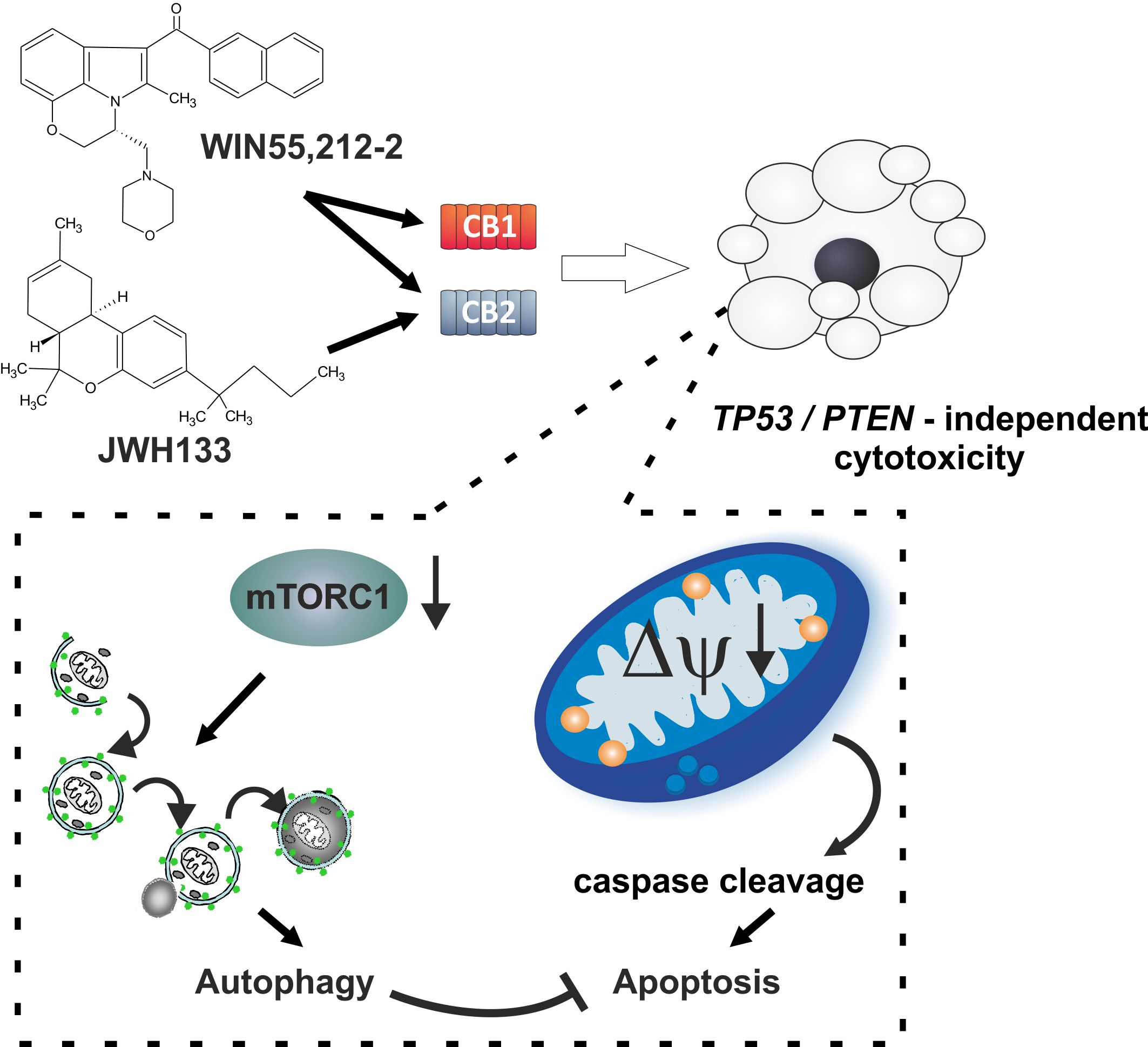
Synthetic Cannabinoids Induce Autophagy and Mitochondrial Apoptotic Pathways in Human Glioblastoma Cells Independently of Deficiency in TP53 or PTEN Tumor Suppressors


Abstract
:Simple Summary
Abstract

1. Introduction
2. Results
2.1. Human Glioblastoma Cells Express CB1 and CB2 Receptors
2.2. Cannabinoids Induce Growth Arrest and Cell Death in p53mut and/or PTENmut Human Glioblastoma Cells
2.3. Cannabinoids Trigger Mitochondrial Pathway of Cell Death and Caspase Activation
2.4. Cannabinoids Induce Autophagy Associated with Inhibition of mTOR Pathway in Glioblastoma Cells
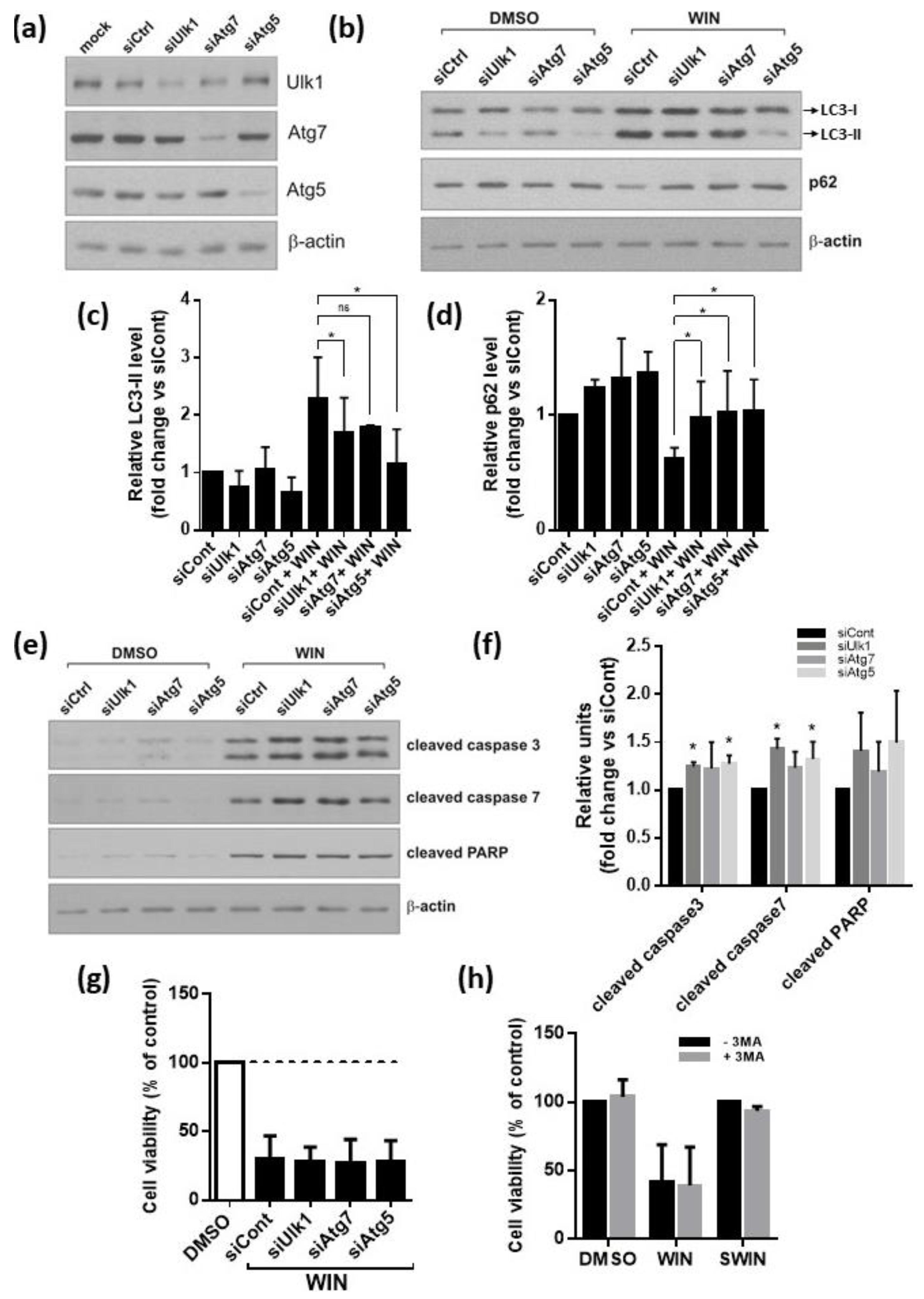
2.5. Inhibition of Autophagy Augments Cannabinoids-Induced Apoptotic Cell Death
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture and Treatment
4.3. Tissue Samples
4.4. Cell Viability—MTT Metabolism Assay
4.5. Quantitative RT-PCR Analysis
4.6. Immunocytochemistry and Acridine Orange Staining
4.7. Transfection with Plasmid DNA for Detection of Autophagy and p53-Responsive Luciferase Reporter Assay
4.8. Silencing of Autophagy Gene Expression with siRNA
4.9. Western Blot Analysis
4.10. TUNEL Staining
4.11. Flow Cytometry
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kleihues, P.; Louis, D.N.; Scheithauer, B.W.; Rorke, L.B.; Reifenberger, G.; Burger, P.C.; Cavenee, W.K. The WHO classification of tumors of the nervous system. J. Neuropathol. Exp. Neurol. 2002, 61, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Hosli, P.; Sappino, A.P.; de Tribolet, N.; Dietrich, P.Y. Malignant glioma: Should chemotherapy be overthrown by experimental treatments? Ann. Oncol. 1998, 9, 589–600. [Google Scholar] [CrossRef] [PubMed]
- Galanis, E.; Buckner, J.C. Chemotherapy of brain tumors. Curr. Opin. Neurol. 2000, 13, 619–625. [Google Scholar] [CrossRef] [PubMed]
- Ohgaki, H.; Kleihues, P. Population-based studies on incidence, survival rates, and genetic alterations in astrocytic and oligodendroglial gliomas. J. Neuropathol. Exp. Neurol. 2005, 64, 479–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roth, W.; Weller, M. Chemotherapy and immunotherapy of malignant glioma: Molecular mechanisms and clinical perspectives. Cell Mol. Life Sci. 1999, 56, 481–506. [Google Scholar] [CrossRef]
- Iwadate, Y.; Fujimoto, S.; Tagawa, M.; Namba, H.; Sueyoshi, K.; Hirose, M.; Sakiyama, S. Association of p53 gene mutation with decreased chemosensitivity in human malignant gliomas. Int. J. Cancer 1996, 69, 236–240. [Google Scholar] [CrossRef]
- Mohri, M.; Nitta, H.; Yamashita, J. Expression of multidrug resistance-associated protein (MRP) in human gliomas. J. Neurooncol. 2000, 49, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, R.; Capparella, J.; Brown, A. Classification of glioblastoma multiforme in adults by molecular genetics. Cancer J. 2003, 9, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Haas-Kogan, D.; Shalev, N.; Wong, M.; Mills, G.; Yount, G.; Stokoe, D. Protein kinase B (PKB/Akt) activity is elevated in glioblastoma cells due to mutation of the tumor suppressor PTEN/MMAC. Curr. Biol. 1998, 8, 1195–1198. [Google Scholar] [CrossRef] [Green Version]
- Kaufmann, S.H.; Vaux, D.L. Alterations in the apoptotic machinery and their potential role in anticancer drug resistance. Oncogene 2003, 22, 7414–7430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steele, R.J.; Thompson, A.M.; Hall, P.A.; Lane, D.P. The p53 tumour suppressor gene. Br. J. Surg. 1998, 85, 1460–1467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishii, N.; Maier, D.; Merlo, A.; Tada, M.; Sawamura, Y.; Diserens, A.C.; Van Meir, E.G. Frequent co-alterations of TP53, p16/CDKN2A, p14ARF, PTEN tumor suppressor genes in human glioma cell lines. Brain Pathol. 1999, 9, 469–479. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Kato, S.; Kumabe, T.; Sonoda, Y.; Yoshimoto, T.; Kato, S.; Han, S.Y.; Suzuki, T.; Shibata, H.; Kanamaru, R.; et al. Functional evaluation of p53 and PTEN gene mutations in gliomas. Clin. Cancer Res. 2000, 6, 3937–3943. [Google Scholar]
- Mayo, L.D.; Dixon, J.E.; Durden, D.L.; Tonks, N.K.; Donner, D.B. PTEN protects p53 from Mdm2 and sensitizes cancer cells to chemotherapy. J. Biol. Chem. 2002, 277, 5484–5489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mackie, K. Cannabinoid receptors as therapeutic targets. Annu. Rev. Pharmacol. Toxicol. 2006, 46, 101–122. [Google Scholar] [CrossRef] [Green Version]
- Guzman, M. Cannabinoids: Potential anticancer agents. Nat. Rev. Cancer 2003, 3, 745–755. [Google Scholar] [CrossRef] [PubMed]
- Ellert-Miklaszewska, A.; Ciechomska, I.A.; Kaminska, B. Cannabinoid Signaling in Glioma Cells. Adv. Exp. Med. Biol. 2020, 1202, 223–241. [Google Scholar] [CrossRef]
- Sanchez, C.; Galve-Roperh, I.; Canova, C.; Brachet, P.; Guzman, M. Delta9-tetrahydrocannabinol induces apoptosis in C6 glioma cells. FEBS Lett. 1998, 436, 6–10. [Google Scholar] [CrossRef] [Green Version]
- Duntsch, C.; Divi, M.K.; Jones, T.; Zhou, Q.; Krishnamurthy, M.; Boehm, P.; Wood, G.; Sills, A.; Ii, B.M. Safety and efficacy of a novel cannabinoid chemotherapeutic, KM-233, for the treatment of high-grade glioma. J. Neurooncol. 2006, 77, 143–152. [Google Scholar] [CrossRef]
- Massi, P.; Vaccani, A.; Ceruti, S.; Colombo, A.; Abbracchio, M.P.; Parolaro, D. Antitumor effects of cannabidiol, a nonpsychoactive cannabinoid, on human glioma cell lines. J. Pharmacol. Exp. Ther. 2004, 308, 838–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galve-Roperh, I.; Sanchez, C.; Cortes, M.L.; del Pulgar, T.G.; Izquierdo, M.; Guzman, M. Anti-tumoral action of cannabinoids: Involvement of sustained ceramide accumulation and extracellular signal-regulated kinase activation. Nat. Med. 2000, 6, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, C.; de Ceballos, M.L.; del Pulgar, T.G.; Rueda, D.; Corbacho, C.; Velasco, G.; Galve-Roperh, I.; Huffman, J.W.; Ramon y Cajal, S.; Guzman, M. Inhibition of glioma growth in vivo by selective activation of the CB(2) cannabinoid receptor. Cancer Res. 2001, 61, 5784–5789. [Google Scholar] [PubMed]
- Guzman, M.; Duarte, M.J.; Blazquez, C.; Ravina, J.; Rosa, M.C.; Galve-Roperh, I.; Sanchez, C.; Velasco, G.; Gonzalez-Feria, L. A pilot clinical study of Delta9-tetrahydrocannabinol in patients with recurrent glioblastoma multiforme. Br. J. Cancer 2006, 95, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Afrin, F.; Chi, M.; Eamens, A.L.; Duchatel, R.J.; Douglas, A.M.; Schneider, J.; Gedye, C.; Woldu, A.S.; Dun, M.D. Can Hemp Help? Low-THC Cannabis and Non-THC Cannabinoids for the Treatment of Cancer. Cancers 2020, 12, 1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carracedo, A.; Lorente, M.; Egia, A.; Blazquez, C.; Garcia, S.; Giroux, V.; Malicet, C.; Villuendas, R.; Gironella, M.; Gonzalez-Feria, L.; et al. The stress-regulated protein p8 mediates cannabinoid-induced apoptosis of tumor cells. Cancer Cell 2006, 9, 301–312. [Google Scholar] [CrossRef] [Green Version]
- Gomez del Pulgar, T.; Velasco, G.; Sanchez, C.; Haro, A.; Guzman, M. De novo-synthesized ceramide is involved in cannabinoid-induced apoptosis. Biochem. J. 2002, 363, 183–188. [Google Scholar] [CrossRef] [Green Version]
- Salazar, M.; Carracedo, A.; Salanueva, I.J.; Hernandez-Tiedra, S.; Lorente, M.; Egia, A.; Vazquez, P.; Blazquez, C.; Torres, S.; Garcia, S.; et al. Cannabinoid action induces autophagy-mediated cell death through stimulation of ER stress in human glioma cells. J. Clin. Investig. 2009, 119, 1359–1372. [Google Scholar] [CrossRef] [Green Version]
- Klionsky, D.J.; Emr, S.D. Autophagy as a regulated pathway of cellular degradation. Science 2000, 290, 1717–1721. [Google Scholar] [CrossRef]
- Kondo, Y.; Kanzawa, T.; Sawaya, R.; Kondo, S. The role of autophagy in cancer development and response to therapy. Nat. Rev. Cancer 2005, 5, 726–734. [Google Scholar] [CrossRef]
- Kang, C.; Avery, L. To be or not to be, the level of autophagy is the question: Dual roles of autophagy in the survival response to starvation. Autophagy 2008, 4, 82–84. [Google Scholar] [CrossRef] [Green Version]
- Ciechomska, I.A.; Gabrusiewicz, K.; Szczepankiewicz, A.A.; Kaminska, B. Endoplasmic reticulum stress triggers autophagy in malignant glioma cells undergoing cyclosporine a-induced cell death. Oncogene 2013, 32, 1518–1529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shrivastava, A.; Kuzontkoski, P.M.; Groopman, J.E.; Prasad, A. Cannabidiol induces programmed cell death in breast cancer cells by coordinating the cross-talk between apoptosis and autophagy. Mol. Cancer Ther. 2011, 10, 1161–1172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stella, N. Cannabinoid signaling in glial cells. Glia 2004, 48, 267–277. [Google Scholar] [CrossRef]
- Ellert-Miklaszewska, A.; Grajkowska, W.; Gabrusiewicz, K.; Kaminska, B.; Konarska, L. Distinctive pattern of cannabinoid receptor type II (CB2) expression in adult and pediatric brain tumors. Brain Res. 2007, 1137, 161–169. [Google Scholar] [CrossRef]
- Ellert-Miklaszewska, A.; Kaminska, B.; Konarska, L. Cannabinoids down-regulate PI3K/Akt and Erk signalling pathways and activate proapoptotic function of Bad protein. Cell Signal. 2005, 17, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Van Meir, E.G.; Kikuchi, T.; Tada, M.; Li, H.; Diserens, A.C.; Wojcik, B.E.; Huang, H.J.; Friedmann, T.; de Tribolet, N.; Cavenee, W.K. Analysis of the p53 gene and its expression in human glioblastoma cells. Cancer Res. 1994, 54, 649–652. [Google Scholar]
- Weller, M.; Winter, S.; Schmidt, C.; Esser, P.; Fontana, A.; Dichgans, J.; Groscurth, P. Topoisomerase-I inhibitors for human malignant glioma: Differential modulation of p53, p21, bax and bcl-2 expression and of CD95-mediated apoptosis by camptothecin and beta-lapachone. Int. J. Cancer 1997, 73, 707–714. [Google Scholar] [CrossRef]
- Lukashchuk, N.; Vousden, K.H. Ubiquitination and degradation of mutant p53. Mol. Cell Biol. 2007, 27, 8284–8295. [Google Scholar] [CrossRef] [Green Version]
- Zarkowska, T.; Mittnacht, S. Differential phosphorylation of the retinoblastoma protein by G1/S cyclin-dependent kinases. J. Biol. Chem. 1997, 272, 12738–12746. [Google Scholar] [CrossRef] [Green Version]
- Weinberg, R.A. The retinoblastoma protein and cell cycle control. Cell 1995, 81, 323–330. [Google Scholar] [CrossRef] [Green Version]
- Kanzawa, T.; Germano, I.M.; Komata, T.; Ito, H.; Kondo, Y.; Kondo, S. Role of autophagy in temozolomide-induced cytotoxicity for malignant glioma cells. Cell Death Differ. 2004, 11, 448–457. [Google Scholar] [CrossRef] [Green Version]
- Shingu, T.; Fujiwara, K.; Bogler, O.; Akiyama, Y.; Moritake, K.; Shinojima, N.; Tamada, Y.; Yokoyama, T.; Kondo, S. Inhibition of autophagy at a late stage enhances imatinib-induced cytotoxicity in human malignant glioma cells. Int. J. Cancer 2009, 124, 1060–1071. [Google Scholar] [CrossRef]
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.A.; Outzen, H.; Overvatn, A.; Bjorkoy, G.; Johansen, T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 2007, 282, 24131–24145. [Google Scholar] [CrossRef] [Green Version]
- De Jesus, M.L.; Hostalot, C.; Garibi, J.M.; Salles, J.; Meana, J.J.; Callado, L.F. Opposite changes in cannabinoid CB1 and CB2 receptor expression in human gliomas. Neurochem. Int. 2010, 56, 829–833. [Google Scholar] [CrossRef]
- Wu, X.; Han, L.; Zhang, X.; Li, L.; Jiang, C.; Qiu, Y.; Huang, R.; Xie, B.; Lin, Z.; Ren, J.; et al. Alteration of endocannabinoid system in human gliomas. J. Neurochem. 2012, 120, 842–849. [Google Scholar] [CrossRef]
- McAllister, S.D.; Chan, C.; Taft, R.J.; Luu, T.; Abood, M.E.; Moore, D.H.; Aldape, K.; Yount, G. Cannabinoids selectively inhibit proliferation and induce death of cultured human glioblastoma multiforme cells. J. Neurooncol. 2005, 74, 31–40. [Google Scholar] [CrossRef]
- Widmer, M.; Hanemann, C.O.; Zajicek, J. High concentrations of cannabinoids activate apoptosis in human U373MG glioma cells. J. Neurosci. Res. 2008. [Google Scholar] [CrossRef]
- Held-Feindt, J.; Dorner, L.; Sahan, G.; Mehdorn, H.M.; Mentlein, R. Cannabinoid receptors in human astroglial tumors. J. Neurochem. 2006, 98, 886–893. [Google Scholar] [CrossRef]
- Molina-Holgado, E.; Vela, J.M.; Arevalo-Martin, A.; Almazan, G.; Molina-Holgado, F.; Borrell, J.; Guaza, C. Cannabinoids promote oligodendrocyte progenitor survival: Involvement of cannabinoid receptors and phosphatidylinositol-3 kinase/Akt signaling. J. Neurosci. 2002, 22, 9742–9753. [Google Scholar] [CrossRef]
- Gong, J.P.; Onaivi, E.S.; Ishiguro, H.; Liu, Q.R.; Tagliaferro, P.A.; Brusco, A.; Uhl, G.R. Cannabinoid CB2 receptors: Immunohistochemical localization in rat brain. Brain Res. 2006, 1071, 10–23. [Google Scholar] [CrossRef]
- Zupanska, A.; Kaminska, B. The diversity of p53 mutations among human brain tumors and their functional consequences. Neurochem. Int. 2002, 40, 637–645. [Google Scholar] [CrossRef]
- Herrera, B.; Carracedo, A.; Diez-Zaera, M.; Gomez del Pulgar, T.; Guzman, M.; Velasco, G. The CB2 cannabinoid receptor signals apoptosis via ceramide-dependent activation of the mitochondrial intrinsic pathway. Exp. Cell Res. 2006, 312, 2121–2131. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Tiedra, S.; Fabrias, G.; Davila, D.; Salanueva, I.J.; Casas, J.; Montes, L.R.; Anton, Z.; Garcia-Taboada, E.; Salazar-Roa, M.; Lorente, M.; et al. Dihydroceramide accumulation mediates cytotoxic autophagy of cancer cells via autolysosome destabilization. Autophagy 2016, 12, 2213–2229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donadelli, M.; Dando, I.; Zaniboni, T.; Costanzo, C.; Dalla Pozza, E.; Scupoli, M.T.; Scarpa, A.; Zappavigna, S.; Marra, M.; Abbruzzese, A.; et al. Gemcitabine/cannabinoid combination triggers autophagy in pancreatic cancer cells through a ROS-mediated mechanism. Cell Death Dis. 2011, 2, e152. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg-Lerner, A.; Bialik, S. Fau—Simon, H.U.; Simon Hu Fau—Kimchi, A.; Kimchi, A. Life and death partners: Apoptosis, autophagy and the cross-talk between them. Cell Death Differ. 2009, 16, 966–975. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, J.L.; Hill, D.S.; McKee, C.S.; Hernandez-Tiedra, S.; Lorente, M.; Lopez-Valero, I.; Eleni Anagnostou, M.; Babatunde, F.; Corazzari, M.; Redfern, C.P.F.; et al. Exploiting cannabinoid-induced cytotoxic autophagy to drive melanoma cell death. J. Investig. Dermatol. 2015, 135, 1629–1637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vara, D.; Salazar, M.; Olea-Herrero, N.; Guzman, M.; Velasco, G.; Diaz-Laviada, I. Anti-tumoral action of cannabinoids on hepatocellular carcinoma: Role of AMPK-dependent activation of autophagy. Cell Death Differ. 2011, 18, 1099–1111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, D.R.; Kroemer, G. Cytoplasmic functions of the tumour suppressor p53. Nature 2009, 458, 1127–1130. [Google Scholar] [CrossRef] [PubMed]
- Kern, S.E.; Pietenpol, J.A.; Thiagalingam, S.; Seymour, A.; Kinzler, K.W.; Vogelstein, B. Oncogenic forms of p53 inhibit p53-regulated gene expression. Science 1992, 256, 827–830. [Google Scholar] [CrossRef] [PubMed]
- Baker, S.J.; Markowitz, S.; Fearon, E.R.; Willson, J.K.; Vogelstein, B. Suppression of human colorectal carcinoma cell growth by wild-type p53. Science 1990, 249, 912–915. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | JWH133 (µM) |
WIN55,212-2 (µM) |
|---|---|---|
| LN18 | 13.82 | 9.65 |
| T98G | 12.15 | 8.61 |
| U87MG | 18.78 | 9.86 |
| U251MG | 32.94 | 7.36 |
| LN229 | 143.20 | 15.70 |
| Cell Line | Source | PTEN Status | TP53 Status |
|---|---|---|---|
| Primary culture | |||
| T3 | GBM (grade IV) |
ND | mut |
| T10 | ND | mut | |
| Established cell line | |||
| LN18 | GBM-derived | wt | mut |
| T98G | mut | mut | |
| U87MG | AA-derived | mut | wt |
| U251MG | mut | mut | |
| LN229 | wt | mut * | |
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
|
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ellert-Miklaszewska, A.; Ciechomska, I.A.; Kaminska, B. Synthetic Cannabinoids Induce Autophagy and Mitochondrial Apoptotic Pathways in Human Glioblastoma Cells Independently of Deficiency in TP53 or PTEN Tumor Suppressors. Cancers 2021, 13, 419. https://doi.org/10.3390/cancers13030419
Ellert-Miklaszewska A, Ciechomska IA, Kaminska B. Synthetic Cannabinoids Induce Autophagy and Mitochondrial Apoptotic Pathways in Human Glioblastoma Cells Independently of Deficiency in TP53 or PTEN Tumor Suppressors. Cancers. 2021; 13(3):419. https://doi.org/10.3390/cancers13030419
Chicago/Turabian StyleEllert-Miklaszewska, Aleksandra, Iwona Anna Ciechomska, and Bozena Kaminska. 2021. "Synthetic Cannabinoids Induce Autophagy and Mitochondrial Apoptotic Pathways in Human Glioblastoma Cells Independently of Deficiency in TP53 or PTEN Tumor Suppressors" Cancers 13, no. 3: 419. https://doi.org/10.3390/cancers13030419