Treatment with Cannabinoids as a Promising Approach for Impairing Fibroblast Activation and Prostate Cancer Progression

Abstract
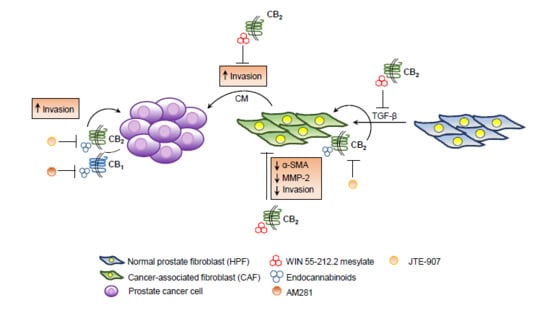
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. WIN 55-212.2 Mesylate Selectively Impairs Prostate Cancer Cells Survival
2.2. CAFs Derived from Aggressive Prostate Cancer Bearing Patients Upregulate CB1 and CB2 Compared to Normal Fibroblasts
2.3. WIN 55-212.2 Mesylate Impacts on CAFs Phenotype and Impairs TGF-β-Induced Activation in Normal Fibroblasts via CB2 Receptor
2.4. The Endocannabinoids Self-Sustain the Migration of Androgen-Insensitive Prostate Cancer Cells and CAFs’ Reactivity by an Autocrine Loop
3. Discussion
4. Materials and Methods
4.1. Antibodies and Reagents
4.2. Cell Models and Cell Cultures
4.3. Conditioned Media Preparation
4.4. Western Blot Analysis
4.5. Crystal Violet Staining
4.6. Gelatin Zymography
4.7. Boyden Chamber Assay
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| α-SMA | Alpha-smooth muscle actin |
| 2-AG | 2-arachydonoyl glycerol |
| 5-HT | 5-hydroxytryptamine |
| AEA | Anandamide |
| CAFs | Cancer-associated fibroblasts |
| CB1 | Cannabinoid receptor 1 |
| CB2 | Cannabinoid receptor 2 |
| CBD | Cannabidiol |
| EMT | Epithelial to mesenchymal transition |
| HPFs | Healthy prostate fibroblasts |
| IL-6 | Interleukin-6 |
| MMP-2 | Matrix metalloproteinase-2 |
| PPAR-γ | Peroxisome proliferator-activated receptor gamma |
| TGF-β | Transforming growth factor beta |
| THC | Delta-9-tetrahydrocannabinol |
| TME | Tumor microenvironment |
| TNF-α | Tumor necrosis factor alpha |
| TRPV1 | Vanilloid receptor 1 |
References
- Hermanson, D.J.; Marnett, L.J. Cannabinoids, endocannabinoids, and cancer. Cancer Metastasis Rev. 2011, 30, 599–612. [Google Scholar] [CrossRef]
- Guzmán, M. Cannabinoids: Potential anticancer agents. Nat. Rev. Cancer 2003, 3, 745–755. [Google Scholar] [CrossRef]
- Velasco, G.; Sánchez, C.; Guzmán, M. Anticancer mechanisms of cannabinoids. Curr. Oncol. 2016, 23, S23–S32. [Google Scholar] [CrossRef] [Green Version]
- Ramer, R.; Hinz, B. Antitumorigenic targets of cannabinoids—Current status and implications. Expert Opin. Ther. Targets 2016, 20, 1219–1235. [Google Scholar] [CrossRef] [PubMed]
- Fraguas-Sánchez, A.; Fernández-Carballido, A.; Torres-Suárez, A. Phyto-, endo- and synthetic cannabinoids: Promising chemotherapeutic agents in the treatment of breast and prostate carcinomas. Expert Opin. Investig. Drugs 2016, 25, 1311–1323. [Google Scholar] [CrossRef] [PubMed]
- Sarfaraz, S.; Afaq, F.; Adhami, V.M.; Mukhtar, H. Cannabinoid Receptor as a Novel Target for the Treatment of Prostate Cancer. Cancer Res. 2005, 65, 1635–1641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orellana-Serradell, O.; Poblete, C.E.; Sanchez, C.; Castellón, E.A.; Gallegos, I.; Huidobro, C.; Llanos, M.N.; Contreras, H.R. Proapoptotic effect of endocannabinoids in prostate cancer cells. Oncol. Rep. 2015, 33, 1599–1608. [Google Scholar] [CrossRef] [Green Version]
- Melck, D.; de Petrocellis, L.; Orlando, P.; Bisogno, T.; Laezza, C.; Bifulco, M.; Di Marzo, V. Suppression of nerve growth factor Trk receptors and prolactin receptors by endocannabinoids leads to inhibition of human breast and prostate cancer cell proliferation. Endocrinology 2000, 141, 118–226. [Google Scholar] [CrossRef]
- Mimeault, M.; Pommery, N.; Wattez, N.; Bailly, C.; Hénichart, J.P. Anti-proliferative and apoptotic effects of anandamide in human prostatic cancer cell lines: Implication of epidermal growth factor receptor down-regulation and ceramide production. Prostate 2003, 56, 1–12. [Google Scholar] [CrossRef]
- Nithipatikom, K.; Endsley, M.P.; Isbell, M.A.; Falck, J.R.; Iwamoto, Y.; Hillard, C.J.; Campbell, W.B. 2-arachidonoylglycerol: A novel inhibitor of androgen-independent prostate cancer cell invasion. Cancer Res. 2004, 64, 8826–8830. [Google Scholar] [CrossRef] [Green Version]
- Sreevalsan, S.; Joseph, S.; Jutooru, I.; Chadalapaka, G.; Safe, S.H. Induction of apoptosis by cannabinoids in prostate and colon cancer cells is phosphatase dependent. Anticancer Res. 2011, 31, 3799–3807. [Google Scholar] [PubMed]
- Endsley, M.P.; Aggarwal, N.; Isbell, M.A.; Wheelock, C.E.; Hammock, B.D.; Falck, J.R.; Campbell, W.B.; Nithipatikom, K. Diverse roles of 2-arachidonoylglycerol in invasion of prostate carcinoma cells: Location, hydrolysis and 12-lipoxygenase metabolism. Int. J. Cancer 2007, 121, 984–991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberto, D.; Klotz, L.H.; Venkateswaran, V. Cannabinoid WIN 55,212-2 induces cell cycle arrest and apoptosis, and inhibits proliferation, migration, invasion, and tumor growth in prostate cancer in a cannabinoid-receptor 2 dependent manner. Prostate 2019, 79, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Giannoni, E.; Bianchini, F.; Masieri, L.; Serni, S.; Torre, E.; Calorini, L.; Chiarugi, P. Reciprocal activation of prostate cancer cells and cancer-associated fibroblasts stimulates epithelial-mesenchymal transition and cancer stemness. Cancer Res. 2010, 70, 6945–6956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiaschi, T.; Marini, A.; Giannoni, E.; Taddei, M.L.; Gandellini, P.; De Donatis, A.; Lanciotti, M.; Serni, S.; Cirri, P.; Chiarugi, P. Reciprocal metabolic reprogramming through lactate shuttle coordinately influences tumor-stroma interplay. Cancer Res. 2012, 72, 5130–5140. [Google Scholar] [CrossRef] [Green Version]
- Giannoni, E.; Taddei, M.L.; Parri, M.; Bianchini, F.; Santosuosso, M.; Grifantini, R.; Fibbi, G.; Mazzanti, B.; Calorini, L.; Chiarugi, P. EphA2-mediated mesenchymal-amoeboid transition induced by endothelial progenitor cells enhances metastatic spread due to cancer-associated fibroblasts. J. Mol. Med. 2013, 91, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Comito, G.; Giannoni, E.; Segura, C.P.; Barcellos-de-Souza, P.; Raspollini, M.R.; Baroni, G.; Lanciotti, M.; Serni, S.; Chiarugi, P. Cancer-associated fibroblasts and M2-polarized macrophages synergize during prostate carcinoma progression. Oncogene 2014, 33, 2423–2431. [Google Scholar] [CrossRef] [Green Version]
- Giannoni, E.; Taddei, M.L.; Morandi, A.; Comito, G.; Calvani, M.; Bianchini, F.; Richichi, B.; Raugei, G.; Wong, N.; Tang, D.; et al. Targeting stromal-induced pyruvate kinase M2 nuclear translocation impairs oxphos and prostate cancer metastatic spread. Oncotarget 2015, 6, 24061–24074. [Google Scholar] [CrossRef] [Green Version]
- Ippolito, L.; Morandi, A.; Taddei, M.L.; Parri, M.; Comito, G.; Iscaro, A.; Raspollini, M.R.; Magherini, F.; Rapizzi, E.; Masquelier, J.; et al. Cancer-associated fibroblasts promote prostate cancer malignancy via metabolic rewiring and mitochondrial transfer. Oncogene 2019, 38, 5339–5355. [Google Scholar] [CrossRef]
- Comito, G.; Iscaro, A.; Bacci, M.; Morandi, A.; Ippolito, L.; Parri, M.; Montagnani, I.; Raspollini, M.R.; Serni, S.; Simeoni, L.; et al. Lactate modulates CD4+ T-cell polarization and induces an immunosuppressive environment, which sustains prostate carcinoma progression via TLR8/miR21 axis. Oncogene 2019, 38, 3681–3695. [Google Scholar] [CrossRef]
- Bifulco, M.; Di Marzo, V. Targeting the endocannabinoid system in cancer therapy: A call for further research. Nat. Med. 2002, 8, 547–550. [Google Scholar] [CrossRef] [PubMed]
- Ramer, R.; Schwarz, R.; Hinz, B. Modulation of the Endocannabinoid System as a Potential Anticancer Strategy. Front. Pharmacol. 2019, 10, 430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuxhorn, J.A.; Ayala, G.E.; Smith, M.J.; Smith, V.C.; Dang, T.D.; Rowley, D.R. Reactive stroma in human prostate cancer: Induction of myofibroblast phenotype and extracellular matrix remodeling. Clin. Cancer Res. 2002, 8, 2912–2923. [Google Scholar]
- Barron, D.A.; Rowley, D.R. The reactive stroma microenvironment and prostate cancer progression. Endocr. Relat. Cancer 2012, 19, R187–R204. [Google Scholar] [CrossRef] [Green Version]
- Doldi, V.; Callari, M.; Giannoni, E.; D’Aiuto, F.; Maffezzini, M.; Valdagni, R.; Chiarugi, P.; Gandellini, P.; Zaffaroni, N. Integrated gene and miRNA expression analysis of prostate cancer associated fibroblasts supports a prominent role for interleukin-6 in fibroblast activation. Oncotarget 2015, 6, 31441–31460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jean-Gilles, L.; Braitch, M.; Latif, M.L.; Aram, J.; Fahey, A.J.; Edwards, L.J.; Robins, R.A.; Tanasescu, R.; Tighe, P.J.; Gran, B.; et al. Effects of pro-inflammatory cytokines on cannabinoid CB1 and CB2 receptors in immune cells. ACTA Physiol. 2015, 214, 63–74. [Google Scholar] [CrossRef]
- Pietrovito, L.; Leo, A.; Gori, V.; Lulli, M.; Parri, M.; Becherucci, V.; Piccini, L.; Bambi, F.; Taddei, M.L.; Chiarugi, P. Bone marrow-derived mesenchymal stem cells promote invasiveness and transendothelial migration of osteosarcoma cells via a mesenchymal to amoeboid transition. Mol. Oncol. 2018, 12, 659–676. [Google Scholar] [CrossRef] [PubMed]
- Peres, F.F.; Lima, A.C.; Hallak, J.E.C.; Crippa, J.A.; Silva, R.H.; Abílio, V.C. Cannabidiol as promising strategy to treat and prevent movement disorders? Front. Pharmacol. 2018, 9, 482. [Google Scholar] [CrossRef] [Green Version]
- Giannoni, E.; Bianchini, F.; Calorini, L.; Chiarugi, P. Cancer associated fibroblasts exploit reactive oxygen species through a proinflammatory signature leading to epithelial mesenchymal transition and stemness. Antioxid. Redox. Signal. 2011, 14, 2361–2371. [Google Scholar] [CrossRef]
- Pistore, C.; Giannoni, E.; Colangelo, T.; Rizzo, F.; Magnani, E.; Muccillo, L.; Giurato, G.; Mancini, M.; Rizzo, S.; Riccardi, M.; et al. DNA methylation variations are required for epithelial-to-mesenchymal transition induced by cancer-associated fibroblasts in prostate cancer cells. Oncogene 2017, 36, 5551–5566. [Google Scholar] [CrossRef]
- Schwarz, R.; Ramer, R.; Hinz, B. Targeting the endocannabinoid system as a potential anticancer approach. Drug Metab. Rev. 2018, 50, 26–53. [Google Scholar] [CrossRef] [PubMed]
- Maccarrone, M.; Bab, I.; Bíró, T.; Cabral, G.A.; Dey, S.K.; Di Marzo, V.; Konje, J.C.; Kunos, G.; Mechoulam, R.; Pacher, P.; et al. Endocannabinoid signaling at the periphery: 50 years after THC. Trends Pharmacol. Sci. 2015, 36, 277–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nithipatikom, K.; Isbell, M.A.; Endsley, M.P.; Woodliffm, J.E.; Campbell, W.B. Anti-proliferative effect of a putative endocannabinoid, 2-arachidonylglyceryl ether in prostate carcinoma cells. Prost. Other. Lipid Mediat. 2011, 94, 34–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carracedo, A.; Lorente, M.; Egia, A.; Blázquez, C.; García, S.; Giroux, V.; Malicet, C.; Villuendas, R.; Gironella, M.; González-Feria, L.; et al. The stress-regulated protein p8 mediates cannabinoid-induced apoptosis of tumor cells. Cancer Cell 2006, 9, 301–312. [Google Scholar] [CrossRef] [Green Version]
- Galve-Roperh, I.; Sánchez, C.; Cortés, M.L.; Gómez del Pulgar, T.; Izquierdo, M.; Guzmán, M. Anti-tumoral action of cannabinoids: Involvement of sustained ceramide accumulation and extracellular signal-regulated kinase activation. Nat. Med. 2000, 6, 313–319. [Google Scholar] [CrossRef]
- Ellert-Miklaszewska, A.; Ciechomska, I.; Kaminska, B. Cannabinoid signaling in glioma cells. Adv. Exp. Med. Biol. 2013, 986, 209–220. [Google Scholar]
- Galve-Roperh, I.; Aguado, T.; Palazuelos, J.; Guzmán, M. Mechanisms of control of neuron survival by the endocannabinoid system. Curr. Pharm. Des. 2008, 14, 2279–2288. [Google Scholar] [CrossRef] [Green Version]
- Salazar, M.; Carracedo, A.; Salanueva, I.J.; Hernández-Tiedra, S.; Lorente, M.; Egia, A.; Vázquez, P.; Blázquez, C.; Torres, S.; García, S.; et al. Cannabinoid action induces autophagy-mediated cell death through stimulation of ER stress in human glioma cells. J. Clin. Invest. 2009, 119, 1359–1372. [Google Scholar] [CrossRef] [Green Version]
- Bifulco, M.; Laezza, C.; Portella, G.; Vitale, M.; Orlando, P.; De Petrocellis, L.; Di Marzo, V. Control by the endogenous cannabinoid system of ras oncogene-dependent tumor growth. FASEB J. 2001, 15, 2745–2747. [Google Scholar] [CrossRef] [Green Version]
- Casanova, M.L.; Blázquez, C.; Martínez-Palacio, J.; Villanueva, C.; Fernández-Aceñero, M.J.; Huffman, J.W.; Jorcano, J.L.; Guzmán, M. Inhibition of skin tumor growth and angiogenesis in vivo by activation of cannabinoid receptors. J. Clin. Invest. 2003, 111, 43–50. [Google Scholar] [CrossRef] [Green Version]
- De Petrocellis, L.; Melck, D.; Palmisano, A.; Bisogno, T.; Laezza, C.; Bifulco, M.; Di Marzo, V. The endogenous cannabinoid anandamide inhibits human breast cancer cell proliferation. Proc. Natl. Acad. Sci. USA 1998, 95, 8375–8380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKallip, R.J.; Nagarkatti, M.; Nagarkatti, P.S. Delta-9-tetrahydrocannabinol enhances breast cancer growth and metastasis by suppression of the antitumor immune response. J. Immunol. 2005, 174, 3281–3289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sánchez, M.G.; Sánchez, A.M.; Ruiz-Llorente, L.; Díaz-Laviada, I. Enhancement of androgen receptor expression induced by (R)-methanandamide in prostate LNCaP cells. FEBS Lett. 2003, 555, 561–566. [Google Scholar]
- Hart, S.; Fischer, O.M.; Ullrich, A. Cannabinoids induce cancer cell proliferation via tumor necrosis factor alpha-converting enzyme (TACE/ADAM17)-mediated transactivation of the epidermal growth factor receptor. Cancer Res. 2004, 64, 1943–1950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bifulco, M.; Laezza, C.; Pisanti, S.; Gazzerro, P. Cannabinoids and cancer: Pros and cons of an antitumour strategy. Br. J. Pharmacol. 2006, 148, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Chiarugi, P. Cancer-associated fibroblasts and macrophages: Friendly conspirators for malignancy. Oncoimmunology 2013, 2, e25563. [Google Scholar] [CrossRef] [Green Version]
- Ramer, R.; Heinemann, K.; Merkord, J.; Rohde, H.; Salamon, A.; Linnebacher, M.; Hinz, B. COX-2 and PPAR-γ confer cannabidiol-induced apoptosis of human lung cancer cells. Mol. Cancer Ther. 2013, 12, 69–82. [Google Scholar] [CrossRef] [Green Version]
- Silva, A.G.; Lopes, C.F.B.; Carvalho Júnior, C.G.; Thomé, R.G.; Dos Santos, H.B.; Reis, R.; Ribeiro, R.I.M.A. WIN55,212-2 induces caspase-independent apoptosis on human glioblastoma cells by regulating HSP70, p53 and Cathepsin, D. Toxicol. Vitro 2019, 57, 233–243. [Google Scholar] [CrossRef]
- Hiebel, C.; Kromm, T.; Stark, M.; Behl, C. Cannabinoid receptor 1 modulates the autophagic flux independent of mTOR- and BECLIN1-complex. J. Neurochem. 2014, 131, 484–497. [Google Scholar] [CrossRef] [Green Version]
- Emery, S.M.; Alotaibi, M.R.; Tao, Q.; Selley, D.E.; Lichtman, A.H.; Gewirtz, D.A. Combined antiproliferative effects of the aminoalkylindole WIN55,212-2 and radiation in breast cancer cells. J. Pharmacol. Exp. Ther. 2014, 348, 293–302. [Google Scholar] [CrossRef]
- Caffarel, M.M.; Andradas, C.; Mira, E.; Pérez-Gómez, E.; Cerutti, C.; Moreno-Bueno, G.; Flores, J.M.; García-Real, I.; Palacios, J.; Mañes, S.; et al. Cannabinoids reduce ErbB2-driven breast cancer progression through Akt inhibition. Mol. Cancer 2010, 9, 196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katchan, V.; David, P.; Shoenfeld, Y. Cannabinoids and autoimmune diseases: A systematic review. Autoimmun. Rev. 2016, 15, 513–528. [Google Scholar] [CrossRef] [PubMed]
- Pyszniak, M.; Tabarkiewicz, J.; Łuszczki, J.J. Endocannabinoid system as a regulator of tumor cell malignancy—Biological pathways and clinical significance. Onco. Targets Ther. 2016, 9, 4323–4336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taddei, M.L.; Giannoni, E.; Morandi, A.; Ippolito, L.; Ramazzotti, M.; Callari, M.; Gandellini, P.; Chiarugi, P. Mesenchymal to amoeboid transition is associated with stem-like features of melanoma cells. Cell Commun. Signal. 2014, 12, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pietrovito, L.; Iozzo, M.; Bacci, M.; Giannoni, E.; Chiarugi, P. Treatment with Cannabinoids as a Promising Approach for Impairing Fibroblast Activation and Prostate Cancer Progression. Int. J. Mol. Sci. 2020, 21, 787. https://doi.org/10.3390/ijms21030787
Pietrovito L, Iozzo M, Bacci M, Giannoni E, Chiarugi P. Treatment with Cannabinoids as a Promising Approach for Impairing Fibroblast Activation and Prostate Cancer Progression. International Journal of Molecular Sciences. 2020; 21(3):787. https://doi.org/10.3390/ijms21030787
Chicago/Turabian StylePietrovito, Laura, Marta Iozzo, Marina Bacci, Elisa Giannoni, and Paola Chiarugi. 2020. "Treatment with Cannabinoids as a Promising Approach for Impairing Fibroblast Activation and Prostate Cancer Progression" International Journal of Molecular Sciences 21, no. 3: 787. https://doi.org/10.3390/ijms21030787