hydrolyze all types of damaged organelles, proteins, lipids, and nucleic acids40,55. A diagram of the autophagy process is shown in Fig. 3.

Mechanisms of autophagy. The mechanisms of autophagy can be divided into four steps, initiation, nucleation, expansion and closure, and fusion and degradation. In mammals, the assembly of the ULK1/2 complex is necessary for the formation of the phagophore assembly site, whereas the ULK1/2 complex is regulated by mTORC1, which is positively regulated by PI3K/AKT and negatively regulated by AMPK. Growth factors activate the PI3K/Akt pathway through receptor tyrosine kinases (RTKs). The Beclin1 complex, which is usually suppressed by Bcl-2, is activated and drives the isolation membrane to nucleation, and the transmembrane protein Atg9 and vesicle membrane protein VMP1 may be involved in the transport of lipids to the isolation membrane. In addition, two ubiquitin-like protein-conjugated systems (Atg12 and LC3 systems) are needed in this process. Subsequently, the autophagosome and lysosome fusion is mediated by SNARE-like protein, and, finally, various lysosomal enzymes hydrolyze all types of damaged organelles, proteins, lipids, and nucleic acids
Necrosis and necroptosis
Unlike apoptosis, necrosis is often considered to be an unregulated and accidental cell death2. However, the identification of programmed necrosis supported the existence of multiple nonapoptotic regulated cell death mechanisms. Several types of programmed necrosis have been reported, including necroptosis56, parthanatos57, ferroptosis58, pyroptosis59, and NETosis60. Here, we focus on necroptosis, a type of regulated necrotic cell death that shares several key signaling pathways with apoptosis. Most of the knowledge regarding necroptosis originated from investigations of TNF signaling. TNF is a pleiotropic cytokine that plays an important role in the process of inflammation61. TNF is also a potent cell death inducer under certain conditions through binding to TNFR12. Although an early study revealed that TNF-induced RIPK1-mediated caspase-independent cell death62, TNF-induced nonapoptotic cell death did not attract much attention, until researchers further uncovered that cells executed necrosis-like death when apoptosis was blocked63,64.
Necroptosis is initiated by the engagement of death receptors, such as TNFR165,66,67 and Toll-like receptors (TLRs)68,69,70. TNF binding to the death receptor TNFR induces the conformational changes of TNFR, which recruits multiple proteins, including TNFR1-associated death domain protein (TRADD), RIPK1, TRAF2, E3 ubiquitin ligases, cIAP1/2, and LUBAC, to form TRADD and the RIPK1-dependent complex I. This multi-protein complex transduces pro-inflammatory and pro-survival signals by recruiting TGF-activated kinase 1 (TAK1)-binding protein (TAB) complexes and Iκb kinase (IKK) complexes consisting of IKK1, IKK2, and NF-κB essential modulator (NEMO)71,72 to activate NF-κB signaling, AP-1 signaling, and mitogen-activated protein kinase signaling. When RIPK1 is deubiquitinated by cylindromatosis lysine 63 deubiquitinase73, complex I becomes unstable and renders the dissociation of RIPK1 and the formation of another complex, termed complex IIa, by interacting with TRADD, FADD, pro-caspase-8, and FLIP73. Pro-caspase-8, together with FLIPL, cleaves RIPK1 to prevent necroptosis and activate apoptosis signaling16,74,75. TNF–TNFR signaling can also induce apoptosis through the formation of complex IIb when the function of IAP (inhibitors of apoptosis)16, TAK176, NEMO, and/or Pellino3 is blocked77. This complex IIb comprises RIPK1, RIPK3, FADD, pro-caspase-8, and FLIPL and causes RIPK1-dependent apoptosis15. However, complex IIb may be further transformed into the necrosome, a microfilament-like complex, when the levels of RIPK3 and mixed lineage kinase domain-like (MLKL) are sufficiently high and the activity of caspase-8 is inhibited78. Oligomerization and phosphorylation of RIPK3 in the necrosome lead to the recruitment and phosphorylation of MLKL78,79, and then MLKL translocates to the plasma membrane and causes membrane damage and necroptosis80. In addition, the necrosome interacts with mitochondrial serine/threonine protein phosphatase PGAM family member 5 on the mitochondrial membrane and activates mitochondrial fission factor dynamin-related protein 1 to induce necroptosis through mitochondrial fragmentation81. As mentioned above, the binding of FasL or TRAIL to the death receptor Fas or TRAILR induces the formation of DISC, activates caspase-8, and executes apoptosis82. However, in the absence of cIAPs or inhibition of caspase-8, RIPK1 translocates to the membrane93 and promotes the formation of complex II-b83 and initiation of necroptosis when Fas/TRAILR is activated 78,79,84,85.
Activation of TLRs induces the formation of a platform that recruits the cytoplasmic adaptor protein TRIF (Toll/IL-1 receptor domain-containing adaptor protein inducing interferon-Β). TRIF is involved in the activation of NF-κB signaling and induction of type I IFNs86. By relying on its RHIM (RIP homotypic interaction motif) domain, TRIF can interact with RIPK1 and RIPK3. In the presence of the apoptosis inhibitor zVAD-fmk, activation of TLR4 by lipopolysaccharide or activation of TLR3 by polyinosine–polycytidylic acid can induce TRIF-mediated necroptosis87, 88, which can be blocked by the inhibition of necrostatin-1 (Nec1) or knockout of RIPK189. These results suggest that the RIPK1-TRIF signaling complex plays an important role in TLR3/4-induced necroptosis. In the absence of RIPK1, TRIF can also induce necroptosis by directly recruiting and activating RIPK387,89.
In addition to death receptor signaling and TLR signaling, DNA-dependent activator of IFN-regulatory factors (DAI), a cytoplasmic viral DNA sensor90,91, can also induce necroptosis. Like TRIF, DAI has the RHIM structure. In response to viral (such as murine cytomegalovirus) double-stranded DNA, DAI can activate NF-κB, induce type I IFNs, and mediate RIPK3-dependent necroptosis91. Moreover, Wei et al. recently reported a novel necrosis mechanism, indicating that the acute cell necrosis induced by cationic nanocarriers occurs through the impairment of Na+/K+-ATPase, which causes a subsequent inflammatory response92. A diagram of the process of classical necroptosis is shown in Fig. 4.

Mechanisms of necroptosis. In TNFR signaling, complex I containing TRADD, RIPK1, TRAF2, E3 ubiquitin ligases, cIAP1/2, and LUBAC is unstable when RIP1K is deubiquitinated by CYLD, leading to the formation of the necrosome together with high levels of RIPK3 and MLKL as well as inhibited caspase-8. Subsequently, RIPK3 in the necrosome oligomerizes and is phosphorylated, leading to the recruitment and phosphorylation of MLKL, and phosphorylated MLKL translocates to the plasma membrane to cause membrane damage and necroptosis, or phosphorylated MLKL interacts with phosphorylase PGAM5 on the mitochondrial membrane and then activates mitochondrial fission factor Drp1 to induce necroptosis. In Fas/TRAILR signaling, when cIAPs are absent and caspase-8 is inhibited, the activation of Fas/TRAILR can induce necroptosis. In TLR3/4 signaling, their activation can induce TRIF-mediated necroptosis in the presence of zVAD-fmk, TLR4 activation by lipopolysaccharide (LPS) or TLR3 activation by polyinosine–polycytidylic acid. In DAI signaling, in response to viral double-stranded DNA, DAI also mediates RIPK3-dependent necroptosis under certain conditions
Associations among the three types of cell death
Association between apoptosis and necroptosis
Apoptosis and necroptosis may occur simultaneously93 or mutually transform because of the interconnection of the downstream death signaling pathways. For example, cells can commit to necrotic cell death when apoptosis is blocked94, and oxidative stress-induced necrotic cell death involves the activation of the apoptosis-associated caspase-8/Bid pathway95. The final form of cell death will depend on the cell type, cell microenvironment, and initial inducers.
Caspase-8 and RIPK1/3
As an apoptotic initiator caspase, caspase-8 interacts with FADD to form DISC, followed by homologous dimerization and proteolytic processing. Activated caspase-8 is then released from DISC and triggers downstream apoptotic signaling96. Meanwhile, caspase-8 can inhibit necroptosis by cleaving and inactivating RIPK1 and RIPK385,97,98. However, when the activity of caspase-8 is inhibited pharmacologically, such as by the pan-caspase inhibitor ZVAD, or genetically, such as in RNAi-mediated knockdown, RIPK1 and RIPK3 become activated through phosphorylation and induce the formation of the necrosome to trigger necroptosis12,99,100.
ATP
ATP plays a crucial role in the decision of cell death fate101. A high level of intracellular ATP often favors apoptosis, whereas a low level often promotes necrosis102. Therefore, excessive consumption of intracellular ATP or the inhibition of ATP synthesis may convert apoptosis to necrosis101,103. For example, substantial DNA damage leads to the activation of poly ADP-ribose polymerase-1 (PARP-1), a nuclear enzyme involved in DNA repair, resulting in the consumption of many NAD+ and ATP molecules and subsequent necrotic death104,105. The mitochondria is the major site that generates ATP; therefore, mitochondria dysfunction can trigger necrosis by ATP depletion. In addition, excessive mitochondrial ROS formation and the onset of the mitochondrial permeability transition are also causally linked to the conversion of apoptosis to necroptosis106.
Association between autophagy and apoptosis
Autophagy is an intracellular catabolic mechanism that involves the degradation and recycling of cytoplasmic undesired components, such as malfunctioning proteins or damaged organelles, to maintain cellular homeostasis107,108,109. Autophagy is a double-edged sword and can either protect cells from apoptosis110 or promote apoptosis111 depending on the cell type, intracellular metabolic activity, extracellular nutrient supply and triggering stimuli.
Beclin1
Mammalian Beclin1 (Atg6 in yeast) cross-regulates autophagy and apoptosis through direct interaction with anti-apoptosis family members112,113,114,115. Beclin1 is a key molecule involved in the autophagosome formation. Beclin1 interacts with class III type PI3KC3/Vps34 and promotes the formation of the Beclin1-Vps34-Vps15 core complex116,117. Beclin1 is also a member of the BH3-only protein family. The antiapoptotic protein Bcl-2 or Bcl-xL combines with Beclin1 through the BH3 domains118 to simultaneously block the process of autophagy by inhibiting Beclin1 activity119 and the occurrence of endogenous apoptosis111,118,120,121. On the other hand, NOXA and other BH3-only family proteins can displace Bcl-2 family members from Beclin1 and promote autophagic cell death122,123. Furthermore, Beclin1 can also be cleaved by several caspase proteins, such as caspase-8 and caspase-3, to shift the cell fate from autophagy to apoptosis14, 93. The C-terminal fragment of Beclin1 can then translocate to the mitochondria and induce mitochondrial membrane permeability and apoptosis124.
Association between autophagy and necroptosis
The interconnection between autophagy and necroptosis has been investigated in several studies with conflicting results. As a protective mechanism, autophagy can unsurprisingly inhibit necroptosis125,126. Interestingly, autophagy appears to facilitate necroptosis in certain instances127. Khan et al.128 reported that palmitic acid triggers Ca2+-dependent autophagy, resulting in the necroptosis of endothelial cells.
mTORC1
mTORC1 is a key sensor of nutrients, growth factors, and stress and controls cell metabolism, growth, and survival. Activation of mTORC1 by growth factors and nutrients can suppress autophagy by phosphorylation of autophagy-related proteins involved in autophagy initiation, such as ULK1 and ATG13, in the ULK complex and ATG14 in the VPS34 complex129. Cellular metabolic and energetic status regulates mTORC1 activity and consequently impacts autophagy induction. At a low-energy status, AMPK signaling is activated in response to an increased AMP/ATP ratio, which inhibits the mTORC1 signaling pathway via the phosphorylation of TSC2 (tuberous sclerosis complex 2), a mTORC1 negative regulator13,41,130,131,132,133, subsequently promoting autophagy141,130,134. Autophagy stimulation by the downregulation of mTORC1 signaling protect cells from programmed cell death, including apoptosis and necroptosis, under nutrient- or energy-deprived conditions135,136,137,138,139,140,141,142. As mentioned above, the activation of PARP-1 in the DNA damage response causes necrosis owing to ATP depletion, which also leads to AMPK activation, mTORC1 inhibition, and autophagy induction as the last protective resort143. The balance between autophagy and necrosis will determine the cell death fate.
Association among apoptosis, autophagy, and necroptosis
Cellular FILCE-like inhibitory protein (cFLIP)
FADD-like interleukin-1β-converting enzyme (FLICE)-like inhibitory proteins (FLIPs) possess caspase-8-like structures but lack proteolytic activity. cFLIP has three major isoforms in humans containing one long protein cFLIPL and two short proteins cFLIPs and cFLIPR144,145,146. cFLIPL possesses a C-terminal caspase-8-like domain but does not have enzymatic activity due to the substitution of several catalytically important amino-acid residues144,145,146,147, whereas the other isoforms cFLIPs and cFLIPR do not possess the caspase-like C-terminal domain. Nevertheless, these three isoforms all contain two death receptor domains at the N-termini, which allow them to interact with the adaptor protein FADD to form the DISC complex.
cFLIP regulates not only the death receptor-mediated extrinsic apoptosis pathway but also death receptor-independent apoptosis pathways. In complex IIb/ripoptosome, homodimeric caspase-8 initiates apoptosis by cleaving RIPK1 and disassembling complex IIb/ripoptosome. When procaspase-8 forms a heterodimer with cFLIPL, not only is necroptosis prevented due to cleavage of RIPK1 but apoptosis is also blocked because activated caspase-8 is not formed148. However, formation of a heterodimer by procaspase-8 with cFLIPS/R triggers necroptosis owing to the lack of proteolytic cleavage of RIPK1149,150,151 and simultaneously fails to induce caspase-8-dependent apoptosis. Therefore, the existence of cFLIP isoforms in the ripoptosome determines whether cells will execute RIPK1-dependent necroptosis or caspase-8-dependent apoptosis.
In addition to the regulation of apoptosis and necroptosis, cFLIP is known to be a negative regulator of autophagy. During autophagosome formation, Atg3 covalently binds the microtubule-associated protein LC3. Strikingly, cFLIP prevents the combination of Atg3 and LC3 by competitively binding Atg3 and consequently inhibiting autophagy152. The process in which cFLIP regulates apoptosis and necroptosis through formation of the ripoptosome occurs at the plasma membrane, but cFLIP inhibits autophagy at the sites where autophagosomes form. Thus, the different subcellular localizations of cFLIP may be important for its actions152. The complex associations among the three types of cell death are summarized in Fig. 5.

The relationship among the three types of cell death. FLIP regulates the modes of cell death by interacting with caspase-8, interfering with the functions of RIPK1, and combining Atg3 and LC3 by competitively binding Atg3. Caspase-8, which is a key factor in apoptosis, inhibits necroptosis by hydrolyzing RIPK1 and RIPK3. The level of intracellular ATP plays a crucial role in the decision of cell fate between apoptosis and necrosis. High levels of intracellular ATP often favor apoptosis, whereas low levels of intracellular ATP often favor necrosis. Beclin1, which is a key molecule required for autophagosome formation, can control the switch between autophagy and apoptosis via several mechanisms, such as by combining with Bcl-2 or Bcl-XL, which are anti-apoptotic proteins, and becoming hydrolyzed by several caspase proteins. mTOR can sense the level of intracellular ATP and relieve the inhibition of autophagy when the level of intracellular ATP is low, triggering necrotic cell death. When activated by growth factor, AKT can induce mTOR signaling to inhibit autophagy. The activation of AKT can inhibit apoptosis by phosphorylating apoptotic factors, such as Bad and caspase-9
Cell death and disease treatment
Cell death in neurodegenerative diseases
Neurodegenerative diseases, including Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, and amyotrophic lateral sclerosis153, involve the loss of structures and functions of nerve cells owing to the accumulation of abnormal proteins in the intracellular and extracellular spaces154. Deregulated cell death has been implicated as a major mechanism in these neurodegenerative diseases. The intervention of cell death pathways is therefore considered a potential therapeutic strategy.
The interaction between β-amyloid accumulation and the death of neurocytes contributes to the progression of Alzheimer’s disease155. β-amyloid accumulates in mitochondria, mediates mitochondrial toxicity, and induces caspase-3-dependent apoptosis156, which, in turn, accelerates the formation of β-amyloid and neurofibrillary tangles157. Activation of caspases also cleaves autophagy-related proteins, such as Beclin1, consequently inhibiting autophagy158.
An overwhelming body of evidence indicates that the inhibition of programmed cell death may be an effective strategy for the treatment of neurodegenerative diseases. For example, the efficacy of minocycline in the treatment of Alzheimer’s disease and Parkinson’s disease has been linked to its anti-apoptosis action by inhibiting oxidative stress, cytochrome c release, and caspase-3 activation and by increasing the expression levels of anti-apoptotic proteins, such as Bcl-2159. Blocking necroptosis by the RIPK1 inhibitor Nec-1 protects cortical neuronal cells of embryonic rats and mouse hippocampal neurons from excitatory toxicosis-induced cell death160,161. Nec-1 can also reduce the death of striatal neurons with Huntingtin (HTT) mutations and retard the process of Huntington’s disease in HTT-mutant transgenic mice162. As a cell survival mechanism, promoting autophagy163,164 by mTOR inhibitors, such as rapamycin and its analogs (CCI-779, RAD001, and AP23573), protects cells from apoptotic and necrotic cell death165,166 and has been proven to be effective in the treatment of neurodegenerative diseases.
Cell death in cancer
A delicate balance between cell death and survival maintains the homeostasis within a cell, a tissue, and an organism. Uncontrolled cell proliferation and escape from cell death have been recognized as the hallmarks of cancer cells167.
Substantial studies have suggested that autophagy plays a dual role in tumorigenesis. At the early stage, autophagy exerts an antitumor effect and curbs chronic tissue damage, inflammation, and genome instability; however, during the late stage, autophagy meets the energy and nutrient requirements to sustain tumor development168. Therefore, autophagy inhibitors, such as chloroquine or hydroxychloroquine, have a therapeutic potential in cancer treatment169,170,171. Autophagy also contributes to the resistance to cancer therapy. For example, autophagy plays a protective role against quercetin or histone deacetylase inhibitor SAHA (suberoylanilide hydroxamic acid)-induced apoptosis, and the combination of autophagy inhibitors and quercetin or SAHA may provide a rational utility of these drugs in the clinic172,173. Similarly, inhibition of autophagy has been reported to enhance the sensitivity of tumor cells to TRAIL agonists and promote apoptosis170.
Cancer cells exhibit aberrant apoptotic signaling, including upregulation of anti-apoptotic molecules and suppression of proapoptotic molecules174,175,176. The high expression level of survivin, a member of the IAP family177, the disrupted balance among the Bcl-2 family members178, or the impaired activity of caspases179, enhances cancer cell survival and is associated with tumor aggressiveness and the survival of cancer patients. Therefore, targeting apoptosis-related proteins has been one of the most active research fields in cancer therapeutics for the long-term and has achieved significant progress recently. The BH3 analog ABT-737 and its derivative ABT-263 act as pan-Bcl-2 inhibitors and simultaneously inhibit several members of Bcl-2 family proteins, including Bcl-2, Bcl-xL, and Bcl-W, to induce cell apoptosis180,181. BH3 analogs have shown therapeutic benefits for solid tumors and hematological malignancies182,183. The negative regulatory role of cFLIP in apoptosis makes it an attractive target molecule to treat cancer. The modulation of cFLIP expression and activity has been linked to the antitumor action of several targeted therapies, including those utilizing mTOR inhibitors and histone deacetylase inhibitors184,185,186.
Cell death-related pathways participate in the cellular stress response. Cancer cells exposed to various stresses (for example, DNA damages, oxidative stress) during oncogenic transformation and adaption to these stresses are required for cancer cells to survive. Oxidative stress results from the accumulation of ROS. ROS collectively include superoxide onion, hydrogen peroxide, and hydroxyl radical and regulate programmed cell death. Activation of death receptor-mediated signaling pathways has been associated with ROS production187,188,189,190. Wang et al.191 demonstrated ROS-mediated degradation of cFLIP, a negative regulator of Fas-induced apoptosis in lung epithelial cells. ROS can also trigger the intrinsic apoptotic cascade by disruption of the mitochondrial membrane potential, promoting cytochrome c detachment from cardiolipin and release to the cytosol and inducing oxidative mitochondrial DNA damage192. Furthermore, ROS can activate apoptotic signaling via ASK1/JNK signaling193. Accumulation of ROS can also activate autophagy. ROS-mediated oxidative modulation of Atg4 inhibited Atg4-delipidating activity, leading to the accumulation of LC3-PE on autophagosomal membranes and facilitating autophagosome formation. The interplay between ROS and autophagy contributes to cancer progression194. In the early stage of cancer initiation, autophagy is proposed to play a tumor suppressor role by reducing ROS accumulation via the degradation of ROS-producing mitochondria, thus limiting genomic instability195. However, at the later stage of tumor progression, autophagy may be exploited by cancer cells to promote their survival and oncogenic mutations196. ROS-mediated programmed cell death makes ROS-based therapies an attractive strategy in cancer treatment. Moreover, although cancer cells have developed redox adaptation mechanisms to survive in a high oxidative environment, extensive studies have suggested that they are more vulnerable to oxidative stress caused by ROS-generating agents than normal cells, providing the selectivity of ROS-based therapies197. Exogenous ROS-generating agents used as a single agent or in combination with other standard therapies have shown promise in pre-clinical studies198,199,200.
Cell death in other diseases
In addition to neurodegenerative diseases and cancer, cell death is associated with other diseases. Autophagy is involved in intestinal homeostasis201, muscular dystrophy, stroke, pancreatitis, heart disease, liver disease, and type II diabetes202,203,204,205. Increasing the activity of Beclin1 has been proposed as a therapeutic strategy for these autophagy-related diseases202. Apoptosis is also associated with ischemic stroke, acute central nervous system injury, heart disease, infectious diseases, autoimmune diseases29. Therefore, targeting apoptotic pathways such as the broad-spectrum caspase inhibitor Q-VD-OPh, has been reported to cure diseases induced by apoptosis206,207,208,209. Necroptosis has been linked to the pathogenesis of ischemia-reperfusion injury210, multiple sclerosis211, myocardial infarction, stroke63,212, inflammatory disease213, acute kidney injury214, and microbial infection184,187,215,216,217. Nec-1, an inhibitor of RIPK1, has shown the efficacy by preventing necroptosis64,164,212,214,218,219,220.
Prospects
Apoptosis, autophagy, and necrosis, three types of cell death, have been studied separately and are considered independent processes. Recent advances in cell death research have changed our perception, leading us to consider these processes as interconnected with overlapping signaling pathways and cross-talk in response to different stresses. The existence of diverse regulated cell death pathways implicates the complexity of cell death programs but also provides novel therapeutic targets. Further studies are required to investigate the linkage within different cell death programs and identify key molecular factors that determine cell death under specific pathological conditions and that can be pharmacologically manipulated.
References
Degterev, A. & Yuan, J. Expansion and evolution of cell death programmes. Nat. Rev. Mol. Cell Biol. 9, 378–390 (2008).
Kroemer, G. et al. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death. Cell Death Differ. 12, 1463–1467 (2005).
Ellis, H. M. & Horvitz, H. R. Genetic control of programmed cell death in the nematode C. elegans. Cell 44, 817–829 (1986).
Miura, M., Zhu, H., Rotello, R., Hartwieg, E. A. & Yuan, J. Induction of apoptosis in fibroblasts by IL-1β-converting enzyme, a mammalian homolog of the C. elegans cell death gene ced-3. Cell 75, 653–660 (1993).
Kerr, J. F., Wyllie, A. H. & Currie, A. R. Apoptosis: a basic biological phenomenon with wideranging implications in tissue kinetics. Br. J. Cancer 26, 239–257 (1972).
Trump, B., Goldblatt, P. & Stowell, R. Studies of necrosis in vitro of mouse hepatic parenchymal cells. Ultrastructural alterations in endoplasmic reticulum, Golgi apparatus, plasma membrane, and lipid droplets. Lab. Invest. 14, 2000–2028 (1965).
Kono, H. & Rock, K. L. How dying cells alert the immune system to danger. Nat. Rev. Immunol. 8, 279–289 (2008).
He, C. & Klionsky, D. J. Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet. 43, 67–93 (2009).
Gump, J. M. & Thorburn, A. Autophagy and apoptosis: what is the connection? Trends Cell Biol. 21, 387–392 (2011).
Mariño, G., Niso-Santano, M., Baehrecke, E. H. & Kroemer, G. Self-consumption: the interplay of autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 15, 81–94 (2014).
Hitomi, J. et al. Identification of a molecular signaling network that regulates a cellular necrotic cell death pathway. Cell 135, 1311–1323 (2008).
Vandenabeele, P., Galluzzi, L., Berghe, T. V. & Kroemer, G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat. Rev. Mol. Cell Biol. 11, 700–714 (2010).
Alers, S., Löffler, A. S., Wesselborg, S. & Stork, B. Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: cross talk, shortcuts, and feedbacks. Mol. Cell Biol. 32, 2–11 (2012).
Luo, S. & Rubinsztein, D. Apoptosis blocks Beclin 1-dependent autophagosome synthesis: an effect rescued by Bcl-xL. Cell Death Differ. 17, 268–277 (2010).
Declercq, W., Berghe, T. V. & Vandenabeele, P. RIP kinases at the crossroads of cell death and survival. Cell 138, 229–232 (2009).
Wang, L., Du, F. & Wang, X. TNF-α induces two distinct caspase-8 activation pathways. Cell 133, 693–703 (2008).
Garrido, C. & Kroemer, G. Life’s smile, death’s grin: vital functions of apoptosis-executing proteins. Curr. Opin. Cell Biol. 16, 639–646 (2004).
Galluzzi, L. et al. No death without life: vital functions of apoptotic effectors. Cell Death Differ. 15, 1113–1123 (2008).
Lockshin, R. A. & Zakeri, Z. Programmed cell death and apoptosis: origins of the theory. Nat. Rev. Mol. Cell Biol. 2, 545–550 (2001).
Degterev, A., Boyce, M. & Yuan, J. A decade of caspases. Oncogene 22, 8543–8567 (2003).
Danial, N. N. & Korsmeyer, S. J. Cell death: critical control points. Cell 116, 205–219 (2004).
Nakagawa, T. et al. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-β. Nature 403, 98–103 (2000).
Lavrik, I., Golks, A. & Krammer, P. H. Death receptor signaling. J. Cell Sci. 118, 265–267 (2005).
Wang, S. & El-Deiry, W. S. TRAIL and apoptosis induction by TNF-family death receptors. Oncogene 22, 8628–8633 (2003).
Duprez, L., Wirawan, E., Berghe, T. V. & Vandenabeele, P. Major cell death pathways at a glance. Microbes Infect. 11, 1050–1062 (2009).
Wang, Y. & Tjandra, N. Structural insights of tBid, the caspase-8-activated Bid, and its BH3 domain. J. Biol. Chem. 288, 35840–35851 (2013).
Fan, T.-J., Han, L.-H., Cong, R.-S. & Liang, J. Caspase family proteases and apoptosis. Acta Biochem. Biophys. Sin. 37, 719–727 (2005).
McIlwain, D. R., Berger, T. & Mak, T. W. Caspase functions in cell death and disease. Cold Spring Harb. Perspect. Biol. 5, a008656 (2013).
Favaloro, B., Allocati, N., Graziano, V., Di Ilio, C. & De Laurenzi, V. Role of apoptosis in disease. Aging 4, 330–349 (2012).
Ricci, J.-E. et al. Disruption of mitochondrial function during apoptosis is mediated by caspase cleavage of the p75 subunit of complex I of the electron transport chain. Cell 117, 773–786 (2004).
Özören, N. & El-Deiry, W. S. Defining characteristics of Types I and II apoptotic cells in response to TRAIL. Neoplasia 4, 551–557 (2002).
Gross, A., McDonnell, J. M. & Korsmeyer, S. J. BCL-2 family members and the mitochondria in apoptosis. Genes Dev. 13, 1899–1911 (1999).
Yuan, S. & Akey, C. W. Apoptosome structure, assembly, and procaspase activation. Structure 21, 501–515 (2013).
Soldani, C. et al. Poly (ADP-ribose) polymerase cleavage during apoptosis: when and where? Exp. Cell Res. 269, 193–201 (2001).
Morishima, N., Nakanishi, K., Takenouchi, H., Shibata, T. & Yasuhiko, Y. An endoplasmic reticulum stress-specific caspase cascade in apoptosis cytochrome c-independent activation of caspase-9 by caspase-12. J. Biol. Chem. 277, 34287–34294 (2002).
Yoneda, T. et al. Activation of caspase-12, an endoplastic reticulum (ER) resident caspase, through tumor necrosis factor receptor-associated factor 2-dependent mechanism in response to the ER stress. J. Biol. Chem. 276, 13935–13940 (2001).
Tan, Y. et al. Ubiquitous calpains promote caspase-12 and JNK activation during endoplasmic reticulum stress-induced apoptosis. J. Biol. Chem. 281, 16016–16024 (2006).
Cheong, H., Nair, U., Geng, J. & Klionsky, D. J. The Atg1 kinase complex is involved in the regulation of protein recruitment to initiate sequestering vesicle formation for nonspecific autophagy in Saccharomyces cerevisiae. Mol. Biol. Cell 19, 668–681 (2008).
Kawamata, T., Kamada, Y., Kabeya, Y., Sekito, T. & Ohsumi, Y. Organization of the pre-autophagosomal structure responsible for autophagosome formation. Mol. Biol. Cell 19, 2039–2050 (2008).
Hamasaki, M. et al. Autophagosomes form at ER–mitochondria contact sites. Nature 495, 389–393 (2013).
Jung, C. H. et al. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol. Biol. Cell 20, 1992–2003 (2009).
He, C. & Klionsky, D. J. Atg9 trafficking in autophagy-related pathways. Autophagy 3, 271–274 (2007).
Molejon, M. I., Ropolo, A. & Vaccaro, M. I. VMP1 is a new player in the regulation of the autophagy-specific phosphatidylinositol 3-kinase complex activation. Autophagy 9, 933–935 (2013).
Ohsumi, Y. Ubiquitin and proteasomes: Molecular dissection of autophagy: two ubiquitin-like systems. Nat. Rev. Mol. Cell Biol. 2, 211–216 (2001).
Mizushima, N. et al. A protein conjugation system essential for autophagy. Nature 395, 395–398 (1998).
Mizushima, N., Noda, T. & Ohsumi, Y. Apg16p is required for the function of the Apg12p–Apg5p conjugate in the yeast autophagy pathway. EMBO J. 18, 3888–3896 (1999).
Shintani, T. et al. Apg10p, a novel protein‐conjugating enzyme essential for autophagy in yeast. EMBO J. 18, 5234–5241 (1999).
Ichimura, Y. et al. A ubiquitin-like system mediates protein lipidation. Nature 408, 488–492 (2000).
Kirisako, T. et al. The reversible modification regulates the membrane-binding state of Apg8/Aut7 essential for autophagy and the cytoplasm to vacuole targeting pathway. J. Cell Biol. 151, 263–276 (2000).
Hanada, T. et al. The Atg12-Atg5 conjugate has a novel E3-like activity for protein lipidation in autophagy. J. Biol. Chem. 282, 37298–37302 (2007).
Diao, J., Li, L., Lai, Y. & Zhong, Q. In vitro reconstitution of autophagosome–lysosome fusion. Methods Enzymol. 587, 365–376 (2017).
Cheng, X. et al. Pacer mediates the function of class III PI3K and HOPS complexes in autophagosome maturation by engaging Stx17. Mol. Cell 65, 1029–1043 (2017).
Itakura, E., Kishi-Itakura, C. & Mizushima, N. The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell 151, 1256–1269 (2012).
Nair, U. et al. SNARE proteins are required for macroautophagy. Cell 146, 290–302 (2011).
Mizushima, N., Yoshimori, T. & Ohsumi, Y. The role of Atg proteins in autophagosome formation. Annu. Rev. Cell Dev. Biol. 27, 107–132 (2011).
Galluzzi, L. et al. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 19, 107–120 (2012).
Xu, J.-C. et al. Cultured networks of excitatory projection neurons and inhibitory interneurons for studying human cortical neurotoxicity. Sci. Transl. Med. 8, 333ra348 (2016).
Shimada, K. et al. Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat. Chem. Biol. 12, 497–503 (2016).
Abe, J.-i & Morrell, C. Pyroptosis as a regulated form of necrosis: PI + /annexin V–/high caspase 1/low caspase 9 activity in cells = pyroptosis? Circ. Res. 118, 1457–1460 (2016).
Dwivedi, N. & Radic, M. Citrullination of autoantigens implicates NETosis in the induction of autoimmunity. Ann. Rheum. Dis. 73, 483–491 (2014).
Popa, C., Netea, M. G., Van Riel, P. L., van der Meer, J. W. & Stalenhoef, A. F. The role of TNF-α in chronic inflammatory conditions, intermediary metabolism, and cardiovascular risk. J. Lipid Res. 48, 751–762 (2007).
Vercammen, D. et al. Inhibition of caspases increases the sensitivity of L929 cells to necrosis mediated by tumor necrosis factor. J. Exp. Med. 187, 1477–1485 (1998).
Degterev, A. et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat. Chem. Biol. 4, 313–321 (2008).
Degterev, A. et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol. 1, 112–119 (2005).
Weinlich, R., Dillon, C. P. & Green, D. R. Ripped to death. Trends Cell Biol. 21, 630–637 (2011).
Oberst, A. & Green, D. R. It cuts both ways: reconciling the dual roles of caspase 8 in cell death and survival. Nat. Rev. Mol. Cell Biol. 12, 757–763 (2011).
Challa, S. & Chan, F. K.-M. Going up in flames: necrotic cell injury and inflammatory diseases. Cell Mol. Life Sci. 67, 3241–3253 (2010).
Kim, S. & Li, J. Caspase blockade induces RIP3-mediated programmed necrosis in Toll-like receptor-activated microglia. Cell Death Dis. 4, e716 (2013).
Seya, T. et al. TLR3/TICAM-1 signaling in tumor cell RIP3-dependent necroptosis. Oncoimmunology 1, 917–923 (2012).
Kaiser, W. J. et al. RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature 471, 368–372 (2011).
Silke, J. & Brink, R. Regulation of TNFRSF and innate immune signalling complexes by TRAFs and cIAPs. Cell Death Differ. 17, 35–45 (2010).
Dempsey, P. W., Doyle, S. E., He, J. Q. & Cheng, G. The signaling adaptors and pathways activated by TNF superfamily. Cytokine Growth Factor Rev. 14, 193–209 (2003).
Reiley, W. W. et al. Deubiquitinating enzyme CYLD negatively regulates the ubiquitin-dependent kinase Tak1 and prevents abnormal T cell responses. J. Exp. Med. 204, 1475–1485 (2007).
Micheau, O. & Tschopp, J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 114, 181–190 (2003).
Kelliher, M. A. et al. The death domain kinase RIP mediates the TNF-induced NF-κB signal. Immunity 8, 297–303 (1998).
Dondelinger, Y. et al. RIPK3 contributes to TNFR1-mediated RIPK1 kinase-dependent apoptosis in conditions of cIAP1/2 depletion or TAK1 kinase inhibition. Cell Death Differ. 20, 1381–1392 (2013).
Yang, S. et al. Pellino3 targets RIP1 and regulates the proapoptotic effects of TNF-α. Nat. Commun. 4, 2583 (2013).
Orozco, S. et al. RIPK1 both positively and negatively regulates RIPK3 oligomerization and necroptosis. Cell Death Differ. 21, 1511–1521 (2014).
Wu, X. et al. Distinct roles of RIP1–RIP3 hetero-and RIP3–RIP3 homo-interaction in mediating necroptosis. Cell Death Differ. 21, 1709–1720 (2014).
Murphy, J. M. et al. The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity 39, 443–453 (2013).
Wang, Z., Jiang, H., Chen, S., Du, F. & Wang, X. The mitochondrial phosphatase PGAM5 functions at the convergence point of multiple necrotic death pathways. Cell 148, 228–243 (2012).
Scheidereit, C. IκB kinase complexes: gateways to NF-κB activation and transcription. Oncogene 25, 6685–6705 (2006).
Feoktistova, M. et al. cIAPs block Ripoptosome formation, a RIP1/caspase-8 containing intracellular cell death complex differentially regulated by cFLIP isoforms. Mol. Cell 43, 449–463 (2011).
Cho, Y. et al. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 137, 1112–1123 (2009).
He, S. et al. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-α. Cell 137, 1100–1111 (2009).
Takeuchi, O. & Akira, S. Pattern recognition receptors and inflammation. Cell 140, 805–820 (2010).
He, S., Liang, Y., Shao, F. & Wang, X. Toll-like receptors activate programmed necrosis in macrophages through a receptor-interacting kinase-3–mediated pathway. Proc. Natl. Acad. Sci. USA 108, 20054–20059 (2011).
Kaiser, W. J. et al. Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J. Biol. Chem. 288, 31268–31279 (2013).
Polykratis, A. et al. Cutting edge: RIPK1 Kinase inactive mice are viable and protected from TNF-induced necroptosis in vivo. J. Immunol. 193, 1539–1543 (2014).
Welz, P.-S. & Pasparakis, M. A way to DAI. Cell Host. Microbe 11, 223–225 (2012).
Upton, J. W., Kaiser, W. J. & Mocarski, E. S. DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host. Microbe 11, 290–297 (2012).
Wei, X. et al. Cationic nanocarriers induce cell necrosis through impairment of Na + /K + -ATPase and cause subsequent inflammatory response. Cell Res. 25, 237–253 (2015).
Zhang, L., Wang, H., Ding, K. & Xu, J. FTY720 induces autophagy-related apoptosis and necroptosis in human glioblastoma cells. Toxicol. Lett. 236, 43–59 (2015).
Leist, M. & Jäättelä, M. Four deaths and a funeral: from caspases to alternative mechanisms. Nat. Rev. Mol. Cell Biol. 2, 589–598 (2001).
Wang, X. et al. Necrotic cell death in response to oxidant stress involves the activation of the apoptogenic caspase-8/bid pathway. J. Biol. Chem. 278, 29184–29191 (2003).
Ashkenazi A., Dixit V. M. Death receptors: signaling and modulation. Science 281, 1305–1308.
Lin, Y., Devin, A., Rodriguez, Y. & Liu, Z.-g. Cleavage of the death domain kinase RIP by caspase-8 prompts TNF-induced apoptosis. Genes Dev. 13, 2514–2526 (1999).
Feng, S. et al. Cleavage of RIP3 inactivates its caspase-independent apoptosis pathway by removal of kinase domain. Cell Signal. 19, 2056–2067 (2007).
Moquin, D. & Chan, F. K.-M. The molecular regulation of programmed necrotic cell injury. Trends Biochem. Sci. 35, 434–441 (2010).
Holler, N. et al. Fas triggers an alternative, caspase-8–independent cell death pathway using the kinase RIP as effector molecule. Nat. Immunol. 1, 489–495 (2000).
Eguchi, Y., Shimizu, S. & Tsujimoto, Y. Intracellular ATP levels determine cell death fate by apoptosis or necrosis. Cancer Res. 57, 1835–1840 (1997).
Leist, M., Single, B., Castoldi, A. F., Kühnle, S. & Nicotera, P. Intracellular adenosine triphosphate (ATP) concentration: a switch in the decision between apoptosis and necrosis. J. Exp. Med. 185, 1481–1486 (1997).
Los, M. et al. Activation and caspase-mediated inhibition of PARP: a molecular switch between fibroblast necrosis and apoptosis in death receptor signaling. Mol. Biol. Cell 13, 978–988 (2002).
Szabó, C. & Dawson, V. L. Role of poly (ADP-ribose) synthetase in inflammation and ischaemia–reperfusion. Trends Pharmacol. Sci. 19, 287–298 (1998).
Sims, J. L., Berger, S. J. & Berger, N. A. Poly (ADP-ribose) polymerase inhibitors preserve oxidized nicotinamide adenine dinucleotide and adenosine 5’-triphosphate pools in DNA-damaged cells: mechanism of stimulation of unscheduled DNA synthesis. Biochemistry 22, 5188–5194 (1983).
Denecker, G. et al. Death receptor-induced apoptotic and necrotic cell death: differential role of caspases and mitochondria. Cell Death Differ. 8, 829–840 (2001).
Yu, L. et al. Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science 304, 1500–1502 (2004). (80-).
Shimizu, S. et al. Role of Bcl-2 family proteins in a nonapoptotic programmed cell death dependent on autophagy genes. Nat. Cell Biol. 6, 1221–1228 (2004).
Bursch, W. The autophagosomal–lysosomal compartment in programmed cell death. Cell Death Differ. 8, 569–581 (2001).
Li, Y. et al. Suppression of autophagy enhanced growth inhibition and apoptosis of interferon-β in human glioma cells. Mol. Neurobiol. 47, 1000–1010 (2013).
Maiuri, M. C., Zalckvar, E., Kimchi, A. & Kroemer, G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 8, 741–752 (2007).
Funderburk, S. F., Wang, Q. J. & Yue, Z. The Beclin 1–VPS34 complex–at the crossroads of autophagy and beyond. Trends Cell Biol. 20, 355–362 (2010).
Cao, Y. & Klionsky, D. J. Physiological functions of Atg6/Beclin 1: a unique autophagy-related protein. Cell Res. 17, 839–849 (2007).
Edinger, A. L. & Thompson, C. B. Death by design: apoptosis, necrosis and autophagy. Curr. Opin. Cell Biol. 16, 663–669 (2004).
Erlich, S. et al. Differential interactions between Beclin 1 and Bcl-2 family members. Autophagy 3, 561–568 (2007).
Kihara, A., Kabeya, Y., Ohsumi, Y. & Yoshimori, T. Beclin–phosphatidylinositol 3‐kinase complex functions at the trans‐Golgi network. EMBO Rep. 2, 330–335 (2001).
Liang, X. H. et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 402, 672–676 (1999).
He, C. & Levine, B. The beclin 1 interactome. Curr. Opin. Cell Biol. 22, 140–149 (2010).
Morselli, E. et al. Anti-and pro-tumor functions of autophagy. BBA-Mol. Cell Res. 1793, 1524–1532 (2009).
Robert, G. et al. The anti-apoptotic Bcl-B protein inhibits BECN1-dependent autophagic cell death. Autophagy 8, 637–649 (2012).
Itakura, E., Kishi, C., Inoue, K. & Mizushima, N. Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol. Biol. Cell 19, 5360–5372 (2008).
Elgendy, M., Sheridan, C., Brumatti, G. & Martin, S. J. Oncogenic Ras-induced expression of Noxa and Beclin-1 promotes autophagic cell death and limits clonogenic survival. Mol. Cell 42, 23–35 (2011).
Maiuri, M. C. et al. Functional and physical interaction between Bcl‐XL and a BH3‐like domain in Beclin‐1. EMBO J. 26, 2527–2539 (2007).
Wirawan, E. et al. Caspase-mediated cleavage of Beclin-1 inactivates Beclin-1-induced autophagy and enhances apoptosis by promoting the release of proapoptotic factors from mitochondria. Cell Death Dis. 1, e18 (2011).
Farkas, T., Daugaard, M. & Jäättelä, M. Identification of small molecule inhibitors of phosphatidylinositol 3-kinase and autophagy. J. Biol. Chem. 286, 38904–38912 (2011).
Bell, B. D. et al. FADD and caspase-8 control the outcome of autophagic signaling in proliferating Tcells. Proc. Natl. Acad. Sci. USA 105, 16677–16682 (2008).
Bonapace, L. et al. Induction of autophagy-dependent necroptosis is required for childhood acute lymphoblastic leukemia cells to overcome glucocorticoid resistance. J. Clin. Invest. 120, 1310–1323 (2010).
Khan, M. J. et al. Inhibition of autophagy rescues palmitic acid-induced necroptosis of endothelial cells. J. Biol. Chem. 287, 21110–21120 (2012).
Kim, Y. C. & Guan, K.-L. mTOR: a pharmacologic target for autophagy regulation. J. Clin. Invest. 125, 25–32 (2015).
Kim, J., Kundu, M., Viollet, B. & Guan, K.-L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 13, 132–141 (2011).
Hosokawa, N. et al. Nutrient-dependent mTORC1 association with the ULK1–Atg13–FIP200 complex required for autophagy. Mol. Biol. Cell 20, 1981–1991 (2009).
Gwinn, D. M. et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 30, 214–226 (2008).
Bolster, D. R., Crozier, S. J., Kimball, S. R. & Jefferson, L. S. AMP-activated protein kinase suppresses protein synthesis in rat skeletal muscle through down-regulated mammalian target of rapamycin (mTOR) signaling. J. Biol. Chem. 277, 23977–23980 (2002).
Egan, D. F. et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 331, 456–461 (2011). (80-).
Yang, J.-C. et al. Selective targeting of breast cancer cells through ROS-mediated mechanisms potentiates the lethality of paclitaxel by a novel diterpene, gelomulide K. Free Radic. Biol. Med. 51, 641–657 (2011).
Hwang, J-w. et al. Cigarette smoke-induced autophagy is regulated by SIRT1–PARP-1-dependent mechanism: implication in pathogenesis of COPD. Arch. Biochem. Biophys. 500, 203–209 (2010).
Alexander, A. et al. ATM signals to TSC2 in the cytoplasm to regulate mTORC1 in response to ROS. Proc. Natl. Acad. Sci. USA 107, 4153–4158 (2010).
Muñoz-Gámez, J. A. et al. PARP-1 is involved in autophagy induced by DNA damage. Autophagy 5, 61–74 (2009).
Huang, Q. & Shen, H.-M. To die or to live: the dual role of poly (ADP-ribose) polymerase-1 in autophagy and necrosis under oxidative stress and DNA damage. Autophagy 5, 273–276 (2009).
Amaravadi, R. K. & Thompson, C. B. The roles of therapy-induced autophagy and necrosis in cancer treatment. Clin. Cancer Res. 13, 7271–7279 (2007).
Albert, J. M. et al. Inhibition of poly (ADP-ribose) polymerase enhances cell death and improves tumor growth delay in irradiated lung cancer models. Clin. Cancer Res. 13, 3033–3042 (2007).
Lum, J. J. et al. Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell 120, 237–248 (2005).
Nikoletopoulou, V., Markaki, M., Palikaras, K. & Tavernarakis, N. Crosstalk between apoptosis, necrosis and autophagy. Biochim. Biophys. Acta 1833, 3448–3459 (2013).
Safa, A. R. & Pollok, K. E. Targeting the anti-apoptotic protein c-FLIP for cancer therapy. Cancers 3, (1639–1671 (2011).
Micheau, O. Cellular FLICE-inhibitory protein: an attractive therapeutic target? Expert. Opin. Ther. Targets 7, 559–573 (2003).
Bagnoli, M., Canevari, S. & Mezzanzanica, D. Cellular FLICE-inhibitory protein (c-FLIP) signalling: a key regulator of receptor-mediated apoptosis in physiologic context and in cancer. Int. J. Biochem. Cell Biol. 42, 210–213 (2010).
Safa, A. R., Day, T. W. & Wu, C.-H. Cellular FLICE-like inhibitory protein (C-FLIP): a novel target for cancer therapy. Curr. Cancer Drug. Targets 8, 37–46 (2008).
Kubli, D. A. & Gustafsson, Å. B. Mitochondria and mitophagy: the yin and yang of cell death control. Circ. Res. 111, 1208–1221 (2012).
Suzuki, Y., Nakabayashi, Y. & Takahashi, R. Ubiquitin-protein ligase activity of X-linked inhibitor of apoptosis protein promotes proteasomal degradation of caspase-3 and enhances its anti-apoptotic effect in Fas-induced cell death. Proc. Natl. Acad. Sci. USA 98, 8662–8667 (2001).
Li, F.-Y., Jeffrey, P. D., Jong, W. Y. & Shi, Y. Crystal structure of a viral flip insights into flip-mediated inhibition of death receptor signaling. J. Biol. Chem. 281, 2960–2968 (2006).
Budd, R. C., Yeh, W.-C. & Tschopp, J. cFLIP regulation of lymphocyte activation and development. Nat. Rev. Immunol. 6, 196–204 (2006).
Lee, J.-S. et al. FLIP-mediated autophagy regulation in cell death control. Nat. Cell Biol. 11, 1355–1362 (2009).
Xilouri, M. & Stefanis, L. Autophagy in the central nervous system: implications for neurodegenerative disorders. CNS Neurol. Disord.-DR 9, 701–719 (2010).
Walker, L. C. & LeVine, H. The cerebral proteopathies. Mol. Neurobiol. 21, 83–95 (2000).
Duyckaerts, C., Delatour, B. & Potier, M.-C. Classification and basic pathology of Alzheimer disease. Acta Neuropathol. 118, 5–36 (2009).
Lustbader, J. W. et al. ABAD directly links Aß to mitochondrial toxicity in Alzheimer’s Disease. Science 304, 448–452 (2004). (80-).
Rohn, T. T. The role of caspases in Alzheimer’s disease; potential novel therapeutic opportunities. Apoptosis 15, 1403–1409 (2010).
Rohn, T. T. et al. Depletion of Beclin-1 due to proteolytic cleavage by caspases in the Alzheimer’s disease brain. Neurobiol. Dis. 43, 68–78 (2011).
Kim, H.-S. & Suh, Y.-H. Minocycline and neurodegenerative diseases. Behav. Brain. Res. 196, 168–179 (2009).
Li, Y., Yang, X., Ma, C., Qiao, J. & Zhang, C. Necroptosis contributes to the NMDA-induced excitotoxicity in rat’s cultured cortical neurons. Neurosci. Lett. 447, 120–123 (2008).
Xu, X. et al. Necrostatin‐1 protects against glutamate‐induced glutathione depletion and caspase‐independent cell death in HT‐22 cells. J. Neurochem. 103, 2004–2014 (2007).
Zhu, S., Zhang, Y., Bai, G. & Li, H. Necrostatin-1 ameliorates symptoms in R6/2 transgenic mouse model of Huntington’s disease. Cell Death Dis. 2, e115 (2011).
Ravikumar, B. et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat. Genet. 36, 585–595 (2004).
Ravikumar, B., Duden, R. & Rubinsztein, D. C. Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum. Mol. Genet. 11, 1107–1117 (2002).
Wu, Y.-T. et al. Autophagy plays a protective role during zVAD-induced necrotic cell death. Autophagy 4, 457–466 (2008).
Ravikumar, B., Berger, Z., Vacher, C., O’kane, C. J. & Rubinsztein, D. C. Rapamycin pre-treatment protects against apoptosis. Hum. Mol. Genet. 15, 1209–1216 (2006).
Hanahan, D. & Weinberg, R. A. Hallmarks of cancer: the next generation. Cell 144, 646–674 (2011).
White, E. Deconvoluting the context-dependent role for autophagy in cancer. Nat. Rev. Cancer 12, 401–410 (2012).
Dikic, I., Johansen, T. & Kirkin, V. Selective autophagy in cancer development and therapy. Cancer Res. 70, 3431–3434 (2010).
Sotelo, J., Briceno, E. & López-González, M. A. Adding chloroquine to conventional treatment for glioblastoma multiforme: a randomized, double-blind, placebo-controlled trial. Ann. Intern. Med. 144, 337–343 (2006).
Kondo, Y. & Kondo, S. Autophagy and cancer therapy. Autophagy 2, 85–90 (2006).
Wang, K. et al. Quercetin induces protective autophagy in gastric cancer cells: involvement of Akt-mTOR-and hypoxia-induced factor 1α-mediated signaling. Autophagy 7, 966–978 (2011).
Li, J. et al. Proteomic analysis revealed association of aberrant ROS signaling with suberoylanilide hydroxamic acid-induced autophagy in Jurkat T-leukemia cells. Autophagy 6, 711–724 (2010).
Ou, W.-B. et al. HDACi inhibits liposarcoma via targeting of the MDM2-p53 signaling axis and PTEN, irrespective of p53 mutational status. Oncotarget 6, 10510–10520 (2015).
He, G. et al. Overexpression of tumor suppressor TSLC1 by a survivin-regulated oncolytic adenovirus significantly inhibits hepatocellular carcinoma growth. J. Cancer Res. Clin. Oncol. 138, 657–670 (2012).
Igney, F. H. & Krammer, P. H. Death and anti-death: tumour resistance to apoptosis. Nat. Rev. Cancer 2, 277–288 (2002).
Ambrosini, G., Adida, C. & Altieri, D. C. A novel anti-apoptosis gene, survivin, expressed in cancer and lymphoma. Nat. Med. 3, 917–921 (1997).
Jäger, U. et al. Follicular lymphomas9 BCL-2/IgH junctions contain templated nucleotide insertions: novel insights into the mechanism of t (14; 18) translocation. Blood 95, 3520–3529 (2000).
Lawrence, M. S. et al. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 505, 495–501 (2014).
Tse, C. et al. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 68, 3421–3428 (2008).
Oltersdorf, T. et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 435, 677–681 (2005).
Tan, N. et al. Navitoclax enhances the efficacy of taxanes in non–small cell lung cancer models. Clin. Cancer Res. 17, 1394–1404 (2011).
Cragg, M. S., Harris, C., Strasser, A. & Scott, C. L. Unleashing the power of inhibitors of oncogenic kinases through BH3 mimetics. Nat. Rev. Cancer 9, 321–326 (2009).
Kerr, E. et al. Identification of an acetylation-dependant Ku70/FLIP complex that regulates FLIP expression and HDAC inhibitor-induced apoptosis. Cell Death Differ. 19, 1317–1327 (2012).
Bangert, A. et al. Histone deacetylase inhibitors sensitize glioblastoma cells to TRAIL-induced apoptosis by c-myc-mediated downregulation of cFLIP. Oncogene 31, 4677–4688 (2012).
Pathil, A. et al. HDAC inhibitor treatment of hepatoma cells induces both TRAIL‐independent apoptosis and restoration of sensitivity to TRAIL. Hepatology 43, 425–434 (2006).
Vercammen, D. et al. Dual signaling of the Fas receptor: initiation of both apoptotic and necrotic cell death pathways. J. Exp. Med. 188, 919–930 (1998).
Gulbins, E. et al. Fas‐induced programmed cell death is mediated by a Ras‐regulated O2– synthesis. Immunology 89, 205–212 (1996).
Sato, T. et al. Fas-mediated apoptosome formation is dependent on reactive oxygen species derived from mitochondrial permeability transition in Jurkat cells. J. Immunol. 173, 285–296 (2004).
Medan, D. et al. Regulation of Fas (CD95)‐induced apoptotic and necrotic cell death by reactive oxygen species in macrophages. J. Cell Physiol. 203, 78–84 (2005).
Wang, L. et al. The Fas death signaling pathway connecting reactive oxygen species generation and FLICE inhibitory protein down-regulation. J. Immunol. 180, 3072–3080 (2008).
Ryter, S. W. et al. Mechanisms of cell death in oxidative stress. Antioxid. Redox Signal. 9, 49–89 (2007).
Matsuzawa, A. & Ichijo, H. Redox control of cell fate by MAP kinase: physiological roles of ASK1-MAP kinase pathway in stress signaling. Biochem. Biophys. Acta 1780, 1325–1336 (2008).
Dewaele, M., Maes, H. & Agostinis, P. ROS-mediated mechanisms of autophagy stimulation and their relevance in cancer therapy. Autophagy 6, 838–854 (2010).
Mathew, R. et al. Autophagy suppresses tumor progression by limiting chromosomal instability. Genes Dev. 21, 1367–1381 (2007).
Degenhardt, K. et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 10, 51–64 (2006).
Trachootham, D., Alexandre, J. & Huang, P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat. Rev. Drug. Discov. 8, 579–591 (2009).
Pignanelli, C. et al. Selective targeting of cancer cells by oxidative vulnerabilities with novel curcumin analogs. Sci. Rep. 7, 1105 (2017).
Ma, D. et al. Cancer cell mitochondria targeting by pancratistatin analogs is dependent on functional complex II and III. Sci. Rep. 7, 42957 (2017).
Raj, L. et al. Selective killing of cancer cells by a small molecule targeting the stress response to ROS. Nature 475, 231–234 (2011).
Patel, K. K. & Stappenbeck, T. S. Autophagy and intestinal homeostasis. Annu. Rev. Physiol. 75, 241–262 (2013).
Levine, B., Packer, M. & Codogno, P. Development of autophagy inducers in clinical medicine. J. Clin. Invest. 125, 14–24 (2015).
Mizushima, N. & Komatsu, M. Autophagy: renovation of cells and tissues. Cell 147, 728–741 (2011).
Fleming, A., Noda, T., Yoshimori, T. & Rubinsztein, D. C. Chemical modulators of autophagy as biological probes and potential therapeutics. Nat. Chem. Biol. 7, 9–17 (2011).
Levine, B. & Kroemer, G. Autophagy in the pathogenesis of disease. Cell 132, 27–42 (2008).
Han, W., Sun, Y., Wang, X., Zhu, C. & Blomgren, K. Delayed, long-term administration of the caspase inhibitor Q-VD-OPh reduced brain injury induced by neonatal hypoxia-ischemia. Dev. Neurosci. 36, 64–72 (2014).
Renolleau, S. et al. Specific caspase inhibitor Q‐VD‐OPh prevents neonatal stroke in P7 rat: a role for gender. J. Neurochem. 100, 1062–1071 (2007).
Carlson, D. L., Maass, D. L., White, J., Sikes, P. & Horton, J. W. Caspase inhibition reduces cardiac myocyte dyshomeostasis and improves cardiac contractile function after major burn injury. J. Appl. Physiol. 103, 323–330 (2007).
Turmel, H. et al. Caspase‐3 activation in 1‐methyl‐4‐phenyl‐1, 2, 3, 6‐tetrahydropyridine (MPTP)‐treated mice. Mov. Disord. 16, 185–189 (2001).
Linkermann, A. et al. Rip1 (receptor-interacting protein kinase 1) mediates necroptosis and contributes to renal ischemia/reperfusion injury. Kidney Int. 81, 751–761 (2012).
Ofengeim, D. et al. Activation of necroptosis in multiple sclerosis. Cell Rep. 10, 1836–1849 (2015).
Smith, C. C. et al. Necrostatin: a potentially novel cardioprotective agent? Cardiovasc, Drug Ther. 21, 227–233 (2007).
Wu, J. et al. Mlkl knockout mice demonstrate the indispensable role of Mlkl in necroptosis. Cell Res. 23, 994–1006 (2013).
Kinsey, G. R. & Okusa, M. D. Pathogenesis of acute kidney injury: foundation for clinical practice. Am. J. Kideny Dis. 58, 291–301 (2011).
Wang, X. et al. Direct activation of RIP3/MLKL-dependent necrosis by herpes simplex virus 1 (HSV-1) protein ICP6 triggers host antiviral defense. Proc. Natl. Acad. Sci. USA 111, 15438–15443 (2014).
Kaiser, W. J., Upton, J. W. & Mocarski, E. S. Viral modulation of programmed necrosis. Curr. Opin. Virol. 3, 296–306 (2013).
Cho, Y., McQuade, T., Zhang, H., Zhang, J. & Chan, F. K.-M. RIP1-dependent and independent effects of necrostatin-1 in necrosis and T cell activation. PLoS ONE 6, e23209 (2011).
Oerlemans, M. I. et al. Inhibition of RIP1-dependent necrosis prevents adverse cardiac remodeling after myocardial ischemia–reperfusion in vivo. Basic. Res. Cardiol. 107, 270 (2012).
Peri, P., Nuutila, K., Vuorinen, T., Saukko, P. & Hukkanen, V. Cathepsins are involved in virus-induced cell death in ICP4 and Us3 deletion mutant herps simplex virus type 1-infected monocytic cells. J. Gen. Virol. 92, 173–180 (2011).
Xu, X. et al. Synergistic protective effects of humanin and necrostatin-1 on hypoxia and ischemia/reperfusion injury. Brain Res. 1355, 189–194 (2010).
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (Nos. 81770176 and 31470071), the New Century 151 Talent Project of Zhejiang Province, the 521 Talent Foundation of Zhejiang Sci-Tech University, and the Open Foundation from Key Laboratory of Tumor Molecular Diagnosis and Individualized Medicine of Zhejiang Province (No. ZJZLSYS004). The funders played no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chen, Q., Kang, J. & Fu, C. The independence of and associations among apoptosis, autophagy, and necrosis. Sig Transduct Target Ther 3, 18 (2018). https://doi.org/10.1038/s41392-018-0018-5
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-018-0018-5
This article is cited by
Regulated cell death and its role in Alzheimer’s disease and amyotrophic lateral sclerosis
Acta Neuropathologica (2024)
Deep in situ microscopy for real-time analysis of mammalian cell populations in bioreactors
Scientific Reports (2023)
Diversity and complexity of cell death: a historical review
Experimental & Molecular Medicine (2023)
Neuroprotective effect of astragalin via activating PI3K/Akt-mTOR-mediated autophagy on APP/PS1 mice
Cell Death Discovery (2023)
Plastic response of macrophages to metal ions and nanoparticles in time mimicking metal implant body environment
Environmental Science and Pollution Research (2023)
