


Abstract
The antitumorigenic mechanism of cannabidiol is still controversial. This study investigates the role of COX-2 and PPAR-γ in cannabidiol's proapoptotic and tumor-regressive action. In lung cancer cell lines (A549, H460) and primary cells from a patient with lung cancer, cannabidiol elicited decreased viability associated with apoptosis. Apoptotic cell death by cannabidiol was suppressed by NS-398 (COX-2 inhibitor), GW9662 (PPAR-γ antagonist), and siRNA targeting COX-2 and PPAR-γ. Cannabidiol-induced apoptosis was paralleled by upregulation of COX-2 and PPAR-γ mRNA and protein expression with a maximum induction of COX-2 mRNA after 8 hours and continuous increases of PPAR-γ mRNA when compared with vehicle. In response to cannabidiol, tumor cell lines exhibited increased levels of COX-2–dependent prostaglandins (PG) among which PGD2 and 15-deoxy-Δ12,14-PGJ2 (15d-PGJ2) caused a translocation of PPAR-γ to the nucleus and induced a PPAR-γ–dependent apoptotic cell death. Moreover, in A549-xenografted nude mice, cannabidiol caused upregulation of COX-2 and PPAR-γ in tumor tissue and tumor regression that was reversible by GW9662. Together, our data show a novel proapoptotic mechanism of cannabidiol involving initial upregulation of COX-2 and PPAR-γ and a subsequent nuclear translocation of PPAR-γ by COX-2–dependent PGs. Mol Cancer Ther; 12(1); 69–82. ©2012 AACR.
Introduction
Within the last decade, evidence has been accumulated to suggest an antitumorigenic action of cannabinoids elicited via induction of apoptosis and alternative anticarcinogenic mechanisms (for review see ref. 1, 2). As the clinical use of cannabinoids is limited by their adverse psychotropic side effects, the interest in the nonpsychoactive Cannabis-derived compound cannabidiol as potential systemic cancer therapeutic has substantially increased in recent years. In fact, cannabidiol has been shown to elicit pronounced proapoptotic or autophagic effects on different types of tumor cells (3–7). In a panel of tumor cell lines, cannabidiol even exhibited the most potent antiproliferative action as compared with a variety of other cannabinoids (3). Moreover, cannabidiol has been proven to exert anti-invasive properties on cervical (8), lung (9, 10), and breast cancer cells (11) in vitro, an antimetastatic action on lung (8, 10) and breast cancer cells (3), as well as tumor-regressive effects on solid tumors in vivo (3, 9, 10). Apart from its direct antitumorigenic action, cannabidiol has recently also been shown to relieve intractable and opioid-insensitive cancer pain (12) and to reduce severe adverse reactions associated with chemotherapy such as neurotoxic and nephrotoxic effects (13, 14).
However, cannabidiol still remains enigmatic with respect to its precise mode of action. In view of the fact that the eicosanoid system has been reported to be involved in biologic effects of cannabinoids (15–17), we were particularly interested in a possible involvement of COX-2 expression and prostaglandin (PG) synthesis in the proapoptotic action of cannabidiol. In fact, recent studies indicate a COX-2–dependent pathway underlying apoptosis elicited by the cannabinoid compounds anandamide and its analog R(+)-methanandamide (18, 19). In addition, COX-2 upregulation has been reported to be involved in the proapoptotic action of stearoylethanolamide (20), the alkylphospholipids edelfosin and perifosin (21, 22), as well as the chemotherapeutics cisplatin, 5-fluorouracil, and paclitaxel (23, 24). About the mechanism underlying COX-2–dependent apoptosis, several studies provided evidence for an activation of the transcription factor PPAR-γ by COX-2–dependent PGs of the D- and J-series (19, 24–26). This view is further corroborated by investigations indicating a pivotal role of PPAR-γ activation in eliciting apoptosis of different tumor cells including non–small cell lung cancer cells (NSCLC; refs. 27, 28).
The present study investigates the contribution of COX-2 and PPAR-γ to the proapoptotic action of cannabidiol on human lung cancer cells in vitro as well as to its tumor-regressive action in vivo. Here, we present evidence for a hitherto unknown cannabidiol-induced proapoptotic pathway involving initial upregulation of COX-2 and PPAR-γ at the expression level and a subsequent activation of PPAR-γ by the de novo synthesized COX-2–dependent PGs PGD2 and 15-deoxy-Δ12,14-PGJ2 (15d-PGJ2).
Materials and Methods
Materials
(−)-Cannabidiol and troglitazone were purchased from Tocris, AM-251, AM-630, capsazepine, and NS-398 from Alexis Deutschland GmbH, PGE2 from Cayman, 15d-PGJ2 and GW9662 from Sigma-Aldrich, and PGD2 from Enzo Life Sciences. Dulbecco's Modified Eagle's Medium (DMEM) with 4 mmol/L l-glutamine and 4.5 g/L glucose was from Cambrex Bio Science Verviers S.p.r.l.. PBS and fetal calf serum (FCS) were obtained from PAN Biotech. Penicillin–streptomycin was obtained from Invitrogen. Chemical structures of the main substances used in this study are given in Fig. 1.
Chemical structures of cannabidiol, AM-251, AM-630, capsazepine, GW9662, NS-398, PGE2, PGD2, and 15d-PGJ2.
Chemical structures of cannabidiol, AM-251, AM-630, capsazepine, GW9662, NS-398, PGE2, PGD2, and 15d-PGJ2.
Cell culture
A549 human lung carcinoma cells were purchased from Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH (DSMZ; A549: DSMZ no.: ACC 107, species confirmation as human with isoelectric focusing of malate dehydrogenase, nucleosid phosphorylase; fingerprint: multiplex PCR of minisatellite markers revealed a unique DNA profile). H460 cells were purchased from ATCC-LGC [American Type Culture Collection (ATCC) number: HTB-177; cell line confirmation by cytogenetic analysis]. Following resuscitation of frozen cultures, none of the cell lines was cultured longer than 6 months. Primary lung tumor cells were obtained from a resection of a brain metastasis of a 67-year-old male Caucasian with NSCLC. The patient had been informed about the establishment of cellular models from his tumor and had given informed consent in written form. The procedure was approved by the Institutional Ethical Committee. Samples from brain metastasis were excised, stored at 4°C in PBS and immediately transferred to the laboratory. Cells were passaged 5 times in DMEM containing 20% FCS, 100 U/mL penicillin, and 100 μg/mL streptomycin. Experiments with primary lung tumor cells were conducted using passages 5 to 8. Primary tumor cells were not authenticated. All incubations were conducted in serum-free DMEM. PBS was used as vehicle for test substances with a final concentration of 0.1% (v/v) ethanol (for cannabidiol) or 0.1% (v/v) dimethyl sulfoxide (DMSO; for AM-251, AM-630, capsazepine, NS-398, GW9662, troglitazone, and PGs).
Quantitative real-time RT-PCR
Total RNA was isolated using the RNeasy total RNA Kit (Qiagen GmbH). β-Actin (internal standard), COX-2, and PPAR-γ mRNA levels were determined by real-time reverse transcriptase PCR (RT-PCR) using the TaqMan RNA-to-CT 1-Step Kit and TaqMan Gene Expression Assays (Applied Biosystems) as described (18, 19, 24).
Western blot analysis
Proteins were isolated and analyzed as described (18, 19, 24). Antibodies were from BD Biosciences (COX-2), Santa Cruz (PPAR-γ), and Calbiochem (β-actin). Membranes were probed with horseradish peroxidase–conjugated Fab-specific anti-mouse immunoglobulin G (IgG) for detection of COX-2 and PPAR-γ (Cell Signaling Technology) and IgM for detection of β-actin (Calbiochem). Densitometric analysis of COX-2 and PPAR-γ band intensities was achieved by optical scanning and quantification using the Quantity One 1-D Analysis Software (Biorad).
Determination of PGs
Cells seeded in 24-well plates at a density of 2 × 105 cells per well and grown to confluence were used for incubations in 300 μL medium. PG concentrations in cell culture supernatants were determined using enzyme immunoassay kits from Cayman (PGE2 and PGD2) and Enzo Life Sciences (15d-PGJ2). PG levels were normalized to cellular protein.
Analysis of cytotoxicity and apoptosis
For analysis of cellular viability, cells seeded at a density of 5 × 103 cells per well in 96-well flat-bottom microplates and grown to confluence thereafter were used for incubations in 100 μL medium without serum. Cell viability was measured by the WST-1 test (Roche Diagnostics). To analyze apoptosis, cells seeded in 24-well plates at a density of 1 × 105 cells per well and grown to confluence were used for incubations in 500 μL medium. To assess apoptosis, adherent cells were harvested by trypsinization and combined with detached cells. Cytoplasmic histone-associated DNA fragments were assessed using the Cell Death Detection ELISAPLUS Kit (Roche Diagnostics). For detection of apoptotic nuclear morphology, fixed cells were stained with 100 ng/mL bisbenzimide as described (18).
Transfections with siRNA
Cells grown to 50% to 80% confluence were transfected with silencing or nonsilencing siRNA using RNAiFect (Qiagen GmbH) as described (18, 19, 24). Final concentration of siRNA or nonsilencing siRNA were 2.5 μg/mL (COX-2 siRNA) and 1.25 μg/mL (PPAR-γ siRNA), respectively. A negative (nonsilencing) control siRNA was from Eurogentec.
Quantification of nuclear PPAR-γ
Cells were grown to 60% to 80% confluence in BD Falcon 4-well culture slides (BD Biosciences). For confocal imaging, fixed cells were incubated with lamin A/C antibody (Cell Signaling Technology) to determine nuclear regions. The PPAR-γ antibody was from Cayman. Secondary antibodies were goat anti-rabbit Alexa Fluor 555 labeled IgG for detection of PPAR-γ and goat anti-mouse Alexa Fluor 488 labeled IgG for lamin A/C (Molecular Probes). Shapes of nuclear regions were merged to images of PPAR-γ–stained cells. Fluorescence intensity of PPAR-γ within lamin A/C–positive spots was quantified for 30 cells per sample using the Quantity One 1-D Analysis Software (Biorad).
Immunohistochemical analysis
Primary antibodies were purchased from Cayman (COX-2, PPAR-γ) and BD Biosciences (CD31). The secondary antibody for PPAR-γ was a polyclonal biotin-labeled goat anti-rabbit IgG (Abcam) and for COX-2 a biotin-SP–conjugated AffiniPure Fcy fragment-specific rabbit anti-mouse IgG (Jackson ImmunoResearch Laboratories Inc.). The secondary antibody for CD31 was a polyclonal biotin-labeled goat anti-rat IgG (BD Biosciences). Visualization of antibody binding was conducted using fuchsin (Dako) for COX-2 and PPAR-γ or the DAB Substrate Kit (BD Biosciences) for CD31 as chromogens. Quantitative evaluation was conducted by counting sharply red (COX-2, PPAR-γ) or brown (CD31) stained cells in microscopic views in an investigator-blinded fashion. For statistical analyses 200 to 300 cells per tumor were analyzed and the percentage of positive cells among the respective total cell number counted was calculated for each tumor.
Induction of A549 xenografts in athymic nude mice
Tumors were induced in female NMRI (nu/nu) mice (Charles River) by subcutaneous inoculation of 1 × 107 A549 cells into the right dorsal flank. Animals were injected intraperitoneally all 72 hours with vehicle, cannabidiol (5 mg/kg body weight) or GW9662 (1 mg/kg body weight). The treatment was started 7 days after tumor induction. GW9662 was injected 0.5 hours before vehicle or cannabidiol. Tumor volume was calculated as (4π/3) × (width/2)2 × (length/2). After 29 days, animals were sacrificed and tumors were explanted for mRNA and immunohistochemical protein analysis. Therefore, tissue parts were quick-frozen in liquid nitrogen for preparation of mRNA using the TRIzol reagent (Invitrogen) and subsequent real-time RT-PCR. Other parts of the tumors were stored in 4% paraformaldehyde. Following dehydratation, sections were embedded in paraffin for subsequent immunohistochemical analyses.
Statistics
Student 2-tailed t test (pairwise comparisons) and ANOVA with post hoc Student–Newman–Keuls test (multiple comparisons) were conducted using GraphPad Prism 5.00 (GraphPad Software). IC50 values were calculated by nonlinear regression of log(inhibitor) versus response.
Results
Role of cannabinoid-activated receptors in cannabidiol-induced apoptotic cell death
About cannabidiol's impact on tumor cell viability in the WST-1 test, experiments revealed IC50 values of 3.47 μmol/L (A549) or 2.80 μmol/L (H460) with a complete loss of viability up from 8 μmol/L (A549) and 7 μmol/L (H460; n = 6 experiments), respectively. The role of cannabinoid-activated receptors in this response was investigated by use of AM-251 (CB1 receptor antagonist), AM-630 (CB2 receptor antagonist), a combination of AM-251 and AM-630 as well as capsazepine (TRPV1 antagonist). However, all of these compounds did not alter cannabidiol-induced apoptotic cell death (Table 1).
Role of COX-2, PPAR-γ, and cannabinoid receptors in cannabidiol-mediated apoptotic cell death
| Viability (mean ± SEM) | DNA fragmentation (mean ± SEM) | |||
|---|---|---|---|---|
| A549 | H460 | A549 | H460 | |
| Vehicle | 100% ± 7% | 100% ± 3% | 100% ± 6% | 100% ± 4% |
| CBD | 49% ± 9%c | 17% ± 2%c | 541% ± 126%b | 391% ± 80%a |
| CBD + AM-251 | 50% ± 2%c,e | 14% ± 2%c,e | 458% ± 65%b,e | 409% ± 88%a,e |
| CBD + AM-630 | 45% ± 5%c,e | 8% ± 2%c,e | 679% ± 89%b,e | 360% ± 6%a,e |
| CBD + AM-251 + AM-630 | 42% ± 3%c,e | 12% ± 2%c,e | 541% ± 35%b,e | 504% ± 118%b,e |
| CBD + capsazepine | 53% ± 3%c,e | 22% ± 2%c,e | 599% ± 152%b,e | 429% ± 67%a,e |
| Vehicle | 100% ± 3% | 100% ± 2% | 100% ± 10% | 100% ± 15% |
| CBD | 24% ± 7%c | 41% ± 3%c | 423% ± 74%c | 573% ± 65%c |
| CBD + NS-398 | 82% ± 9%d | 67% ± 7%d | 81% ± 6%d | 170% ± 35%d |
| CBD + GW9662 | 76% ± 4%d | 67% ± 8%d | 81% ± 7%d | 168% ± 30%d |
| CBD + NS-398 + GW9662 | 112% ± 8%d,f,i | 90% ± 7%d,g,h | 56% ± 7%d | 57% ± 2%d |
| NS-398 | 112% ± 3% | 100% ± 1% | 90% ± 10% | 115% ± 16% |
| GW9662 | 104% ± 4% | 95% ± 4% | 114% ± 27% | 98% ± 18% |
| Viability (mean ± SEM) | DNA fragmentation (mean ± SEM) | |||
|---|---|---|---|---|
| A549 | H460 | A549 | H460 | |
| Vehicle | 100% ± 7% | 100% ± 3% | 100% ± 6% | 100% ± 4% |
| CBD | 49% ± 9%c | 17% ± 2%c | 541% ± 126%b | 391% ± 80%a |
| CBD + AM-251 | 50% ± 2%c,e | 14% ± 2%c,e | 458% ± 65%b,e | 409% ± 88%a,e |
| CBD + AM-630 | 45% ± 5%c,e | 8% ± 2%c,e | 679% ± 89%b,e | 360% ± 6%a,e |
| CBD + AM-251 + AM-630 | 42% ± 3%c,e | 12% ± 2%c,e | 541% ± 35%b,e | 504% ± 118%b,e |
| CBD + capsazepine | 53% ± 3%c,e | 22% ± 2%c,e | 599% ± 152%b,e | 429% ± 67%a,e |
| Vehicle | 100% ± 3% | 100% ± 2% | 100% ± 10% | 100% ± 15% |
| CBD | 24% ± 7%c | 41% ± 3%c | 423% ± 74%c | 573% ± 65%c |
| CBD + NS-398 | 82% ± 9%d | 67% ± 7%d | 81% ± 6%d | 170% ± 35%d |
| CBD + GW9662 | 76% ± 4%d | 67% ± 8%d | 81% ± 7%d | 168% ± 30%d |
| CBD + NS-398 + GW9662 | 112% ± 8%d,f,i | 90% ± 7%d,g,h | 56% ± 7%d | 57% ± 2%d |
| NS-398 | 112% ± 3% | 100% ± 1% | 90% ± 10% | 115% ± 16% |
| GW9662 | 104% ± 4% | 95% ± 4% | 114% ± 27% | 98% ± 18% |
NOTE: Cells were incubated with cannabidiol (3 μmol/L) or vehicle for 48 hours (WST-1 test) or 18 hours (DNA fragmentation) following a 1-hour pretreatment with AM-251 (CB1 antagonist; 1 μmol/L), AM-630 (CB2 antagonist; 1 μmol/L), capsazepine (TRPV1 antagonist; 1 μmol/L), NS-398 (COX-2 inhibitor; 1 μmol/L), or GW9662 (PPAR-γ antagonist; 10 μmol/L). Values are mean ± SEM of n = 6–12 (WST), n = 3–8 (DNA fragmentation of GW9662- and NS-398–treated cells), n = 8 (DNA fragmentation of cells treated with AM-251, AM-630, and capsazepine) experiments.
Abbreviation: CBD, cannabidiol.
aP < 0.05.
bP < 0.01.
cP < 0.001 vs. vehicle.
dP < 0.001 vs. CBD.
enot significant vs. CBD.
fP < 0.05.
gP < 0.01 for CBD + NS-398 + GW9662 vs. CBD + NS-398.
hP < 0.05.
iP < 0.01 for CBD + NS-398 + GW9662 vs. CBD + GW9662.
Impact of cannabidiol on COX-2 and PPAR-γ
Initial experiments revealed increased COX-2 and PPAR-γ protein levels in A549 and H460 cells following an 8-hour incubation with cannabidiol at 1 and 3 μmol/L (Fig. 2A and B, left). In view of the more pronounced upregulation by cannabidiol at 3 μmol/L, this concentration was used for subsequent experiments. In A549 and H460, cannabidiol caused a time-dependent prolonged increase of PPAR-γ mRNA and protein. In A549, COX-2 mRNA was upregulated transiently with a peak at 8 hours (Fig. 2A), whereas in H460 COX-2 mRNA induction by cannabidiol remained highly significant even after an 18-hour incubation (Fig. 2B). COX-2 protein levels seemed delayed as compared with the respective mRNA levels.
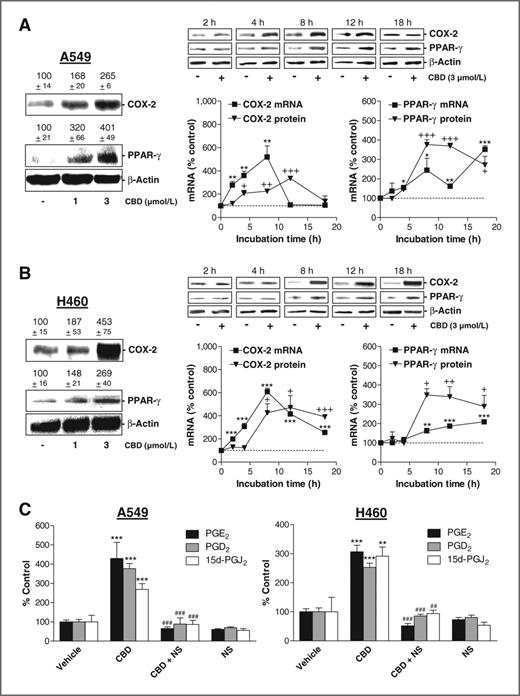
Effect of cannabidiol (CBD) on COX-2 and PPAR-γ expression and on PG synthesis. A and B, real-time RT-PCR and Western blot analyses of the effect of cannabidiol on COX-2 and PPAR-γ mRNA and protein expression in A549 (A) and H460 (B) cells. β-Actin was used as loading control for Western blot analysis. Videodensitometric evaluations of Western blot analyses are given as percentage of vehicle control above the blots or in the charts. Cells were incubated with the indicated concentrations of cannabidiol or vehicle for 8 hours (A and B, left) or with 3 μmol/L cannabidiol or vehicle for the indicated times (A and B, middle and right). C, effect of 1 μmol/L NS-398 on cannabidiol (3 μmol/L)-induced PG synthesis. NS-398 was added to cells 1 hour before cannabidiol or vehicle and the incubation was continued for 18 hours. Values are mean ± SEM of n = 3–4 (COX-2 mRNA in A and B and values in C), n = 3 (A, left), n = 4 (PPAR-γ mRNA in A and B), n = 6 (B, left) or n = 3–6 (Western blot analyses of time-courses in A and B) experiments. +, P < 0.05; ++, P < 0.01; +++, P < 0.001 (for comparison of protein levels); *, P < 0.05; **, P < 0.01; ***, P < 0.001 versus vehicle; ##, P < 0.01; ###, P < 0.001 versus cannabidiol.
Effect of cannabidiol (CBD) on COX-2 and PPAR-γ expression and on PG synthesis. A and B, real-time RT-PCR and Western blot analyses of the effect of cannabidiol on COX-2 and PPAR-γ mRNA and protein expression in A549 (A) and H460 (B) cells. β-Actin was used as loading control for Western blot analysis. Videodensitometric evaluations of Western blot analyses are given as percentage of vehicle control above the blots or in the charts. Cells were incubated with the indicated concentrations of cannabidiol or vehicle for 8 hours (A and B, left) or with 3 μmol/L cannabidiol or vehicle for the indicated times (A and B, middle and right). C, effect of 1 μmol/L NS-398 on cannabidiol (3 μmol/L)-induced PG synthesis. NS-398 was added to cells 1 hour before cannabidiol or vehicle and the incubation was continued for 18 hours. Values are mean ± SEM of n = 3–4 (COX-2 mRNA in A and B and values in C), n = 3 (A, left), n = 4 (PPAR-γ mRNA in A and B), n = 6 (B, left) or n = 3–6 (Western blot analyses of time-courses in A and B) experiments. +, P < 0.05; ++, P < 0.01; +++, P < 0.001 (for comparison of protein levels); *, P < 0.05; **, P < 0.01; ***, P < 0.001 versus vehicle; ##, P < 0.01; ###, P < 0.001 versus cannabidiol.
In both cell lines, cannabidiol induced significant upregulations of PGE2, PGD2, and 15d-PGJ2 with all increases being sensitive to the selective COX-2 inhibitor NS-398 (Fig. 2C). Even in A549 cells in which COX-2 protein was transiently upregulated, increased PG levels were still detected after 18 hours. PGD2 levels in cell culture media (mean ± SEM of n = 4 experiments) of cannabidiol-treated cells were 0.52 ± 0.20 nmol/L in A549 cells and 0.14 ± 0.01 nmol/L in H460 cells. 15d-PGJ2 levels (mean ± SEM of n = 4 experiments) were 0.94 ± 0.12 nmol/L in A549 and 0.23 ± 0.05 nmol/L in H460 cells.
Influence of NS-398 and GW9662 on cannabidiol-induced apoptotic cell death
Next, the impact of the COX-2 inhibitor NS-398 and the PPAR-γ antagonist GW9662 on the cannabidiol-induced loss of viability and induction of apoptosis was investigated. According to Table 1, the cannabidiol-induced DNA fragmentation and loss of viability was inhibited by NS-398 and GW9662 in A549 and H460 cells. A combined treatment of cells with NS-398 and GW9662 was shown to fully restore control conditions. A statistical analysis between this combination group versus the groups treated with cannabidiol and the respective inhibitor alone revealed significant differences in viability rates but not on the level of DNA fragmentation.
Influence of COX-2 and PPAR-γ siRNA on cannabidiol-induced apoptotic cell death
SiRNA experiments were carried out to exclude off-target effects of NS-398 and GW9662. Transfection of cells with COX-2 and PPAR-γ siRNA was shown to interfere with cannabidiol-induced COX-2 and PPAR-γ protein levels (Fig. 3A) and significantly inhibited toxicity (Fig. 3B) and apoptosis (Fig. 3C) elicited by cannabidiol in both cell lines.
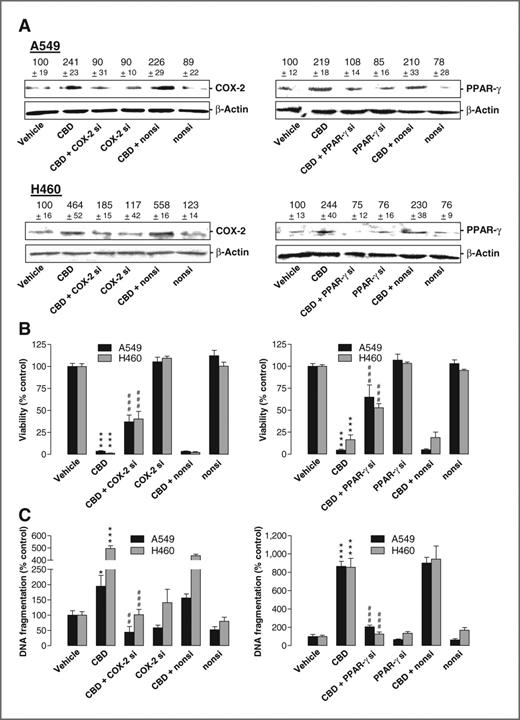
Impact of COX-2 and PPAR-γ siRNA on cannabidiol (CBD)-induced apoptosis. A–C, effect of COX-2 and PPAR-γ siRNA on COX-2 and PPAR-γ protein expression (A), cellular viability (B), and DNA fragmentation (C) in the presence or absence of 3 μmol/L cannabidiol. Incubation times were 8 hours (A, Western blot analyses), 48 hours (B, WST-1 test), and 18 hours (C, DNA fragmentation), respectively. β-Actin was used as loading control for Western blot analysis. Values above the blots represent videodensitometric analysis given as percentage of vehicle control (A). Values are mean ± SEM of n = 4 (A, top left, bottom right), n = 3 (A, bottom left, top right), n = 11–12 (B, left), n = 12 (B, right, A549), n = 18 (B, right, H460), n = 3–4 (C). *, P < 0.05; ***, P < 0.001 versus vehicle; ##, P < 0.01; ###, P < 0.001 versus cannabidiol.
Impact of COX-2 and PPAR-γ siRNA on cannabidiol (CBD)-induced apoptosis. A–C, effect of COX-2 and PPAR-γ siRNA on COX-2 and PPAR-γ protein expression (A), cellular viability (B), and DNA fragmentation (C) in the presence or absence of 3 μmol/L cannabidiol. Incubation times were 8 hours (A, Western blot analyses), 48 hours (B, WST-1 test), and 18 hours (C, DNA fragmentation), respectively. β-Actin was used as loading control for Western blot analysis. Values above the blots represent videodensitometric analysis given as percentage of vehicle control (A). Values are mean ± SEM of n = 4 (A, top left, bottom right), n = 3 (A, bottom left, top right), n = 11–12 (B, left), n = 12 (B, right, A549), n = 18 (B, right, H460), n = 3–4 (C). *, P < 0.05; ***, P < 0.001 versus vehicle; ##, P < 0.01; ###, P < 0.001 versus cannabidiol.
Influence of exogenous PGs on lung cancer cell apoptosis
To investigate a link between cannabidiol-induced elevation of COX-2–dependent PGs and subsequent apoptosis, the impact of PGs on cancer cell viability was evaluated. Incubation of cells with PGD2 and 15d-PGJ2, but not PGE2, was associated with a partial but concentration-dependent loss of viability attaining a plateau in most cases (Fig. 4A).

Impact of COX-2–dependent PGs on apoptosis and PPAR-γ activation. A, concentration-dependent effect of PGD2, 15d-PGJ2, and PGE2 on the viability of A549 and H460 cells following a 48-hour incubation with the respective compound. Data represent mean ± SEM of n = 5–12 experiments. B, evaluation of nuclear PPAR-γ by confocal microscopy following an 18-hour incubation with PGD2, 15d-PGJ2, PGE2, and the PPAR-γ agonist troglitazone as positive control (all compounds tested at 10 μmol/L). Densitometric quantification of PPAR-γ in nuclear regions is indicated below confocal views. C, effect of GW9662 (10 μmol/L) on DNA fragmentation by PGD2 and 15d-PGJ2. GW9662 was added to cells 1 hour before the respective PGs (10 μmol/L) or vehicle and the incubation was continued for 18 hours. Values are mean ± SEM of n = 6 (A), n = 4 (C, left), or n = 7–18 (C, right) experiments. **, P < 0.01; ***, P < 0.001 vs. vehicle; †††, P < 0.001 vs. 15d-PGJ2; ‡‡‡, P < 0.001 vs. PGD2.
Impact of COX-2–dependent PGs on apoptosis and PPAR-γ activation. A, concentration-dependent effect of PGD2, 15d-PGJ2, and PGE2 on the viability of A549 and H460 cells following a 48-hour incubation with the respective compound. Data represent mean ± SEM of n = 5–12 experiments. B, evaluation of nuclear PPAR-γ by confocal microscopy following an 18-hour incubation with PGD2, 15d-PGJ2, PGE2, and the PPAR-γ agonist troglitazone as positive control (all compounds tested at 10 μmol/L). Densitometric quantification of PPAR-γ in nuclear regions is indicated below confocal views. C, effect of GW9662 (10 μmol/L) on DNA fragmentation by PGD2 and 15d-PGJ2. GW9662 was added to cells 1 hour before the respective PGs (10 μmol/L) or vehicle and the incubation was continued for 18 hours. Values are mean ± SEM of n = 6 (A), n = 4 (C, left), or n = 7–18 (C, right) experiments. **, P < 0.01; ***, P < 0.001 vs. vehicle; †††, P < 0.001 vs. 15d-PGJ2; ‡‡‡, P < 0.001 vs. PGD2.
Confocal microscopic analyses revealed a translocation of PPAR-γ to nuclear regions of A549 and H460 cells upon treatment with PGD2 and 15d-PGJ2, whereas PGE2 was inactive in this respect (Fig. 4B).
To prove a role of PPAR-γ activation in apoptosis by PGD2 and 15d-PGJ2, DNA fragmentation by the respective PG was assessed in the presence of GW9662. As shown for cannabidiol-induced apoptosis before, GW9662 led to a significant inhibition of the proapoptotic effects of PGD2 and 15d-PGJ2 in both cell lines (Fig. 4C).
Influence of cannabidiol-induced COX-2–dependent PGs on total and nuclear PPAR-γ levels
To elucidate a possible connection between cannabidiol-induced COX-2 activity and PPAR-γ, experiments were carried out to clarify whether a combination of cannabidiol and the COX-2 inhibitor NS-398 may abrogate the cannabidiol-induced PPAR-γ expression and/or activation.
As shown in Fig. 5A, NS-398 did not interfere with the cannabidiol-induced PPAR-γ protein expression in A549 and H460 cells. However, according to the confocal views of A549 (Fig. 5B) and a densitometric analysis (Fig. 5C), PPAR-γ was much more restricted to nuclear regions in cells treated with cannabidiol as compared with cells treated with cannabidiol in combination with NS-398.
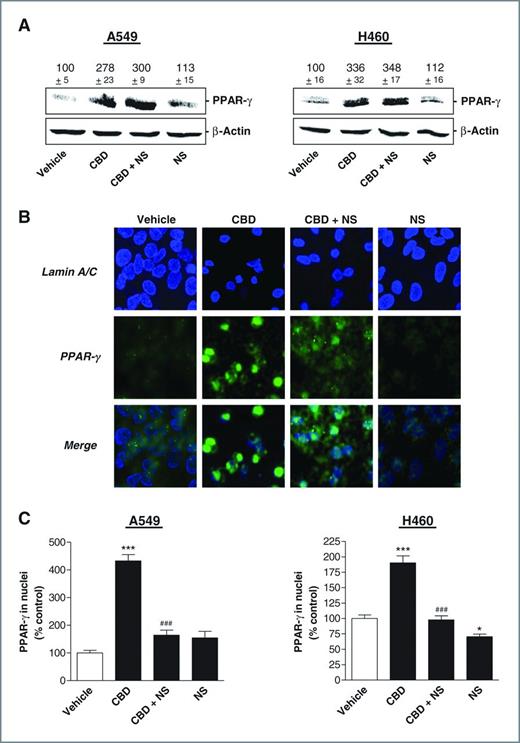
Impact of COX-2 on PPAR-γ expression and activation. A, PPAR-γ protein levels in cells treated with 3 μmol/L cannabidiol (CBD) in the presence or absence of 1 μmol/L NS-398 for 8 hours. β-Actin was used as loading control for Western blot analysis. Values above the blots represent videodensitometric analysis given as percentage of vehicle control. B, evaluation of nuclear PPAR-γ by confocal microscopy in cells incubated with 3 μmol/L cannabidiol in the presence or absence of 1 μmol/L NS-398 for 18 hours. C, densitometric quantification of PPAR-γ in nuclear regions. In all cases NS-398 was added to cells 1 hour before cannabidiol or vehicle and the incubation was continued for the indicated time. Values are mean ± SEM of n = 4 (A, left) or n = 3 (A, right) experiments. *, P < 0.05; ***, P < 0.001 vs. vehicle; ###, P < 0.001 vs. cannabidiol.
Impact of COX-2 on PPAR-γ expression and activation. A, PPAR-γ protein levels in cells treated with 3 μmol/L cannabidiol (CBD) in the presence or absence of 1 μmol/L NS-398 for 8 hours. β-Actin was used as loading control for Western blot analysis. Values above the blots represent videodensitometric analysis given as percentage of vehicle control. B, evaluation of nuclear PPAR-γ by confocal microscopy in cells incubated with 3 μmol/L cannabidiol in the presence or absence of 1 μmol/L NS-398 for 18 hours. C, densitometric quantification of PPAR-γ in nuclear regions. In all cases NS-398 was added to cells 1 hour before cannabidiol or vehicle and the incubation was continued for the indicated time. Values are mean ± SEM of n = 4 (A, left) or n = 3 (A, right) experiments. *, P < 0.05; ***, P < 0.001 vs. vehicle; ###, P < 0.001 vs. cannabidiol.
Confirmation of the proapoptotic mechanism of cannabidiol in primary lung tumor cells
Viability analyses revealed a concentration-dependent cytotoxic action of cannabidiol in primary tumor cells obtained from a brain metastasis of a patient with lung cancer that became even significant at a concentration as low as 0.001 μmol/L (Fig. 6A). Interestingly, in primary tumor cells cannabidiol's impact on viability exerted a bottom plateau at 36.4% ± 3.2% as compared with vehicle control (100%). A nonlinear regression within this viability range revealed an IC50 value of 0.124 μmol/L. Cannabidiol also elicited an induction of COX-2 and PPAR-γ in primary tumor cells on mRNA and protein levels (Fig. 6B). Using bisbenzimide staining, small condensed nuclei as characteristic feature of apoptotic cell death were observed in cells treated with cannabidiol, whereas similar nuclear morphologies were found in cells treated with vehicle or cannabidiol in the presence of NS-398 or GW9662 (Fig. 6C). In agreement with this finding, cannabidiol-induced DNA fragmentation and cytotoxicity was inhibited by NS-398 and GW9662 (Fig. 6D and E).

Cannabidiol (CBD)-induced apoptotic death of primary lung tumor cells. A, concentration-dependent action of cannabidiol on the viability of primary tumor cells. Cells were incubated with cannabidiol or its vehicle for 48 hours. B, effect of cannabidiol on COX-2 and PPAR-γ mRNA (left) and protein (right) expression following an 8 hour incubation (mRNA) or an 18 hour incubation (protein) with vehicle or cannabidiol at 3 μmol/L. β-Actin was used as loading control for Western blot analysis. Values above the blots represent videodensitometric analysis given as percentage of vehicle control. C–E, evaluation of the impact of NS-398 (1 μmol/L) and GW9662 (10 μmol/L) on cannabidiol-induced apoptotic cell death. NS-398 and GW9662 were added to cells 1 hour before 3 μmol/L cannabidiol or vehicle and the incubation was continued for 24 hours (C, bisbenzimide staining), 48 hours (D, WST-1 test), or 18 hours (E, DNA fragmentation). F, concentration-dependent action of COX-2–dependent PGs on the viability of primary tumor cells. Cells were incubated with the respective PG at the indicated concentrations or its vehicle for 18 hours. G and H, effect of GW9662 (10 μmol/L) on viability (G) and DNA fragmentation (H) by PGD2 and 15d-PGJ2. GW9662 (10 μmol/L) was added 1 hour before the respective PG (10 nmol/L) or vehicle and the incubation was continued for 18 hours. Values are mean ± SEM of n = 6 (A), n = 3–4 (B), n = 6–12 (D), n = 4–10 (F), n = 4 (E, G, and H) experiments. **, P < 0.01; ***, P < 0.001 vs. vehicle; ###, P < 0.001 vs. cannabidiol; ‡‡‡, P < 0.001 vs. PGD2; †††, P < 0.001 vs. 15d-PGJ2.
Cannabidiol (CBD)-induced apoptotic death of primary lung tumor cells. A, concentration-dependent action of cannabidiol on the viability of primary tumor cells. Cells were incubated with cannabidiol or its vehicle for 48 hours. B, effect of cannabidiol on COX-2 and PPAR-γ mRNA (left) and protein (right) expression following an 8 hour incubation (mRNA) or an 18 hour incubation (protein) with vehicle or cannabidiol at 3 μmol/L. β-Actin was used as loading control for Western blot analysis. Values above the blots represent videodensitometric analysis given as percentage of vehicle control. C–E, evaluation of the impact of NS-398 (1 μmol/L) and GW9662 (10 μmol/L) on cannabidiol-induced apoptotic cell death. NS-398 and GW9662 were added to cells 1 hour before 3 μmol/L cannabidiol or vehicle and the incubation was continued for 24 hours (C, bisbenzimide staining), 48 hours (D, WST-1 test), or 18 hours (E, DNA fragmentation). F, concentration-dependent action of COX-2–dependent PGs on the viability of primary tumor cells. Cells were incubated with the respective PG at the indicated concentrations or its vehicle for 18 hours. G and H, effect of GW9662 (10 μmol/L) on viability (G) and DNA fragmentation (H) by PGD2 and 15d-PGJ2. GW9662 (10 μmol/L) was added 1 hour before the respective PG (10 nmol/L) or vehicle and the incubation was continued for 18 hours. Values are mean ± SEM of n = 6 (A), n = 3–4 (B), n = 6–12 (D), n = 4–10 (F), n = 4 (E, G, and H) experiments. **, P < 0.01; ***, P < 0.001 vs. vehicle; ###, P < 0.001 vs. cannabidiol; ‡‡‡, P < 0.001 vs. PGD2; †††, P < 0.001 vs. 15d-PGJ2.
WST-1 tests showed a profound toxicity by PGD2 and 15d-PGJ2 with a significant loss of viability at a concentration as low as 0.1 nmol/L of both PGs (Fig. 6F). Again, PGE2 left viability virtually unaltered even at a concentration of 10 μmol/L (data not shown). Apoptotic cell death caused by 10 nmol/L PGD2 or 15d-PGJ2 was inhibited by GW9662 (Fig. 6G and H).
Tumor-regressive action of cannabidiol in xenografted nude mice—role of PPAR-γ
Finally, the impact of the PPAR-γ inhibitor GW9662 on the tumor-regressive effect of cannabidiol was tested in athymic nude mice xenografted with A549 cells. According to Fig. 7A, cannabidiol was found to reduce tumor volume significantly as compared with vehicle-treated animals.
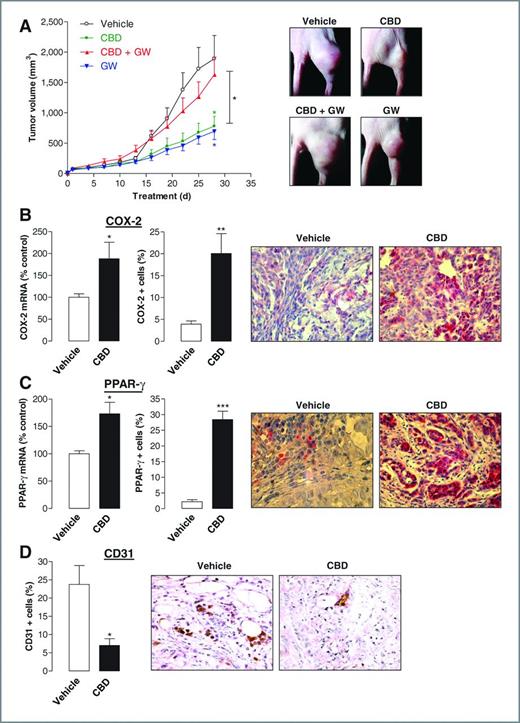
Tumor-regressive action of cannabidiol (CBD) in vivo. Animals were treated with either vehicle or cannabidiol every 72 hours (5 mg/kg) in the presence or absence of GW9662 (1 mg/kg). A, tumor volumes are mean ± SEM of n = 5–7 animals per group. *, P < 0.05 for cannabidiol versus vehicle (green asterisk), GW9662 versus vehicle (blue asterisk), cannabidiol versus cannabidiol + GW9662 (black asterisk). B–D, immunhistochemical protein analyses were obtained from animals treated with cannabidiol or vehicle. Quantification of COX-2 and PPAR-γ mRNA was conducted by real-time RT-PCR (B and C, left). Quantification of COX-2, PPAR-γ, and CD31 (vascularization marker) protein levels (B and C, middle; D, left) was achieved by counting positively stained cells from different animals. Data represent mean of n = 3–4 (mRNA), n = 4 (CD31), or n = 5 (COX-2, PPAR-γ, protein) different tumors per group. *, P < 0.05; **, P < 0.01; ***, P < 0.001 vs. vehicle.
Tumor-regressive action of cannabidiol (CBD) in vivo. Animals were treated with either vehicle or cannabidiol every 72 hours (5 mg/kg) in the presence or absence of GW9662 (1 mg/kg). A, tumor volumes are mean ± SEM of n = 5–7 animals per group. *, P < 0.05 for cannabidiol versus vehicle (green asterisk), GW9662 versus vehicle (blue asterisk), cannabidiol versus cannabidiol + GW9662 (black asterisk). B–D, immunhistochemical protein analyses were obtained from animals treated with cannabidiol or vehicle. Quantification of COX-2 and PPAR-γ mRNA was conducted by real-time RT-PCR (B and C, left). Quantification of COX-2, PPAR-γ, and CD31 (vascularization marker) protein levels (B and C, middle; D, left) was achieved by counting positively stained cells from different animals. Data represent mean of n = 3–4 (mRNA), n = 4 (CD31), or n = 5 (COX-2, PPAR-γ, protein) different tumors per group. *, P < 0.05; **, P < 0.01; ***, P < 0.001 vs. vehicle.
GW9662 was administered at a dose of 1 mg/kg, which has been reported to inhibit PPAR-γ activity in vivo (29). The tumor-regressive action of cannabidiol was significantly reduced by pretreatment with GW9662. Interestingly, GW9662 alone exerted a significant tumor-regressive effect comparable with that caused by cannabidiol.
Evaluation of COX-2 and PPAR-γ levels in tumor tissue and slices revealed significant increases of both mRNA and protein in cannabidiol-treated as compared with vehicle-treated animals (Fig. 7B and C) yielding a 5.1-fold induction of COX-2 and a 13.1-fold induction of PPAR-γ–positive cells. Because of several studies suggesting COX-2 as an important trigger of tumor angiogenesis (30, 31), the impact of cannabidiol on tumor vascularization was addressed by staining of the vascularization marker CD31. However, the number of CD31-positive cells was even significantly downregulated in tumors from mice treated with cannabidiol (Fig. 7D).
Discussion
The present study shows induction of COX-2 and PPAR-γ as a novel mechanism underlying the proapoptotic action of cannabidiol. Following upregulation by cannabidiol at the expression level, both proteins converge in a coordinated fashion via activation of PPAR-γ by the COX-2–derived products PGD2 and 15d-PGJ2.
There are several experimental evidence supporting the aforementioned pathway. First, cannabidiol caused an upregulation of COX-2 and PPAR-γ mRNA and protein expression in 2 NSCLC lines as well as in primary lung tumor cells. Second, elevations of PGE2, PGD2, and 15d-PGJ2 elicited by cannabidiol in cell lines were sensitive to the selective COX-2 inhibitor NS-398, thus confirming that cannabidiol-induced COX-2 expression was associated with functional relevant increases of enzyme activity. Third, suppression of COX-2 activity by NS-398, inhibition of PPAR-γ by GW9662 as well as knockdown of COX-2 and PPAR-γ by siRNA led to profound reductions of both cannabidiol's proapoptotic and cytotoxic action. Fourth, immunocytochemical analyses of the subcellular compartimentalization of PPAR-γ revealed a significant cannabidiol-induced accumulation of PPAR-γ within nuclear regions that has been described as marker of PPAR-γ activation (32). This effect was mimicked by PGD2, 15d-PGJ2, and troglitazone and inhibited by NS-398 indicating an involvement of cannabidiol-induced COX-2 activity in PPAR-γ activation. In addition, PGD2 and 15d-PGJ2, but not PGE2, were shown to exert apoptotic cell death in A549, H460, and primary lung tumor cells via activation of PPAR-γ. This finding is in line with previous studies showing a PPAR-γ–dependent proapoptotic action of PGD2 and 15d-PGJ2 in several cancer cell types (19, 24–26).
Noteworthy, cannabidiol caused a significant reduction of primary tumor cell viability at a concentration as low as 0.001 μmol/L, which is even lower than plasma peaks of 0.036 μmol/L that were analyzed following a 6-week oral treatment with cannabidiol at doses of 10 mg/kg/d (33). Because of differences in experimental settings of the assays used in the current project, a precise correlation between PGs measured in cell culture media and exogenously added PGs is critical. However, given that a significant toxicity by both PGD2 and 15d-PGJ2 was even observed at concentrations as low as 0.001 μmol/L, an approximate correlation between toxic concentrations exogenously added and the range of PGs synthesized endogenously upon cannabidiol treatment seems to exist in A549 and H460 cells.
A possible induction of COX-2 by PPAR-γ as proposed by others (34) was not supported in view of an approximate concurrent upregulation of both mRNAs. Vice versa, a potential stimulatory action of COX-2 on PPAR-γ was excluded by data showing that inhibition of COX-2 activity by NS-398 did not alter cannabidiol-induced PPAR-γ expression within the 8-hour incubation period evaluated here.
Although cannabidiol shares the PG-dependent activation of PPAR-γ with several other substances (19, 21–24), there are also data showing a direct binding of cannabidiol to PPAR-γ independent of endogenous PGs followed by an increase of its transcriptional activity (35). In our hands, NS-398 or GW9662 alone profoundly but in most instances not completely reversed the cytotoxic and proapoptotic action of cannabidiol, whereas a combined treatment of cannabidiol-exposed cells with NS-398 and GW9662 fully restored control conditions. However, given that these experiments revealed no statistical significance on the level of DNA fragmentation when both inhibitors in combination with cannabidiol were compared with cannabidiol- and NS-398–treated cells, no definite conclusion about an additional direct and PG-independent PPAR-γ activation by cannabidiol can be drawn at present.
There are several lines of evidence indicating the presented in vitro mechanism to take also place under in vivo conditions. Accordingly, a profound induction of both COX-2 and PPAR-γ mRNA and protein levels was observed in A549 xenografts of cannabidiol-treated athymic nude mice. A causal link between the tumor-regressive effect of cannabidiol and PPAR-γ activation is implied by data showing a complete reversal of the tumor-regressive action of cannabidiol by the PPAR-γ antagonist GW9662. This observation is supported by a previous report showing a comparable effect of the PPAR-γ agonists troglitazone and pioglitazone in A549 tumor-bearing severe combined immunodeficient mice (27). Despite the fact that GW9662 fully reversed cannabidiol's inhibitory effect on A549 xenograft growth, the substance caused a pronounced tumor-regressive action when administered alone. Similar ambiguous findings have been reported in vitro where the PPAR-γ agonist rosiglitazone and the PPAR-γ antagonist GW9662 inhibited the growth of breast cancer cells to similar extents (36). In fact, PPAR-γ antagonists have been shown to confer PPAR-γ–independent antiproliferative, antiadhesive, and antimetastatic actions (for review see ref. 37). However, a specific binding to PPAR-γ and therefore preventing GW9662 from off-targets conferring apoptosis is expected when PPAR-γ levels are substantially elevated as was the case with tumor tissues of cannabidiol-treated mice in which a 13.1-fold induction of PPAR-γ–positive cells was observed.
Some issues merit special consideration. First, it cannot be excluded that key players of cannabidiol's antitumorigenic effects reported for other experimental systems may also be part of cannabidiol's proapoptotic impact on lung cancer cells. Accordingly, cannabidiol was shown to elicit intrinsic apoptotic pathways via elevation of reactive oxygen species in diverse cancer cell types (3–5, 7) and to activate autophagic signaling pathways (6).
Second, studies on the impact of cannabinoids on COX enzymes have yielded controversial results. In fact, an upregulation of COX-2 expression by diverse cannabinoids including R(+)-methanandamide, Δ9-tetrahydrocannabinol or anandamide has been repeatedly shown in different cell types (16–19, 38). However, there are only a few in vivo investigations addressing cannabidiol's impact on COX-2 in cancer tissue showing no interference with COX-2 protein levels in experimental glioma (39) and colon cancer (40). About the impact of cannabinoids on COX-2 activity in cell-free assays, several cannabinoids including cannabidiolic acid (41), cannabigerol, and cannabigerolic acid (42) have been shown to profoundly inhibit COX-2, whereas cannabidiol tested at 100 μmol/L (41) or 300 μmol/L (42) failed to elicit such response or only slightly suppressed COX-2 at 100 μmol/L (39).
Third, our results support several other investigations pointing to a pivotal role of PPAR-γ activated by COX-2–dependent PGs of the D- and J-series in the proapoptotic action of several substances including well-established chemotherapeutics (19, 21–24), thereby challenging the traditional view considering COX-2 inhibition as an anticancer strategy. In line with these data, overexpression of COX-2 decreased proliferation and increased apoptosis of osteosarcoma cells (43) and protected rather than sensitized animals to experimental skin tumor development (44). In addition, clinical data revealed a correlation between COX-2 expression and a less aggressive breast cancer phenotype (45). On the other hand, a rather protumorigenic function of COX-2 is supported by investigations that showed COX-2–derived PGs to decrease tumor cell apoptosis (46) and to increase tumor cell proliferation (47), invasion (47), and angiogenesis (30, 31). It is noteworthy that a major basis of the view suggesting tumor-promoting properties of COX-2 has been derived from preclinical data pointing to antiproliferative and proapoptotic properties of COX-2 inhibitors at rather high concentrations that, however, retain their proapoptotic properties in cells lacking COX-2 (for review see ref. 48). In agreement with this notion, there are several studies reporting NS-398 to elicit significant proapoptotic and cytotoxic responses when used at concentrations far above those needed for full inhibition of PG synthesis [e.g., 30 μmol/L in HeLa (24); 100 μmol/L in A549 (49)]. The experiments presented here did not reveal an influence of NS-398 at a concentration of 1 μmol/L on the viability of A549 and H460, which is in line with another report indicating no significant toxicity of NS-398 on A549 and H460 at concentrations up to 20 μmol/L (50).
Collectively, our data strengthen the notion that activation of PPAR-γ may present a promising target for lung cancer therapy. In addition and to the best of our knowledge, this is the first report to provide an inhibitor-proven tumor-regressive mechanism of cannabidiol in vivo as well as a proapoptotic mechanism confirmed by use of primary lung tumor cells. Against this background and considering recent findings supporting a profound antimetastatic action of cannabidiol, this cannabinoid may represent a promising anticancer drug.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Authors' Contributions
Conception and design: R. Ramer, B. Hinz
Development of methodology: R. Ramer, J. Merkord, A. Salamon, B. Hinz
Acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.): R. Ramer, K. Heinemann, J. Merkord, A. Salamon, M. Linnebacher
Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): R. Ramer, B. Hinz
Writing, review, and/or revision of the manuscript: R. Ramer, B. Hinz
Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): R. Ramer, K. Heinemann, H. Rohde, A. Salamon, B. Hinz
Study supervision: B. Hinz
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.