
Abstract
The development of hepatocellular carcinoma (HCC) in persons who are persistently infected with hepatitis C virus (HCV) is a growing problem worldwide. Current antiviral therapies are not effective in many patients with chronic hepatitis C, and a greater understanding of the factors leading to progression of HCC will be necessary to design novel approaches to prevention of HCV-associated HCC. The lack of a small animal model of chronic HCV infection has hampered understanding of these factors. As HCV is an RNA virus with little potential for integration of its genetic material into the host genome, the mechanisms underlying HCV promotion of cancer are likely to differ from other models of viral carcinogenesis. In patients persistently infected with HCV, chronic inflammation resulting from immune responses against infected hepatocytes is associated with progressive fibrosis and cirrhosis. Cirrhosis is an important risk factor for HCC independent of HCV infection, and a majority of HCV-associated HCC arises in the setting of cirrhosis. However, a significant minority arises in the absence of cirrhosis, indicating that cirrhosis is not a prerequisite for cancer. Other lines of evidence suggest that direct, virus-specific mechanisms may be involved. Transgenic mice expressing HCV proteins develop cancer in the absence of inflammation or immune recognition of the transgene. In vitro studies have revealed multiple interactions of HCV-encoded proteins with cell cycle regulators and tumor suppressor proteins, raising the possibility that HCV can disrupt control of cellular proliferation, or impair the cell's response to DNA damage. A combination of virus-specific, host genetic, environmental and immune-related factors are likely to determine the progression to HCC in patients who are chronically infected with HCV. Here, we summarize current knowledge of the virus-specific mechanisms that may contribute to HCV-associated HCC.
Similar content being viewed by others

Introduction
Liver cancer is the third leading cause of cancer deaths worldwide. Although incidence rates are stable or declining for many types of cancers, they have increased substantially for hepatocellular carcinoma (HCC) in recent years in both Japan and the United States. In Japan, the age-adjusted annual death rate due to primary liver cancer rose from ∼10/100 000 persons in 1975 to a peak of 27.5 in 2002, attributable in large part to increases in the prevalence of hepatitis C virus (HCV) infection that occurred decades earlier, in the aftermath of World War II (Tanaka et al., 2002; Yoshizawa, 2002; Umemura et al., 2009). HCV infection now accounts for ∼70% of cases of liver cancer in Japan. The incidence of liver cancer has also undergone a remarkable increase in the United States, where extensive spread of HCV occurred ∼30 years later (Tanaka et al., 2002). Between 2001–2006, the incidence of microscopically-confirmed HCC increased at an average annual rate of 9.1% among persons aged 50–59 years (Centers for Disease Control and Prevention (CDC), 2010). Increased rates of HCC are particularly striking among African American and Hispanic males, reflecting racial disparities in the prevalence of chronic HCV infection, now the leading cause of HCC within the United States (Armstrong et al., 2006; Altekruse et al., 2009). Although there are good statistics for the United States and Japan, it is difficult to estimate the global burden of HCV-associated liver cancer. In one analysis, HCV infection accounted for 155 000 liver cancer deaths in 2002 (Perz and Alter, 2006), but such a figure is likely to significantly underestimate the numbers of HCV-associated HCC deaths today.
Despite advances in therapy for HCC, 1-year survival rates remain less than 50% in the United States (Altekruse et al., 2009). Even in Japan, where early diagnosis and potentially curative interventions are common, recurrence is frequent and the long-term prognosis is poor (Masuzaki et al., 2008). Antiviral therapies may prevent progression to HCC in HCV-infected persons, but current interferon-based standard-of-care therapies eliminate the virus in only half of those treated (McHutchison and Fried, 2003). Moreover, the preventive benefits of interferon therapy are limited in the absence of virus eradication (Lok et al., 2009). Newer, direct-acting antiviral agents are on the horizon and will significantly improve treatment outcomes, but rapid selection of resistant viruses will mandate continued reliance on interferon as the foundation of therapy, at least for the near term future (Shimakami et al., 2009). Importantly, only a small proportion of infected persons are likely to have access to these therapies in most countries. The population of persons within the United States who have been infected with HCV for more than 2 decades and who are at highest risk of HCC is thus likely to continue to grow over the next 20 years, fueling continued increases in HCC incidence (Davis et al., 2010). Worldwide, 130–170 million people worldwide are chronically infected with HCV (Lavanchy, 2009).
Direct vs indirect carcinogenic mechanisms
Most persons who are infected with HCV fail to clear the virus, and become persistently infected and thus at long-term risk for progressive hepatic fibrosis, cirrhosis, or HCC (reviewed in Lemon et al., 2007). HCV is a positive-strand RNA virus that replicates in the cytoplasm and has little potential for integration of its genome into cellular DNA. The presence of cirrhosis and chronic hepatic inflammation with associated oxidative stress and accompanying potential for cellular DNA damage are unquestionably important contributing causes to HCV-associated HCC (Nakamoto et al., 1998; Okuda et al., 2002; Bartsch and Nair, 2004). However, there are several lines of evidence supporting a direct role for HCV in cancer promotion. First, although direct comparisons are limited, the incidence of HCC appears to be greater in HCV-associated cirrhosis (7–13% over 5 years) (Degos et al., 2000; Lok et al., 2009) than in cirrhosis resulting from autoimmune hepatitis (Yeoman et al., 2008; Teufel et al., 2009). Moreover, there is an increasing recognition that HCV-associated HCC may arise in the absence of cirrhosis, albeit at a much lower rate than in patients with cirrhosis (Bralet et al., 2000; Lok et al., 2009; Yeh et al., 2010). The occurrence of HCC in some lineages of transgenic mice also provides compelling evidence that HCV protein expression may be directly oncogenic (Moriya et al., 1998; Lerat et al., 2002).
An important question is whether cancer arises in an infected hepatocyte, or in uninfected bystander hepatocytes that are present in far greater numbers in the HCV-infected liver. Estimates on the basis of observed genome copy numbers in the livers of HCV-infected chimpanzees suggest that <30% of hepatocytes contain replicating virus (Bigger et al., 2004). This is consistent with the 2–20% of cells in which we observed HCV antigen expression by two-photon microscopy of frozen sections of non-malignant liver tissue (Liang et al., 2009). The proportion of hepatocytes expressing the Ki67 proliferation marker is increased in advanced hepatitis C (Farinati et al., 1996; Koskinas et al., 2005), and a case-control study suggests that patients with a high proportion of Ki67-positive hepatocytes are at increased risk for HCC (Dutta et al., 1998). However, it is not known whether the proliferating hepatocytes are infected with HCV, or alternatively, whether they are uninfected cells proliferating in response to the loss of infected cells because of virus-induced apoptosis or immunologically-mediated cell death. Such questions are difficult to answer because of limitations in experimental systems and the technical difficulties inherent in detecting the low abundance of HCV antigen expressed by infected cells in human liver (Liang et al., 2009).
A related question is whether the malignant cells in established HCC are productively infected with HCV. The presence of viral RNA in HCC tissue is not a reliable marker of infection in cancer cells, because potentially high numbers of virions circulate in the blood and thus may be expected to be present in any vascular tissue. Importantly, most established human hepatoma cell lines do not support the replication of HCV in cell culture. Such a lack of permissiveness for HCV replication does not exclude a direct role for the virus in the events leading to cancer, however, as the capacity of transformed hepatocytes to support viral replication could be lost because of reduced expression of cellular co-factors required for replication of HCV. As explained below, microRNA 122 (miR-122) is one such co-factor, and it is often expressed at low levels in HCC. These observations do, however, indicate that the malignant phenotype is unlikely to be driven directly by continued viral protein expression, as in the case of papillomaviruses. Rather, they point to a cancer-promoting effect of HCV infection.
Although progression to HCC is variable among patients with chronic hepatitis C, suggesting that cancer arises because of a complex interplay between host, viral and environmental factors, a wealth of studies suggest that epigenetic changes in HCV-infected hepatocytes may underlie the development of HCC. Here, we summarize the evidence that HCV infection leads to critical stresses on hepatocellular homeostasis and altered cellular response pathways that promote the development of cancer above and beyond the non-specific pro-carcinogenic effects of cirrhosis and general hepatic inflammation. Such a hypothesis suggests that cancer arises in infected cells and not in uninfected bystander hepatocytes, and is consistent with the exceptionally high rate of HCC observed in HCV-related cirrhosis.
The HCV lifecycle
The HCV genome consists of a single-stranded positive-sense RNA ∼9.6 kb in length with untranslated RNA segments (UTRs) at both ends and a single large open reading frame encoding a 327 kDa polyprotein that is processed into 10 mature viral proteins (Figure 1) (for a review, see Lemon et al., 2007). Most if not all of these viral proteins are multifunctional. Indeed, the membrane-bound replicase complexes in which viral RNA is synthesized contain <5% of the total complement of non-structural proteins expressed in cells (Quinkert et al., 2005). In addition to their basic functions in supporting viral genome amplification and the production of new virions, viral proteins interact with host proteins in ways that facilitate genome amplification, antagonize host immune responses, or otherwise alter the cellular environment to favor virus replication. Replication of the virus is also dependent upon at least one cellular microRNA, miR-122, as discussed in greater detail below. Many specific details are lacking, but viral RNA replication appears to be an exclusively cytoplasmic process. This suggests that the mechanisms by which HCV infection leads to cancer are likely to differ substantially from other models of viral carcinogenesis. Nonetheless, HCV replication is associated with altered abundance or localization of some typical nuclear proteins, including the retinoblastoma protein (Rb) and DDX5 (p68) (Goh et al., 2004; Munakata et al., 2007).

Hepatitis C virus (HCV) genome structure. (top) Organization of the positive-sense RNA genome of HCV. The 5′ and 3′ untranslated RNA segments (UTRs) contain cis-acting elements required for viral protein expression and RNA synthesis, including an internal ribosome entry site (IRES) in the 5′UTR that directs cap-independent translation of the viral polyprotein. The polyprotein (large box) is processed by both host- and virus-encoded proteases to produce the individual proteins required for viral genome replication, virus assembly and production of infectious progeny virus (reviewed in Moradpour et al., 2007). The structural proteins (blue) that form the HCV virion include core (nucleocapsid), E1 and E2 (envelope glycoproteins). Non-structural proteins of HCV serve a variety of functions including RNA genome replication, virus assembly and maturation: those from NS3-NS5B (pink) are required for genome replication. NS3 has distinct proteinase and helicase domains, whereas NS4A is cofactor that intercalates with NS3 and is required for full expression of NS3 protease activity. NS4B is involved in the formation of the membranous web: a cytoplasmic structure associated with the viral RNA replicase. NS5A is an essential co-factor for virus replication and assembly, whereas NS5B is the viral RNA-dependent RNA polymerase. The p7 and NS2 (green) are additional non-structural proteins that are not required for viral RNA synthesis, but contribute to virion assembly and egress. (bottom) Organization of a dicistronic, selectable HCV ‘replicon’. The 5′ and 3′UTRs are as in the viral genome. A truncated core-protein sequence is fused to the neomycin phosphotransferase gene, followed downstream by a heterologous picornaviral IRES driving translation of the non-structural proteins. This RNA is capable of autonomous amplification in permissive cells, but does not generate infectious virus.
Approaches to the study of HCV pathogenesis
Model systems
HCV is an hepatotropic virus and it replicates primarily if not exclusively within hepatocytes. Some studies have suggested that there may be low level replication in lymphocytes, and this has been well documented in cell culture (Shimizu et al., 1998). However, it is controversial whether this occurs in infected persons. There appear to be multiple blocks to replication in peripheral blood mononuclear cells (Marukian et al., 2008). Other than humans, the only animal species that is fully permissive for HCV infection is the chimpanzee, Pan troglodytes. HCV infection of the chimpanzee recapitulates most if not all features of hepatitis C in humans. Although relatively small numbers of these primates have been infected with HCV since the discovery of the virus, at least one chimpanzee has developed HCC in the setting of chronic infection (Lanford et al., 2011). This animal had multiple tumors, but no cirrhosis and only minimal portal fibrosis. The chimpanzee represents the only animal species in which viral pathogenesis can be accurately modeled, but its availability at present for such studies is extremely limited.
There are no robust small animal models of HCV infection. A chimeric mouse model has been developed in which human hepatocytes are transplanted into SCID/Alb-uPA mice (Mercer et al., 2001). In this system, liver-specific expression of the urokinase plasminogen activator is toxic to the murine hepatocytes, allowing transplanted human hepatocytes to repopulate the mouse liver. The human hepatocytes resident in the resulting chimeric liver can be infected with HCV and this system has proven useful in studying certain aspects of HCV replication. However, as the mice are immunodeficient, it is not possible to study most host immune responses to the virus in these mice. In addition, this system is technically challenging, the mice are not long-lived and they do not develop cancer. As a surrogate for a small animal model of infection, a considerable number of transgenic mouse lines have been developed that express various HCV proteins. These are discussed separately below.
Given the absence of a small animal that is permissive for replication of the virus, it is not surprising that the influence of HCV protein expression on cellular homeostasis has been studied mainly using in vitro systems. These include transient expression studies, the use of cell lines containing HCV RNA replicons (that is, cells that contain autonomously replicating, selectable sub-genomic or genome-length viral RNA, but do not produce infectious virus) (Lohmann et al., 1999) (Figure 1) and cell culture-infectious virus systems (Lindenbach and Rice, 2005; Wakita et al., 2005; Yi et al., 2006). Aside from the fact that all of these systems are in vitro, each has its limitations. HCV proteins may be expressed by replicons at high levels that are not physiologically relevant, and most replicon cell lines have been generated from Huh-7 cells, which express a mutant p53 (Bressac et al., 1990). Moreover, the techniques used to select such cell lines are likely to bias cell growth and survival properties. More recently developed systems allowing the propagation of cell culture-produced HCV (HCVcc) provide the most authentic in vitro system for studying the effects of HCV on cellular homeostasis, but only two viral strains replicate well enough to study. Furthermore, most cell lines that are known to be permissive for HCVcc replication are derived from Huh-7 hepatoma cells. Despite these limitations, the cell culture-infectious virus systems have allowed significant advances in understanding the role of HCV proteins in viral replication and pathogenesis.
HCV transgenic mice and HCC
Transgenic mouse models of HCV pathogenesis typically use liver-specific promoters to drive expression of either individual or multiple HCV proteins. Although they lack viral RNA replication, these transgenic mice provide insights concerning the potential contribution of individual HCV proteins to liver disease and carcinogenesis. Table 1 summarizes HCV transgenic mouse lineages for which the phenotype has been determined up until at least 12 months of age. Of these, only some lineages appear to be at risk for HCC. The propensity of HCV transgenic mice to develop cancer varies with the mouse genetic background, with C57BL/6 mice appearing to be most susceptible (Klopstock et al., 2009). In contrast, HCC has not been described in FVB mice expressing similar HCV transgenes. The promoter used to drive expression of the HCV transgene, the nature of the transgene, as well as the abundance of the HCV protein expressed also are likely to contribute to the presence or absence of a cancer phenotype.
Most transgenic mouse lines express one or more of the HCV structural proteins. Two transgenic lineages with high-level, liver-specific expression of the core protein developed hepatic steatosis with progression to adenomas and HCC (Moriya et al., 1998). No hepatic inflammation was observed in these mice, suggesting a direct role for the core protein in carcinogenesis. However, the level of transgene expression was very high in these mice, and probably not reflective of the expression levels in most infected human livers. A separate study demonstrated the development of cancer in mice transgenic for both core as well as the two envelope proteins, E1 and E2 (Naas et al., 2005). Although the promoter driving transgene expression in these mice was not liver-specific, transgene mRNA levels were highest in the liver. Steatosis was also observed in the livers of these mice, with the degree of steatosis increasing with age. In older mice, hepatocellular adenomas and carcinomas were observed, as well as tumors of lymphoid origin (Naas et al., 2005).
In contrast to these mice, the FL-N/35 lineage expresses the entire HCV polyprotein under the control of the albumin promoter (Lerat et al., 2002). These mice have liver-specific expression of the entire HCV polyprotein at very low abundance and thus may represent a more physiologically relevant model of HCV-associated HCC. HCV proteins are detectable in these mice only using very sensitive immunohistochemical methods (Keasler et al., 2006), and the expression level is likely to be closer to what it is in infected human tissues. Steatosis was observed in these animals, but no inflammation, and liver cancer developed in older male animals at a relatively low, but statistically significant rate (Lerat et al., 2002). Interestingly, a companion transgenic mouse line, which expressed a greater abundance of only the structural proteins, core, E1, E2 and p7, did not have a significantly increased incidence of cancer compared with non-transgenic animals, suggesting a possible role for the non-structural proteins in carcinogenesis (Lerat et al., 2002). However, it is not possible to exclude differences in the transgene integration sites underlying this difference. There are few other reports of transgenic mice that express the non-structural proteins of HCV, but mice transgenic for the NS5A protein also develop significant steatosis and HCC (Wang et al., 2009a).
In addition to possible direct carcinogenic effects of HCV protein expression, studies with transgenic mice have demonstrated that HCV protein expression may enhance susceptibility to various non-chemical carcinogens. For example, the incidence of liver cancer in FL-N/35 polyprotein-expressing mice, is increased by iron overloading (Furutani et al., 2006), co-expression of the hepatitis B virus X protein (Keasler et al., 2006), or intestinal colonization with Helicobacter hepaticus (Fox et al., 2010). Alcohol promoted the development of HCC in NS5A transgenic mice that normally do not develop cancer (Majumder et al., 2003; Machida et al., 2009b). The synergistic effects of alcohol and NS5A expression on the rate of tumor formation were related to induction of TLR4 expression and a downstream mediator, Nanog. These findings are particularly interesting because alcohol ingestion increases the risk of HCC in patients with chronic hepatitis C. In another series of experiments, a transgenic mouse line expressing HCV core under control of the serum amyloid P component promoter developed cancer only after repeated carbon tetrachloride-induced liver injury (Kato et al., 2003). In this case, tumor development was dependent upon both repeated liver injury and core protein expression. In contrast, transgenic expression of the viral structural proteins resulted in an increase in tumor size, but not frequency, following exposure to diethylnitrosamine (Kamegaya et al., 2005).
In summary, cancer phenotypes in HCV transgenic mice suggest roles for both structural and non-structural HCV proteins in hepatocellular carcinogenesis. The lack of detectable inflammation in transgenic mice that develop cancer supports a direct role for HCV proteins in carcinogenesis, whereas other evidence suggests that HCV protein expression may have broader, co-carcinogenic effects. However the variation evident in the phenotypes reported for HCV transgenic mice is disconcerting. No single viral protein has been shown to consistently cause liver cancer when expressed at a low abundance comparable with that present in most patients with HCV-related liver disease.
General consequences of HCV infection
Inflammation, fibrosis and cirrhosis
Persistent HCV infections are typically associated with inflammatory and wound healing responses within the liver. Activation of the NF-κB pathway has a central role in these inflammatory responses and may be important in carcinogenesis (reviewed in Sun and Karin, 2008). Chronic inflammation related to HCV infection drives fibrogenesis, with increased deposition of extracellular matrix proteins leading to fibrotic scarring and ultimately cirrhosis. Activation of hepatic stellate cells is known to be important in this process and may be cytokine-driven, but the specific mechanisms by which HCV infection induces hepatic stellate cell activation are not well defined. Oxidative stress and apoptosis of infected hepatocytes are likely to be contributory factors (Brenner, 2009). Other studies suggest a pro-fibrotic role for specific viral proteins (Bataller et al., 2004; Mazzocca et al., 2005) while yet others point to immune responses as an important trigger (Baroni et al., 1999).
The sensing of HCV infection by pathogen recognition receptors of the innate immune system likely contributes to these processes. Viral double-stranded RNA replication intermediates produced in infected hepatocytes are sensed by retinoic acid-inducible gene I and Toll-like receptor 3, leading to activation of interferon regulatory factor 3 and NF-κB (Saito et al., 2008; Wang et al., 2009b). Although HCV has evolved redundant mechanisms to counter these responses (reviewed in Lemon, 2010), many patients as well as chimpanzees with persistent HCV infection show marked transcriptional upregulation of interferon-stimulated genes. This may reflect the sensing of infected hepatocytes by plasmacytoid dendritic cells through a TLR7-dependent pathway (Takahashi et al., 2010). In addition to these sensing mechanisms, other studies suggest that HCV-induced endoplasmic reticulum stress (Waris et al., 2002) or simply the presence of HCV-encoded proteins may activate NF-κB signaling and the expression of pro-inflammatory cytokines (Waris et al., 2003; Dolganiuc et al., 2004; Sato et al., 2006). Core and NS3 stimulate IL-1 receptor-associated kinase activity in multiple cell types, along with phosphorylation of p38 and activation of extracellular regulated kinase and c-jun N-terminal kinase, through a TLR2-dependent pathway (Dolganiuc et al., 2004).
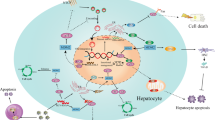
We have recently demonstrated that there is a patchy distribution of HCV infection within the human liver, with infected cells generally appearing in clusters consistent with cell-to-cell spread of virus (Liang et al., 2009) (Figure 2). Such cells do not always appear to be in close proximity to inflammatory cells, and interferons and other soluble mediators may largely control spread through the liver. Nonetheless, virus-specific CD8+ cytotoxic T-cells are present, and have a key role in controlling the infection (reviewed in Bowen and Walker, 2005). Cytotoxic T cells and virus-induced inflammatory responses are likely to result in repeated cycles of hepatocyte destruction and regeneration. This pattern of chronic inflammation and increased hepatocellular proliferation provides an environment that is highly conducive to the development of cancer, and is likely to be common to the development of HCC because of many other causes. The link between chronic inflammation and HCC is well established from transgenic models of hepatitis B (Nakamoto et al., 1998), and it seems certain that this link extends to HCV-associated cancer as well. However, several complementary lines of evidence suggest that HCV also has a direct role in development of HCC.

HCV core antigen visualized in frozen liver tissue from a 50-year-old female with genotype 3 HCV infection, cirrhosis and a serum viral RNA load of 3.3 × 105 IU/ml. Tissue was collected at the time of resection of hepatocellular carcinoma (HCC). Antigen was visualized by multiphoton microscopy following tissue labeling with a monoclonal antibody to core conjugated to a fluorescent quantum dot probe (Liang et al., 2009).
Apoptosis
HCV infection may influence pathways that regulate programmed cell death. Numerous overexpression studies have suggested both pro- and anti-apoptotic functions of individual HCV proteins and have generated a large body of sometimes conflicting data. For example, some reports purport that core protein expression promotes apoptosis (Zhu et al., 1998; Honda et al., 2000; Chou et al., 2005), whereas others suggest an anti-apoptotic effect of core expression (Ray et al., 1998; Lu et al., 1999; Sacco et al., 2003). In addition, E2 (Lee et al., 2005), NS2 (Erdtmann et al., 2003), NS3 (Tanaka et al., 2006) and NS5A (Gale et al., 1999; Lan et al., 2002) have all been reported to exert an anti-apoptotic effect when expressed in cultured cells, whereas other studies suggest that NS3 (Prikhod’ko et al., 2004) and NS4A (Nomura-Takigawa et al., 2006) may promote apoptosis. Most of these studies have evaluated the effects of transient expression of individual HCV proteins at levels that may be considerably higher than in infected hepatocytes in vivo. Core is a highly basic protein that has been reported to have numerous pleiotropic effects when transiently overexpressed. Moreover, the expression of individual HCV proteins in the absence of the remainder of the polyprotein may lead to different localization or post-translational modifications than expressed as a component of the viral polyprotein. Consequently, the biological significance of these findings is not clear.
On the other hand, apoptosis has been demonstrated in response to HCV infection in more physiologically relevant systems that have become available in recent years. For example, recent reports have been consistent in demonstrating apoptosis in Huh-7 cells infected with HCVcc (Deng et al., 2008; Mateu et al., 2008; Walters et al., 2009). Furthermore, in the chimeric SCID/Alb-uPA mouse model, apoptosis was observed in the transplanted human hepatocytes following intrahepatic injection with replication-competent HCV RNA, but not with a replication-defective mutant (Joyce et al., 2009). Although HCV may replicate to a typically high levels both in Huh7 cells as well as human hepatocytes within chimeric livers of severely immunocompromised SCID/Alb-uPA mice, these studies suggest that HCV infection may be inherently pro-apoptotic.
Steatosis
Hepatic steatosis, characterized by the intracellular accumulation of lipids in hepatocytes, is frequently found in patients with chronic HCV infection (for a review, see Cross et al., 2010). Metabolic factors, such as high body mass index and diabetes, influence the development of steatosis in chronic hepatitis C. However, there is a particularly strong association between steatosis and infection with genotype 3 HCV, indicating that steatosis must result, at least in part, from effects on the cell that are virus-specific. Consistent with this, transgenic mice expressing genotype 1 HCV proteins, particularly core, develop steatosis (Moriya et al., 1997; Lerat et al., 2002). Changes in the expression of lipid metabolism genes also have been observed in chimeric SCID/Alb-uPA mice infected with genotype 1 virus (Joyce et al., 2009). Whether directly virus-induced or metabolic in origin, steatosis may contribute to liver cancer through increased oxidative stress or by promoting fibrosis.
Oxidative stress
Both microarray and proteomics studies have demonstrated increased expression of oxidative stress response genes in HCV-associated fibrosis and cirrhosis (Shackel et al., 2002; Diamond et al., 2007). Although no single causative mechanism has been unequivocally identified, increased oxidative stress has the potential to cause DNA damage, potentially leading to the accumulation of mutations, and may also activate hepatic stellate cell thereby promoting fibrosis (Brenner, 2009). Furthermore, chronic oxidative stress may lead to activation of cellular signaling pathways that can contribute to cellular transformation (Waris et al., 2005).
Oxidative stress has been detected in several models of HCV pathogenesis and is generated, at least in part, by the inflammatory response to chronic infection. However, there may also be a direct effect of HCV proteins, particularly core, on intracellular levels of reactive oxygen species. Oxidative stress was observed in cultured cells following the conditional expression of core protein under control of a tetracycline-regulated promoter (Okuda et al., 2002), and this led to increased expression of cellular antioxidant genes (Li et al., 2002). Transgenic mice expressing the core protein also show an increased abundance of both reactive oxygen species and lipid peroxidation products (Korenaga et al., 2005; Moriya et al., 2001). When expressed in cultured cells, core protein localizes in part to mitochondria, and this may promote increased reactive oxygen species production through inhibition of mitochondrial electron transport (Korenaga et al., 2005). Other studies suggest that overexpression of NS5A may also cause oxidative stress (Gong et al., 2001), but the caveats described above for protein overexpression studies must be considered in interpreting these results.
Epigenetic changes in gene expression accompanying HCV infection
The dependence of HCV upon a number of cellular proteins and even a microRNA for its replication reflects extensive adaptation of the virus to its human host, a process shaped by an extraordinarily high rate of virus production (on the order of 1012 new virus particles produced every day over decades of persistent infection in the typical patient) (Neumann et al., 1998) and a highly error-prone RNA replicase. In addition to disrupting signal transduction pathways involved in immune responses, these accessory functions of HCV proteins appear to include a number of interactions with tumor suppressor proteins. Such interactions are likely to have evolved because they promote a favorable cellular environment for virus replication and thus survival of the virus. However, they may also have coincidental, pathologic consequences for the host.
Retinoblastoma protein
The retinoblastoma tumor suppressor protein (Rb) is a common target of oncoproteins expressed by DNA tumor viruses, including adenovirus (Whyte et al., 1988), simian virus 40 (DeCaprio et al., 1988) and human papillomavirus (Dyson et al., 1989). The downregulation of Rb promotes cell cycle entry, activating cellular DNA synthetic pathways required for replication of these viruses. Surprisingly, Rb is also targeted by HCV, an RNA virus, and strongly, negatively regulated by HCV infection in cultured cells (Munakata et al., 2005, 2007; McGivern et al., 2009). This is due to NS5B, the viral RNA-dependent RNA polymerase, which forms a cytoplasmic complex with Rb (Figure 3) and recruits to it the E6-associated protein (E6AP). This leads to polyubiquitination of Rb and Rb degradation through the proteasome (Munakata et al., 2005, 2007). The end result is activation of E2F-responsive promoters, which would be expected to stimulate entry into the S phase of the cell cycle (Munakata et al., 2005). In a fascinating parallel with DNA viruses, the interaction with Rb is dependent upon an LxCxE motif in NS5B that has homology to pRb-binding domains in DNA virus oncoproteins (Munakata et al., 2005).

Laser-scanning confocal microscopy of FT3-7 (Huh7) cells infected with cell culture-produced HCV (HCVcc) (HJ3-5 virus) leads to reductions in nuclear Rb expression (left panel, arrow) and transient accumulation of Rb in the cytoplasm (arrows, right panel). HCV antigens (red) were labeled with polyclonal human anti-HCV antibodies, whereas Rb was labeled with a murine monoclonal antibody (green) (McGivern et al., 2009). Nuclei were counterstained with DAPI.
These findings are unique among RNA viruses and suggest a novel theoretical framework for the origins of liver cancer. In addition to controlling the G1 to S phase transition in the cell cycle, in part through repressive effects on E2F transcription factors (Chellappan et al., 1991; Takahashi et al., 2000; Classon and Harlow, 2002), Rb also regulates DNA damage responses, the mitotic spindle checkpoint, and apoptosis (Classon and Harlow, 2002; Lentini et al., 2002; Hernando et al., 2004; Khidr and Chen, 2006). Although the virus might benefit from enhanced hepatocellular proliferation (which might favor virus replication based on observations in cell culture), the disruption of Rb-mediated regulatory functions may also restrict DNA damage responses. Such a defect would be particularly important in a liver with ongoing inflammation and oxidative stress. Expression of the core protein has also been suggested recently to uncouple the mitotic spindle checkpoint and induce chromosomal polyploidy in transgenic mice and cultured hepatocytes through defective Rb signaling (Machida et al., 2009a). In this case, Rb expression was thought to be restricted at the transcriptional level.
However, the analogy with the DNA tumor viruses only goes so far. Although the p16-cyclin D-Rb pathway is frequently disrupted in HCC, including tumors associated with HCV (Azechi et al., 2001; Edamoto et al., 2003), this does not result from continued virus replication or NS5B expression. If it has a role, NS5B-induced loss of Rb expression seems likely to be an early event in the development of cancer, occurring while hepatocytes remain sufficiently well differentiated to support virus replication, and before the heterogeneous chromosomal mutations that are found commonly in HCC (Thorgeirsson and Grisham, 2002).
p53 pathway
Overexpression studies suggest that HCV proteins, including core, may deregulate the p53 pathway. Again, however, the available experimental systems are limited and published reports conflicting with respect to the effects on p53 activity. Some results suggest that core activates p53-dependent gene expression (Lu et al., 1999; Otsuka et al., 2000) whereas others show repression (Ray et al., 1998). It is possible that this reflects varied levels of expression, with low levels of core expression activating p53 and high levels resulting in repression of p53 activity (Kao et al., 2004). Other in vitro evidence suggests that NS3 may interact with p53 (Ishido and Hotta, 1998) and repress p53-dependent transcription (Kwun et al., 2001). NS3 expression blocks actinomycin D-induced apoptosis; this activity of NS3 was found to be sequence dependent and correlated with p53 interaction (Deng et al., 2006; Tanaka et al., 2006). Yet a third HCV protein, NS5A, has been reported to interact with p53, resulting in its redistribution to the perinuclear membrane in HepG2 hepatoma cells (Majumder et al., 2001). NS5A also interacts with p53 in the rat hepatoma cell line FAO, and inhibits p53-dependent transcription in HCT116 cells (Qadri et al., 2002). Anchorage-independent growth of NIH3T3 cells is promoted by NS5A (Ghosh et al., 1999). Finally, while ectopic expression of p53 in the p53-null Hep3B cell line induces apoptosis, this effect is blocked by NS5A (Lan et al., 2002).
It is possible that HCV infection may also impact p53 function indirectly. As discussed in greater detail below, there is good evidence that core interacts with the cellular RNA helicase DDX3 (Figure 4), a candidate tumor suppressor protein that regulates activity of the p21 (waf1/cip1) promoter (Chao et al., 2006). In addition, NS5B interacts with DDX5 (p68), another RNA helicase, resulting in its relocalization from the nucleus to the cytoplasm (Goh et al., 2004) (Figure 5). DDX5 is a transcriptional co-activator of p53 (Bates et al., 2005), a function that would be impeded by its relocalization to the cytoplasm. Interactions of HCV proteins with the p53 pathway may have evolved to prevent stress-induced growth arrest or apoptosis, both of which would be counter to survival of the virus. On the other hand, recent studies have revealed surprising roles for p53 and DDX3 in Toll-like receptor 3 and retinoic acid-inducible gene I-mediated induction of interferon synthesis (Dharel et al., 2008; Schroder et al., 2008; Taura et al., 2008; Oshiumi et al., 2010), and it is possible that such interactions might reflect yet another mechanism by which the virus escapes innate immune defenses.

Confocal microscopic image showing aggregation of DDX3 in association with HCV core protein in the cytoplasm of HCVcc-infected cells. Huh7 cells were infected with HJ3-5 virus 11 days previously. Labeling was with rabbit anti-core (green) and murine anti-DDX3 (red) antibodies (gifts of Dr Arvind Patel, Medical Research Council Virology Unit, Glasgow). Nuclear counterstain was with DAPI. Mock, mock infected.

DDX5 is redirected from the nucleus to the cytoplasm in Huh7 cells infected with HCVcc (JFH1 strain). Cells were labeled with murine anti-DDX5 (red) and human polyclonal anti-HCV (green) antibodies. Nuclei were counterstained with DAPI. Mock, mock infected.
Many of the immortalized cell lines used to study the impact of HCV protein expression on p53 have defects in the p53 pathway. Huh-7 cells, which are commonly used for propagation of HCV, overexpress a functionally defective p53 mutant (Bressac et al., 1990). To date, this has precluded a direct analysis of the effects of viral infection on p53 function in cell culture. Similarly, NIH3T3, COS-7 and HeLa cells all express viral oncoproteins that directly interact with the p53 protein. In addition, consideration is that the level of protein overexpression in many of these studies may be much higher than in the HCV-infected liver. Therefore, while these findings may be relevant to the development of HCV-associated HCC, they need to be interpreted with considerable caution.
Wnt/β-catenin
The Wnt pathway is a key signal transduction pathway in animal development. In the canonical Wnt pathway, Wnt ligands bind to their receptor to activate a signal transduction cascade resulting in inhibition of a β-catenin degradation complex. Components of the Wnt pathway are frequently mutated in liver cancer, resulting in β-catenin stabilization. Stabilization of β-catenin allows it to enter the nucleus and interact with proteins that regulate transcription of genes that influence cell survival and proliferation.
NS5A may indirectly regulate the Wnt pathway through its interaction with the p85 regulatory subunit of phosphoinositide 3-kinase, which results in activation of the p110 catalytic subunit of phosphoinositide 3-kinase and initiates signaling that activates the downstream kinase, Akt (Street et al., 2004). This leads in turn to phosphorylation and inactivation of glycogen synthase kinase-3β, a key component of the multiprotein complex that normally targets β-catenin for proteasomal degradation (Street et al., 2004). Thus, NS5A expression results in the stabilization of β-catenin and increased β-catenin-dependent transcription (Park et al., 2009; Street et al., 2005). Increases in β-catenin abundance have been observed in cells containing autonomously replicating HCV replicons, and to some extent in cells infected with HCVcc. More recent data, however, suggest that β-catenin may be activated through a direct interaction with the NS5A protein (Park et al., 2009; Milward et al., 2010). The functional significance of the activation of β-catenin within HCV-infected cells is uncertain. Although dysregulation of Wnt signaling does not appear to be independently capable of causing malignant transformation of hepatocytes, β-catenin abundance is increased in most HCC and may promote tumor growth (reviewed in Whittaker et al., 2010).
ATM and Chk2
The ataxia telangiectasia mutated kinase (ATM) is a tumor suppressor protein that detects double-strand DNA breaks and regulates signal transduction pathways controlling the DNA damage checkpoint. An interaction with the HCV NS3/4A protease results in partial relocalization of ATM from the nucleus to the perinuclear region of the cytoplasm (Ariumi et al., 2008; Lai et al., 2008). NS3/4A may also interact with another DNA damage sensor, checkpoint kinase 2 (Chk2) (Ariumi et al., 2008). Knockdown of either ATM or Chk2 impaired viral RNA replication and reduced virus yields, suggesting that these interactions may have evolved to promote replication. Impaired ATM and Chk2 functions may explain why NS3/4A expression results in abnormal responses to DNA damage following ionizing radiation (Lai et al., 2008).
DDX3
Among its many putative interactions, yeast two-hybrid screens and co-immunoprecipitation studies have revealed a now well-documented interaction of the core protein with the DEAD box RNA helicase, DDX3 (Mamiya and Worman, 1999; Owsianka and Patel, 1999; You et al., 1999). This results in a profound change in the cellular distribution of DDX3, from what is normally a diffuse, predominantly cytoplasmic distribution to punctate cytoplasmic foci that co-localize with core protein in HCV-infected cells (Figure 4). DDX3 knockdown studies suggest that it may specifically facilitate HCV replication (Ariumi et al., 2007; Randall et al., 2007). It is difficult to distinguish virus-specific effects of gene knockdown or overexpression, however, from general pleiotropic effects on the cell that may indirectly influence replication, as shown recently in studies of another cellular helicase, DDX6 (Rck/p54), which also interacts with core (Jangra et al., 2010a). At any rate, the ability of DDX3 to modulate HCV replication is independent of its core-binding activity (Angus et al., 2010). Like many DEAD-box helicases, DDX3 appears to be multifunctional. As described above, it regulates activity of the p21 (waf1/cip1) promoter and represses cap-dependent translation through inhibition of eIF4E (Chao et al., 2006; Shih et al., 2008), and may have significant tumor suppressor activity (Chang et al., 2006; Botlagunta et al., 2008). However, DDX3 is also required for efficient induction of interferons in response to virus infections (Schroder et al., 2008; Oshiumi et al., 2010), and this may be the function underlying its targeting by HCV.
Innate immune signaling
Like many other viruses, HCV has evolved mechanisms to antagonize host innate immune signaling pathways (reviewed in Lemon, 2010). These pathways are closely linked to tumor suppressor functions, as alluded to above, and the long-term targeting of innate immune signaling pathways by viral proteins during persistent infection may result in the promotion of cell growth and inhibition of apoptosis. The dsRNA-activated protein kinase (PKR) is interferon-inducible and downregulates translation through phosphorylation of eIF-2α, thereby inhibiting cell growth and promoting apoptosis to restrict virus infection. These properties confer tumor-suppressor activity on PKR (Meurs et al., 1993). In vitro data suggest that the NS5A protein can bind to and functionally repress PKR (Gale et al., 1997; Gale and Katze, 1998), possibly contributing to oncogenesis through this pathway (Gale et al., 1999). In addition to the effects of HCV on PKR, the NS3/4A protease targets essential adapter molecules in both the Toll-like receptor 3 and retinoic acid-inducible gene I-signaling pathways, TIR-domain containing inducer of interferon-β and mitochondrial antiviral signaling protein (MAVS; also known as interferon β promoter stimulator 1, IPS-1), thereby interfering with activation of interferon regulatory factor 3 (Li et al., 2005a, 2005b; Meylan et al., 2005; Wang et al., 2009b), a transcription factor with strong anti-proliferative properties (Kim et al., 2003). These interactions evolved to protect the virus from the antiviral actions of numerous interferon regulatory factor 3 dependent genes, including type I interferons, but may also contribute to the cancer-promoting effects of HCV.
Growth factor signaling
Growth factor signaling pathways have important roles in the initiation and possibly maintenance of HCC (reviewed in Whittaker et al., 2010). The transforming growth factor-β signaling pathway exerts both an anti-proliferative and pro-apoptotic influence, and is important in regulating the expansion of progenitor cells in regenerating liver (Thenappan et al., 2010). The HCV core protein has been suggested to interact with Smad3, a transcriptional modulator that is activated through the transforming growth factor-β pathway, thereby impairing the tumor suppressive properties of transforming growth factor-β (Cheng et al., 2004; Pavio et al., 2005; Battaglia et al., 2009). In addition, NS5A has the capacity to interact with Src homology 3 domains that are found in many proteins involved in signal transduction. Such interactions may involve Grb2, an adaptor protein involved in growth factor signaling (Tan et al., 1999), the p85 subunit of phosphoinositide 3-kinase, as described above (Street et al., 2004), and some members of the Src tyrosine kinase family (Macdonald et al., 2004).
miRNAs and HCV-associated cancer
The miRNAs are likely to have important roles in both the causation and maintenance of cancers, including HCC (Coulouarn et al., 2009; Pineau et al., 2010). Of 940 recognized human miRNAs, miR-122 is uniquely expressed at high abundance in the adult liver, representing ∼50% of miRNAs and with 50 000 or more copies per cell (Chang et al., 2004). It is developmentally regulated and expressed almost exclusively in liver, controlling the expression of numerous hepatocyte-specific genes and promoting hepatocellular differentiation (Chang et al., 2004; Krutzfeldt et al., 2005; Elmen et al., 2008).
HCV replication is critically dependent upon miR-122 (Jopling et al., 2005; Jangra et al., 2010b). The miR-122 binds to two conserved, tandem sites in the HCV 5′UTR that are complementary to its ‘seed’ sequence (Jopling et al., 2008). This interaction is required for effective genome amplification, but available data do not suggest that it directly enhances viral RNA synthesis (Jopling et al., 2005; Norman and Sarnow, 2010). Instead, miR-122 binding promotes cap-independent translation of the viral RNA (Henke et al., 2008; Jangra et al., 2010b). However, the magnitude of this effect appears insufficient to completely explain the dependence of HCV on miR-122, and thus the requirement for miR-122 as a host factor for HCV replication remains incompletely explained. Importantly, however, HCV replication was potently suppressed in chimpanzees after therapeutic silencing of miR-122 by administration of an antisense locked nucleic acid oligonucleotide against miR-122 (Lanford et al., 2010). These recent data have fueled enthusiasm for miR-122 antagomirs as potential antiviral agents, making a better understanding of the role of miR-122 in carcinogenesis imperative.
There is some evidence that miR-122 may have tumor suppressor properties. Expression of miR-122 is low or undetectable in the human hepatoma cell lines, Hep3B and HepG2 cells, in which its overexpression inhibits anchorage-independent growth, migration, invasion and tumor formation in nude mice (Bai et al., 2009). IL-1 α is one of many genes regulated by miR-122, and polymorphisms in the miR-122 binding site in the IL-1α 3′UTR confer an increased risk for HCC (Gao et al., 2009). Cyclin G1 is also regulated by miR-122, influencing the stability of p53 and affecting the growth properties of HCC-derived cells (Fornari et al., 2009). Although a number of studies have profiled miRNA expression in HCC, it remains unclear whether miR-122 abundance is altered in HCV-associated HCC. Overall, miR-122 expression is reduced in 50–70% of HCC (Kutay et al., 2006; Gramantieri et al., 2007; Ura et al., 2009). However, it may be increased in a subset of HCC with mutations in β-catenin (Pineau et al., 2010) and in some, but not all studies suggest that miR-122 expression is preserved specifically in HCV-associated cancers (Varnholt et al., 2008; Coulouarn et al., 2009; Ura et al., 2009). This is an important question that deserves further study, as loss of miR-122 would limit the ability of HCV to replicate in HCC cells.
Despite the unique relationship between HCV and miR-122, few studies have examined how HCV infection influences miR-122 expression. Acute infection of Huh-7 cells with HCVcc was reported recently to cause a significant decline in miR-122 abundance (Liu et al., 2010). Many other miRNAs were differentially regulated by HCV infection in this study, however, and it is difficult to predict how these results relate to infection of normal hepatocytes in vivo. The miRNA biogenesis is modulated by p53 through an interaction with the Drosha processing complex that involves an association with DDX5 (Suzuki et al., 2009). As various HCV proteins interact with both p53 and DDX5, as described above, it is not surprising to find that HCV infection might alter global miRNA expression profiles. However, the lack of normal p53 function in Huh-7 cells mandates a great deal of caution in interpreting the results of such studies. There are large gaps in our understanding of how HCV regulates miRNA expression, and the role of miRNAs in development of HCC. Future studies should be directed at addressing these questions.
Summary and conclusions: a model of HCV-associated hepatocarcinogenesis
Although HCV is increasingly associated with HCC in the United States and probably many other countries, attempts to understand the underlying pathogenetic mechanisms are limited by the absence of good cellular and animal models of HCV pathogenesis. Those data that are available suggest that HCV-associated HCC is likely to be caused by a combination of environmental, epigenetic and genetic factors. In patients who are chronically infected with HCV, cancer typically develops in the setting of cirrhosis. Repeated cycles of immune-mediated destruction of infected hepatocytes and virus-induced apoptosis together with regeneration of damaged tissue cause disturbances in the normal cellular homeostasis of the liver. Furthermore, inflammatory responses associated with persistent infection cause oxidative stress that can damage chromosomal DNA, leading to heritable changes in the genome. These processes mirror the development of HCC because of many other causes. However, the occurrence of HCV-associated HCC in the absence of cirrhosis, the particularly high rate of HCC in cirrhosis caused by chronic HCV infection, and the development of HCC in HCV transgenic mice in the absence of inflammation or fibrosis, suggest that persistent HCV infection and viral protein expression is likely to have a direct cancer-promoting effect. Numerous interactions between viral proteins and cellular tumor suppressor pathways may act not only to further viral replication, but also to deregulate normal control of the cell cycle and cellular responses to DNA damage. These cancer-promoting actions of HCV may be eliminated largely if not completely by effective antiviral therapy. Antiviral therapies remain limited in efficacy, however, and a better understanding of the effects of HCV infection on these regulatory pathways may suggest new opportunities for preventive measures that may be taken in the absence of virus eradication.
References
Centers for Disease Control and Prevention (CDC) (2010). Hepatocellular carcinoma-United States, 2001–2006. MMWR Morb Mortal Wkly Rep 59: 517–520.
Altekruse SF, McGlynn KA, Reichman ME . (2009). Hepatocellular carcinoma incidence, mortality, and survival trends in the United States from 1975 to 2005. J Clin Oncol 27: 1485–1491.
Angus AG, Dalrymple D, Boulant S, McGivern DR, Clayton RF, Scott MJ et al. (2010). Requirement of cellular DDX3 for hepatitis C virus replication is unrelated to its interaction with the viral core protein. J Gen Virol 91: 122–132.
Ariumi Y, Kuroki M, Abe K, Dansako H, Ikeda M, Wakita T et al. (2007). DDX3 DEAD-box RNA helicase is required for hepatitis C virus RNA replication. J Virol 81: 13922–13926.
Ariumi Y, Kuroki M, Dansako H, Abe K, Ikeda M, Wakita T et al. (2008). The DNA damage sensors ataxia-telangiectasia mutated kinase and checkpoint kinase 2 are required for hepatitis C virus RNA replication. J Virol 82: 9639–9646.
Armstrong GL, Wasley A, Simard EP, McQuillan GM, Kuhnert WL, Alter MJ . (2006). The prevalence of hepatitis C virus infection in the United States, 1999 through 2002. Ann Intern Med 144: 705–714.
Azechi H, Nishida N, Fukuda Y, Nishimura T, Minata M, Katsuma H et al. (2001). Disruption of the p16/cyclin D1/retinoblastoma protein pathway in the majority of human hepatocellular carcinomas. Oncology 60: 346–354.
Bai S, Nasser MW, Wang B, Hsu SH, Datta J, Kutay H et al. (2009). MicroRNA-122 inhibits tumorigenic properties of hepatocellular carcinoma cells and sensitizes these cells to sorafenib. J Biol Chem 284: 32015–32027.
Baroni GS, Pastorelli A, Manzin A, Benedetti A, Marucci L, Solforosi L et al. (1999). Hepatic stellate cell activation and liver fibrosis are associated with necroinflammatory injury and Th1-like response in chronic hepatitis C. Liver 19: 212–219.
Bartsch H, Nair J . (2004). Oxidative stress and lipid peroxidation-derived DNA-lesions in inflammation driven carcinogenesis. Cancer Detect Prev 28: 385–391.
Bataller R, Paik YH, Lindquist JN, Lemasters JJ, Brenner DA . (2004). Hepatitis C virus core and nonstructural proteins induce fibrogenic effects in hepatic stellate cells. Gastroenterology 126: 529–540.
Bates GJ, Nicol SM, Wilson BJ, Jacobs AM, Bourdon JC, Wardrop J et al. (2005). The DEAD box protein p68: a novel transcriptional coactivator of the p53 tumour suppressor. EMBO J 24: 543–553.
Battaglia S, Benzoubir N, Nobilet S, Charneau P, Samuel D, Zignego AL et al. (2009). Liver cancer-derived hepatitis C virus core proteins shift TGF-Beta responses from tumor suppression to epithelial-mesenchymal transition. PLoS ONE 4: e4355.
Bigger CB, Guerra KM, Brasky KM, Hubbard G, Beard MR, Luxon B et al. (2004). Intrahepatic gene expression during chronic hepatitis C virus infection in chimpanzees. J Virol 78: 13779–13792.
Botlagunta M, Vesuna F, Mironchik Y, Raman A, Lisok A, Winnard Jr P et al. (2008). Oncogenic role of DDX3 in breast cancer biogenesis. Oncogene 27: 3912–3922.
Bowen DG, Walker CM . (2005). Adaptive immune responses in acute and chronic hepatitis C virus infection. Nature 436: 946–952.
Bralet MP, Regimbeau JM, Pineau P, Dubois S, Loas G, Degos F et al. (2000). Hepatocellular carcinoma occurring in nonfibrotic liver: epidemiologic and histopathologic analysis of 80 French cases. Hepatology 32: 200–204.
Brenner DA . (2009). Molecular pathogenesis of liver fibrosis. Trans Am Clin Climatol Assoc 120: 361–368.
Bressac B, Galvin KM, Liang TJ, Isselbacher KJ, Wands JR, Ozturk M . (1990). Abnormal structure and expression of p53 gene in human hepatocellular carcinoma. Proc Natl Acad Sci USA 87: 1973–1977.
Chang J, Nicolas E, Marks D, Sander C, Lerro A, Buendia MA et al. (2004). miR-122, a mammalian liver-specific microRNA, is processed from hcr mRNA and may downregulate the high affinity cationic amino acid transporter CAT-1. RNA Biol 1: 106–113.
Chang PC, Chi CW, Chau GY, Li FY, Tsai YH, Wu JC et al. (2006). DDX3, a DEAD box RNA helicase, is deregulated in hepatitis virus-associated hepatocellular carcinoma and is involved in cell growth control. Oncogene 25: 1991–2003.
Chao CH, Chen CM, Cheng PL, Shih JW, Tsou AP, Lee YH . (2006). DDX3, a DEAD box RNA helicase with tumor growth-suppressive property and transcriptional regulation activity of the p21waf1/cip1 promoter, is a candidate tumor suppressor. Cancer Res 66: 6579–6588.
Chellappan SP, Hiebert S, Mudryj M, Horowitz JM, Nevins JR . (1991). The E2F transcription factor is a cellular target for the RB protein. Cell 65: 1053–1061.
Cheng PL, Chang MH, Chao CH, Lee YH . (2004). Hepatitis C viral proteins interact with Smad3 and differentially regulate TGF-beta/Smad3-mediated transcriptional activation. Oncogene 23: 7821–7838.
Chou AH, Tsai HF, Wu YY, Hu CY, Hwang LH, Hsu PI et al. (2005). Hepatitis C virus core protein modulates TRAIL-mediated apoptosis by enhancing Bid cleavage and activation of mitochondria apoptosis signaling pathway. J Immunol 174: 2160–2166.
Classon M, Harlow E . (2002). The retinoblastoma tumour suppressor in development and cancer. Nat Rev Cancer 2: 910–917.
Coulouarn C, Factor VM, Andersen JB, Durkin ME, Thorgeirsson SS . (2009). Loss of miR-122 expression in liver cancer correlates with suppression of the hepatic phenotype and gain of metastatic properties. Oncogene 28: 3526–3536.
Cross TJ, Rashid MM, Berry PA, Harrison PM . (2010). The importance of steatosis in chronic hepatitis C infection and its management: a review. Hepatol Res 40: 237–247.
Davis GL, Alter MJ, El-Serag H, Poynard T, Jennings LW . (2010). Aging of hepatitis C virus (HCV)-infected persons in the United States: a multiple cohort model of HCV prevalence and disease progression. Gastroenterology 138: 513–521, 521 e511-516.
DeCaprio JA, Ludlow JW, Figge J, Shew JY, Huang CM, Lee WH et al. (1988). SV40 large tumor antigen forms a specific complex with the product of the retinoblastoma susceptibility gene. Cell 54: 275–283.
Degos F, Christidis C, Ganne-Carrie N, Farmachidi JP, Degott C, Guettier C et al. (2000). Hepatitis C virus related cirrhosis: time to occurrence of hepatocellular carcinoma and death. Gut 47: 131–136.
Deng L, Adachi T, Kitayama K, Bungyoku Y, Kitazawa S, Ishido S et al. (2008). Hepatitis C virus infection induces apoptosis through a Bax-triggered, mitochondrion-mediated, caspase 3-dependent pathway. J Virol 82: 10375–10385.
Deng L, Nagano-Fujii M, Tanaka M, Nomura-Takigawa Y, Ikeda M, Kato N et al. (2006). NS3 protein of Hepatitis C virus associates with the tumour suppressor p53 and inhibits its function in an NS3 sequence-dependent manner. J Gen Virol 87: 1703–1713.
Dharel N, Kato N, Muroyama R, Taniguchi H, Otsuka M, Wang Y et al. (2008). Potential contribution of tumor suppressor p53 in the host defense against hepatitis C virus. Hepatology 47: 1136–1149.
Diamond DL, Jacobs JM, Paeper B, Proll SC, Gritsenko MA, Carithers Jr RL et al. (2007). Proteomic profiling of human liver biopsies: hepatitis C virus-induced fibrosis and mitochondrial dysfunction. Hepatology 46: 649–657.
Dolganiuc A, Oak S, Kodys K, Golenbock DT, Finberg RW, Kurt-Jones E et al. (2004). Hepatitis C core and nonstructural 3 proteins trigger toll-like receptor 2-mediated pathways and inflammatory activation. Gastroenterology 127: 1513–1524.
Dutta U, Kench J, Byth K, Khan MH, Lin R, Liddle C et al. (1998). Hepatocellular proliferation and development of hepatocellular carcinoma: a case-control study in chronic hepatitis C. Hum Pathol 29: 1279–1284.
Dyson N, Howley PM, Munger K, Harlow E . (1989). The human papilloma virus-16 E7 oncoprotein is able to bind to the retinoblastoma gene product. Science 243: 934–937.
Edamoto Y, Hara A, Biernat W, Terracciano L, Cathomas G, Riehle HM et al. (2003). Alterations of RB1, p53 and Wnt pathways in hepatocellular carcinomas associated with hepatitis C, hepatitis B and alcoholic liver cirrhosis. Int J Cancer 106: 334–341.
Elmen J, Lindow M, Schutz S, Lawrence M, Petri A, Obad S et al. (2008). LNA-mediated microRNA silencing in non-human primates. Nature 452: 896–899.
Erdtmann L, Franck N, Lerat H, Le Seyec J, Gilot D, Cannie I et al. (2003). The hepatitis C virus NS2 protein is an inhibitor of CIDE-B-induced apoptosis. J Biol Chem 278: 18256–18264.
Farinati F, Cardin R, D'Errico A, De Maria N, Naccarato R, Cecchetto A et al. (1996). Hepatocyte proliferative activity in chronic liver damage as assessed by the monoclonal antibody MIB1 Ki67 in archival material: the role of etiology, disease activity, iron, and lipid peroxidation. Hepatology 23: 1468–1475.
Fornari F, Gramantieri L, Giovannini C, Veronese A, Ferracin M, Sabbioni S et al. (2009). MiR-122/cyclin G1 interaction modulates p53 activity and affects doxorubicin sensitivity of human hepatocarcinoma cells. Cancer Res 69: 5761–5767.
Fox JG, Feng Y, Theve EJ, Raczynski AR, Fiala JL, Doernte AL et al. (2010). Gut microbes define liver cancer risk in mice exposed to chemical and viral transgenic hepatocarcinogens. Gut 59: 88–97.
Furutani T, Hino K, Okuda M, Gondo T, Nishina S, Kitase A et al. (2006). Hepatic iron overload induces hepatocellular carcinoma in transgenic mice expressing the hepatitis C virus polyprotein. Gastroenterology 130: 2087–2098.
Gale Jr M, Katze MG . (1998). Molecular mechanisms of interferon resistance mediated by viral-directed inhibition of PKR, the interferon-induced protein kinase. Pharmacol Ther 78: 29–46.
Gale Jr M, Kwieciszewski B, Dossett M, Nakao H, Katze MG . (1999). Antiapoptotic and oncogenic potentials of hepatitis C virus are linked to interferon resistance by viral repression of the PKR protein kinase. J Virol 73: 6506–6516.
Gale Jr MJ, Korth MJ, Tang NM, Tan SL, Hopkins DA, Dever TE et al. (1997). Evidence that hepatitis C virus resistance to interferon is mediated through repression of the PKR protein kinase by the nonstructural 5A protein. Virology 230: 217–227.
Gao Y, He Y, Ding J, Wu K, Hu B, Liu Y et al. (2009). An insertion/deletion polymorphism at miRNA-122-binding site in the interleukin-1alpha 3′ untranslated region confers risk for hepatocellular carcinoma. Carcinogenesis 30: 2064–2069.
Ghosh AK, Steele R, Meyer K, Ray R, Ray RB . (1999). Hepatitis C virus NS5A protein modulates cell cycle regulatory genes and promotes cell growth. J Gen Virol 80 (Part 5): 1179–1183.
Goh PY, Tan YJ, Lim SP, Tan YH, Lim SG, Fuller-Pace F et al. (2004). Cellular RNA helicase p68 relocalization and interaction with the hepatitis C virus (HCV) NS5B protein and the potential role of p68 in HCV RNA replication. J Virol 78: 5288–5298.
Gong G, Waris G, Tanveer R, Siddiqui A . (2001). Human hepatitis C virus NS5A protein alters intracellular calcium levels, induces oxidative stress, and activates STAT-3 and NF-kappa B. Proc Natl Acad Sci USA 98: 9599–9604.
Gramantieri L, Ferracin M, Fornari F, Veronese A, Sabbioni S, Liu CG et al. (2007). Cyclin G1 is a target of miR-122a, a microRNA frequently down-regulated in human hepatocellular carcinoma. Cancer Res 67: 6092–6099.
Henke JI, Goergen D, Zheng J, Song Y, Schuttler CG, Fehr C et al. (2008). microRNA-122 stimulates translation of hepatitis C virus RNA. EMBO J 27: 3300–3310.
Hernando E, Nahle Z, Juan G, Diaz-Rodriguez E, Alaminos M, Hemann M et al. (2004). Rb inactivation promotes genomic instability by uncoupling cell cycle progression from mitotic control. Nature 430: 797–802.
Honda A, Arai Y, Hirota N, Sato T, Ikegaki J, Koizumi T et al. (1999). Hepatitis C virus structural proteins induce liver cell injury in transgenic mice. J Med Virol 59: 281–289.
Honda M, Kaneko S, Shimazaki T, Matsushita E, Kobayashi K, Ping LH et al. (2000). Hepatitis C virus core protein induces apoptosis and impairs cell-cycle regulation in stably transformed Chinese hamster ovary cells. Hepatology 31: 1351–1359.
Ishido S, Hotta H . (1998). Complex formation of the nonstructural protein 3 of hepatitis C virus with the p53 tumor suppressor. FEBS Lett 438: 258–262.
Jangra RK, Yi M, Lemon SM . (2010a). DDX6 (Rck/p54) is required for efficient hepatitis C virus replication but not IRES-directed translation. J Virol 84: 6810–6824.
Jangra RK, Yi M, Lemon SM . (2010b). miR-122 regulation of hepatitis C virus translation and infectious virus production. J Virol 84: 6615–6625.
Jopling CL, Yi M, Lancaster AM, Lemon SM, Sarnow P . (2005). Modulation of hepatitis C virus RNA abundance by a liver-specific MicroRNA. Science 309: 1577–1581.
Jopling CL, Schutz S, Sarnow P . (2008). Position-dependent function for a tandem microRNA miR-122-binding site located in the hepatitis C virus RNA genome. Cell Host Microbe 4: 77–85.
Joyce MA, Walters KA, Lamb SE, Yeh MM, Zhu LF, Kneteman N et al. (2009). HCV induces oxidative and ER stress, and sensitizes infected cells to apoptosis in SCID/Alb-uPA mice. PLoS Pathogens 5: e1000291.
Kamegaya Y, Hiasa Y, Zukerberg L, Fowler N, Blackard JT, Lin W et al. (2005). Hepatitis C virus acts as a tumor accelerator by blocking apoptosis in a mouse model of hepatocarcinogenesis. Hepatology 41: 660–667.
Kao CF, Chen SY, Chen JY, Wu Lee YH . (2004). Modulation of p53 transcription regulatory activity and post-translational modification by hepatitis C virus core protein. Oncogene 23: 2472–2483.
Kato T, Miyamoto M, Date T, Yasui K, Taya C, Yonekawa H et al. (2003). Repeated hepatocyte injury promotes hepatic tumorigenesis in hepatitis C virus transgenic mice. Cancer Sci 94: 679–685.
Kawamura T, Furusaka A, Koziel MJ, Chung RT, Wang TC, Schmidt EV et al. (1997). Transgenic expression of hepatitis C virus structural proteins in the mouse. Hepatology 25: 1014–1021.
Keasler VV, Lerat H, Madden CR, Finegold MJ, McGarvey MJ, Mohammed EM et al. (2006). Increased liver pathology in hepatitis C virus transgenic mice expressing the hepatitis B virus X protein. Virology 347: 466–475.
Khidr L, Chen PL . (2006). RB, the conductor that orchestrates life, death and differentiation. Oncogene 25: 5210–5219.
Kim TY, Lee KH, Chang S, Chung C, Lee HW, Yim J et al. (2003). Oncogenic potential of a dominant negative mutant of interferon regulatory factor 3. J Biol Chem 278: 15272–15278.
Klopstock N, Katzenellenbogen M, Pappo O, Sklair-Levy M, Olam D, Mizrahi L et al. (2009). HCV tumor promoting effect is dependent on host genetic background. PloS One 4: e5025.
Koike K, Moriya K, Ishibashi K, Matsuura Y, Suzuki T, Saito I et al. (1995). Expression of hepatitis C virus envelope proteins in transgenic mice. J Gen Virol 76 (Part 12): 3031–3038.
Korenaga M, Wang T, Li Y, Showalter LA, Chan T, Sun J et al. (2005). Hepatitis C virus core protein inhibits mitochondrial electron transport and increases reactive oxygen species (ROS) production. J Biol Chem 280: 37481–37488.
Koskinas J, Petraki K, Kavantzas N, Rapti I, Kountouras D, Hadziyannis S . (2005). Hepatic expression of the proliferative marker Ki-67 and p53 protein in HBV or HCV cirrhosis in relation to dysplastic liver cell changes and hepatocellular carcinoma. J Viral Hepat 12: 635–641.
Krutzfeldt J, Rajewsky N, Braich R, Rajeev KG, Tuschl T, Manoharan M et al. (2005). Silencing of microRNAs in vivo with ′antagomirs′. Nature 438: 685–689.
Kutay H, Bai S, Datta J, Motiwala T, Pogribny I, Frankel W et al. (2006). Downregulation of miR-122 in the rodent and human hepatocellular carcinomas. J Cell Biochem 99: 671–678.
Kwun HJ, Jung EY, Ahn JY, Lee MN, Jang KL . (2001). p53-dependent transcriptional repression of p21(waf1) by hepatitis C virus NS3. J Gen Virol 82: 2235–2241.
Lai CK, Jeng KS, Machida K, Cheng YS, Lai MM . (2008). Hepatitis C virus NS3/4A protein interacts with ATM, impairs DNA repair and enhances sensitivity to ionizing radiation. Virology 370: 295–309.
Lan KH, Sheu ML, Hwang SJ, Yen SH, Chen SY, Wu JC et al. (2002). HCV NS5A interacts with p53 and inhibits p53-mediated apoptosis. Oncogene 21: 4801–4811.
Lanford RE, Hildebrandt-Eriksen ES, Petri A, Persson R, Lindow M, Munk ME et al. (2010). Therapeutic silencing of microRNA-122 in primates with chronic hepatitis C virus infection. Science 327: 198–201.
Lanford RE, Lemon SM, Walker C . (2011). The chimpanzee model of hepatitis C infections and small animal surrogates. In: He Y, Tan T (eds). Hepatitis C Antiviral Drug Discovery & Development. Horizons Scientific Press: Norwich. pp 99–132.
Lavanchy D . (2009). The global burden of hepatitis C. Liver Int 29 (Suppl 1): 74–81.
Lee SH, Kim YK, Kim CS, Seol SK, Kim J, Cho S et al. (2005). E2 of hepatitis C virus inhibits apoptosis. J Immunol 175: 8226–8235.
Lemon SM, Walker C, Alter MJ, Yi M . (2007). Hepatitis C viruses. In: Knipe D, Howley P, Griffin DE, Martin MA, Lamb RA, Roizman B et al (eds). Fields Virology, 5th edn. Lippincott, Williams & Wilkins: Philadelphia. pp 1253–1304.
Lemon SM . (2010). Induction and evasion of innate antiviral responses by hepatitis C virus. J Biol Chem 285: 22741–22747.
Lentini L, Pipitone L, Di LA . (2002). Functional inactivation of pRB results in aneuploid mammalian cells after release from a mitotic block. Neoplasia 4: 380–387.
Lerat H, Honda M, Beard MR, Loesch K, Sun J, Yang Y et al. (2002). Steatosis and liver cancer in transgenic mice expressing the structural and nonstructural proteins of hepatitis C virus. Gastroenterology 122: 352–365.
Li K, Foy E, Ferreon JC, Nakamura M, Ferreon AC, Ikeda M et al. (2005a). Immune evasion by hepatitis C virus NS3/4A protease-mediated cleavage of the Toll-like receptor 3 adaptor protein TRIF. Proc Natl Acad Sci USA 102: 2992–2997.
Li K, Prow T, Lemon SM, Beard MR . (2002). Cellular response to conditional expression of hepatitis C virus core protein in Huh7 cultured human hepatoma cells. Hepatology 35: 1237–1246.
Li XD, Sun L, Seth RB, Pineda G, Chen ZJ . (2005b). Hepatitis C virus protease NS3/4A cleaves mitochondrial antiviral signaling protein off the mitochondria to evade innate immunity. Proc Natl Acad Sci USA 102: 17717–17722.
Liang Y, Shilagard T, Xiao SY, Snyder N, Lau D, Cicalese L et al. (2009). Visualizing hepatitis C virus infections in human liver by two-photon microscopy. Gastroenterology 137: 1448–1458.
Lindenbach BD, Rice CM . (2005). Unravelling hepatitis C virus replication from genome to function. Nature 436: 933–938.
Liu X, Wang T, Wakita T, Yang W . (2010). Systematic identification of microRNA and messenger RNA profiles in hepatitis C virus-infected human hepatoma cells. Virology 398: 57–67.
Lohmann V, Korner F, Koch J, Herian U, Theilmann L, Bartenschlager R . (1999). Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 285: 110–113.
Lok AS, Seeff LB, Morgan TR, di Bisceglie AM, Sterling RK, Curto TM et al. (2009). Incidence of hepatocellular carcinoma and associated risk factors in hepatitis C-related advanced liver disease. Gastroenterology 136: 138–148.
Lu W, Lo SY, Chen M, Wu K, Fung YK, Ou JH . (1999). Activation of p53 tumor suppressor by hepatitis C virus core protein. Virology 264: 134–141.
Macdonald A, Crowder K, Street A, McCormick C, Harris M . (2004). The hepatitis C virus NS5A protein binds to members of the Src family of tyrosine kinases and regulates kinase activity. J Gen Virol 85: 721–729.
Machida K, Liu JC, McNamara G, Levine A, Duan L, Lai MM . (2009a). Hepatitis C virus causes uncoupling of mitotic checkpoint and chromosomal polyploidy through the Rb pathway. J Virol 83: 12590–12600.
Machida K, Tsukamoto H, Mkrtchyan H, Duan L, Dynnyk A, Liu HM et al. (2009b). Toll-like receptor 4 mediates synergism between alcohol and HCV in hepatic oncogenesis involving stem cell marker Nanog. Proc Natl Acad Sci USA 106: 1548–1553.
Majumder M, Ghosh AK, Steele R, Ray R, Ray RB . (2001). Hepatitis C virus NS5A physically associates with p53 and regulates p21/waf1 gene expression in a p53-dependent manner. J Virol 75: 1401–1407.
Majumder M, Steele R, Ghosh AK, Zhou XY, Thornburg L, Ray R et al. (2003). Expression of hepatitis C virus non-structural 5A protein in the liver of transgenic mice. FEBS Lett 555: 528–532.
Mamiya N, Worman HJ . (1999). Hepatitis C virus core protein binds to a DEAD box RNA helicase. J Biol Chem 274: 15751–15756.
Marukian S, Jones CT, Andrus L, Evans MJ, Ritola KD, Charles ED et al. (2008). Cell culture-produced hepatitis C virus does not infect peripheral blood mononuclear cells. Hepatology 48: 1843–1850.
Masuzaki R, Yoshida H, Tateishi R, Shiina S, Omata M . (2008). Hepatocellular carcinoma in viral hepatitis: improving standard therapy. Best Pract Res Clin Gastroenterol 22: 1137–1151.
Mateu G, Donis RO, Wakita T, Bukh J, Grakoui A . (2008). Intragenotypic JFH1 based recombinant hepatitis C virus produces high levels of infectious particles but causes increased cell death. Virology 376: 397–407.
Mazzocca A, Sciammetta SC, Carloni V, Cosmi L, Annunziato F, Harada T et al. (2005). Binding of hepatitis C virus envelope protein E2 to CD81 up-regulates matrix metalloproteinase-2 in human hepatic stellate cells. J Biol Chem 280: 11329–11339.
McGivern DR, Villanueva RA, Chinnaswamy S, Kao CC, Lemon SM . (2009). Impaired replication of hepatitis C virus containing mutations in a conserved NS5B retinoblastoma protein-binding motif. J Virol 83: 7422–7433.
McHutchison JG, Fried MW . (2003). Current therapy for hepatitis C: pegylated interferon and ribavirin. Clin Liver Dis 7: 149–161.
Mercer DF, Schiller DE, Elliott JF, Douglas DN, Hao C, Rinfret A et al. (2001). Hepatitis C virus replication in mice with chimeric human livers. Nat Med 7: 927–933.
Meurs EF, Galabru J, Barber GN, Katze MG, Hovanessian AG . (1993). Tumor suppressor function of the interferon-induced double-stranded RNA-activated protein kinase. Proc Natl Acad Sci USA 90: 232–236.
Meylan E, Curran J, Hofmann K, Moradpour D, Binder M, Bartenschlager R et al. (2005). Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 437: 1167–1172.
Milward A, Mankouri J, Harris M . (2010). Hepatitis C virus NS5A protein interacts with beta-catenin and stimulates its transcriptional activity in a phosphoinositide-3 kinase-dependent fashion. J Gen Virol 91: 373–381.
Moradpour D, Penin F, Rice CM . (2007). Replication of hepatitis C virus. Nat Rev Microbiol 5: 453–463.
Moriya K, Fujie H, Shintani Y, Yotsuyanagi H, Tsutsumi T, Ishibashi K et al. (1998). The core protein of hepatitis C virus induces hepatocellular carcinoma in transgenic mice. Nat Med 4: 1065–1067.
Moriya K, Nakagawa K, Santa T, Shintani Y, Fujie H, Miyoshi H et al. (2001). Oxidative stress in the absence of inflammation in a mouse model for hepatitis C virus-associated hepatocarcinogenesis. Cancer Res 61: 4365–4370.
Moriya K, Yotsuyanagi H, Shintani Y, Fujie H, Ishibashi K, Matsuura Y et al. (1997). Hepatitis C virus core protein induces hepatic steatosis in transgenic mice. J Gen Virol 78: 1527–1531.
Munakata T, Liang Y, Kim S, McGivern DR, Huibregtse JM, Nomoto A et al. (2007). Hepatitis C virus induces E6AP-dependent degradation of the retinoblastoma protein. PLoS Pathog 3: 1335–1347.
Munakata T, Nakamura M, Liang Y, Li K, Lemon SM . (2005). Down-regulation of the retinoblastoma tumor suppressor by the hepatitis C virus NS5B RNA-dependent RNA polymerase. Proc Natl Acad Sci USA 102: 18159–18164.
Naas T, Ghorbani M, Alvarez-Maya I, Lapner M, Kothary R, De Repentigny Y et al. (2005). Characterization of liver histopathology in a transgenic mouse model expressing genotype 1a hepatitis C virus core and envelope proteins 1 and 2. J Gen Virol 86: 2185–2196.
Nakamoto Y, Guidotti LG, Kuhlen CV, Fowler P, Chisari FV . (1998). Immune pathogenesis of hepatocellular carcinoma. J ExpMed 188: 341–350.
Neumann AU, Lam NP, Dahari H, Gretch DR, Wiley TE, Layden TJ et al. (1998). Hepatitis C viral dynamics in vivo and the antiviral efficacy of interferon-alpha therapy. Science 282: 103–107.
Nomura-Takigawa Y, Nagano-Fujii M, Deng L, Kitazawa S, Ishido S, Sada K et al. (2006). Non-structural protein 4A of Hepatitis C virus accumulates on mitochondria and renders the cells prone to undergoing mitochondria-mediated apoptosis. J Gen Virol 87: 1935–1945.
Norman KL, Sarnow P . (2010). Modulation of hepatitis C virus RNA abundance and the isoprenoid biosynthesis pathway by microRNA miR-122 involves distinct mechanisms. J Virol 84: 666–670.
Okuda M, Li K, Beard MR, Showalter LA, Scholle F, Lemon SM et al. (2002). Mitochondrial injury, oxidative stress, and antioxidant gene expression are induced by hepatitis C virus core protein. Gastroenterology 122: 366–375.
Oshiumi H, Sakai K, Matsumoto M, Seya T . (2010). DEAD/H BOX 3 (DDX3) helicase binds the RIG-I adaptor IPS-1 to up-regulate IFN-beta-inducing potential. Eur J Immunol 40: 940–948.
Otsuka M, Kato N, Lan K, Yoshida H, Kato J, Goto T et al. (2000). Hepatitis C virus core protein enhances p53 function through augmentation of DNA binding affinity and transcriptional ability. J Biol Chem 275: 34122–34130.
Owsianka AM, Patel AH . (1999). Hepatitis C virus core protein interacts with a human DEAD box protein DDX3. Virology 257: 330–340.
Park CY, Choi SH, Kang SM, Kang JI, Ahn BY, Kim H et al. (2009). Nonstructural 5A protein activates beta-catenin signaling cascades: implication of hepatitis C virus-induced liver pathogenesis. J Hepatol 51: 853–864.
Pasquinelli C, Shoenberger JM, Chung J, Chang KM, Guidotti LG, Selby M et al. (1997). Hepatitis C virus core and E2 protein expression in transgenic mice. Hepatology 25: 719–727.
Pavio N, Battaglia S, Boucreux D, Arnulf B, Sobesky R, Hermine O et al. (2005). Hepatitis C virus core variants isolated from liver tumor but not from adjacent non-tumor tissue interact with Smad3 and inhibit the TGF-beta pathway. Oncogene 24: 6119–6132.
Perz JF, Alter MJ . (2006). The coming wave of HCV-related liver disease: dilemmas and challenges. J Hepatol 44: 441–443.
Pineau P, Volinia S, McJunkin K, Marchio A, Battiston C, Terris B et al. (2010). miR-221 overexpression contributes to liver tumorigenesis. Proc Natl Acad Sci USA 107: 264–269.
Prikhod'ko EA, Prikhod'ko GG, Siegel RM, Thompson P, Major ME, Cohen JI . (2004). The NS3 protein of hepatitis C virus induces caspase-8-mediated apoptosis independent of its protease or helicase activities. Virology 329: 53–67.
Qadri I, Iwahashi M, Simon F . (2002). Hepatitis C virus NS5A protein binds TBP and p53, inhibiting their DNA binding and p53 interactions with TBP and ERCC3. Biochim Biophys Acta 1592: 193–204.
Quinkert D, Bartenschlager R, Lohmann V . (2005). Quantitative analysis of the hepatitis C virus replication complex. J Virol 79: 13594–13605.
Randall G, Panis M, Cooper JD, Tellinghuisen TL, Sukhodolets KE, Pfeffer S et al. (2007). Cellular cofactors affecting hepatitis C virus infection and replication. Proc Natl Acad Sci USA 104: 12884–12889.
Ray RB, Steele R, Meyer K, Ray R . (1998). Hepatitis C virus core protein represses p21WAF1/Cip1/Sid1 promoter activity. Gene 208: 331–336.
Sacco R, Tsutsumi T, Suzuki R, Otsuka M, Aizaki H, Sakamoto S et al. (2003). Antiapoptotic regulation by hepatitis C virus core protein through up-regulation of inhibitor of caspase-activated DNase. Virology 317: 24–35.
Saito T, Owen DM, Jiang F, Marcotrigiano J, Gale Jr M . (2008). Innate immunity induced by composition-dependent RIG-I recognition of hepatitis C virus RNA. Nature 454: 523–527.
Sato Y, Kato J, Takimoto R, Takada K, Kawano Y, Miyanishi K et al. (2006). Hepatitis C virus core protein promotes proliferation of human hepatoma cells through enhancement of transforming growth factor alpha expression via activation of nuclear factor-kappaB. Gut 55: 1801–1808.
Schroder M, Baran M, Bowie AG . (2008). Viral targeting of DEAD box protein 3 reveals its role in TBK1/IKKepsilon-mediated IRF activation. Embo J 27: 2147–2157.
Shackel NA, McGuinness PH, Abbott CA, Gorrell MD, McCaughan GW . (2002). Insights into the pathobiology of hepatitis C virus-associated cirrhosis: analysis of intrahepatic differential gene expression. Am J Pathol 160: 641–654.
Shih JW, Tsai TY, Chao CH, Wu Lee YH . (2008). Candidate tumor suppressor DDX3 RNA helicase specifically represses cap-dependent translation by acting as an eIF4E inhibitory protein. Oncogene 27: 700–714.
Shimakami T, Lanford RE, Lemon SM . (2009). Hepatitis C: recent successes and continuing challenges in the development of improved treatment modalities. Curr Opin Pharmacol 9: 537–544.
Shimizu YK, Igarashi H, Kiyohara T, Shapiro M, Wong DC, Purcell RH et al. (1998). Infection of a chimpanzee with hepatitis C virus grown in cell culture. J Gen Virol 79: 1383–1386.
Street A, Macdonald A, Crowder K, Harris M . (2004). The Hepatitis C virus NS5A protein activates a phosphoinositide 3-kinase-dependent survival signaling cascade. J Biol Chem 279: 12232–12241.
Street A, Macdonald A, McCormick C, Harris M . (2005). Hepatitis C virus NS5A-mediated activation of phosphoinositide 3-kinase results in stabilization of cellular beta-catenin and stimulation of beta-catenin-responsive transcription. J Virol 79: 5006–5016.
Sun B, Karin M . (2008). NF-kappaB signaling, liver disease and hepatoprotective agents. Oncogene 27: 6228–6244.
Suzuki HI, Yamagata K, Sugimoto K, Iwamoto T, Kato S, Miyazono K . (2009). Modulation of microRNA processing by p53. Nature 460: 529–533.
Takahashi K, Asabe S, Wieland S, Garaigorta U, Gastaminza P, Isogawa M et al. (2010). Plasmacytoid dendritic cells sense hepatitis C virus-infected cells, produce interferon, and inhibit infection. Proc Natl Acad Sci USA 107: 7431–7436.
Takahashi Y, Rayman JB, Dynlacht BD . (2000). Analysis of promoter binding by the E2F and pRB families in vivo: distinct E2F proteins mediate activation and repression. Genes Dev 14: 804–816.
Tan SL, Nakao H, He Y, Vijaysri S, Neddermann P, Jacobs BL et al. (1999). NS5A, a nonstructural protein of hepatitis C virus, binds growth factor receptor-bound protein 2 adaptor protein in a Src homology 3 domain/ligand-dependent manner and perturbs mitogenic signaling. Proc Natl Acad Sci USA 96: 5533–5538.
Tanaka M, Nagano-Fujii M, Deng L, Ishido S, Sada K, Hotta H . (2006). Single-point mutations of hepatitis C virus NS3 that impair p53 interaction and anti-apoptotic activity of NS3. Bio Biophys Res Comms 340: 792–799.
Tanaka Y, Hanada K, Mizokami M, Yeo AE, Shih JW, Gojobori T et al. (2002). Inaugural Article: a comparison of the molecular clock of hepatitis C virus in the United States and Japan predicts that hepatocellular carcinoma incidence in the United States will increase over the next two decades. Proc Natl Acad Sci USA 99: 15584–15589.
Taura M, Eguma A, Suico MA, Shuto T, Koga T, Komatsu K et al. (2008). p53 regulates Toll-like receptor 3 expression and function in human epithelial cell lines. Mol Cell Biol 28: 6557–6567.
Teufel A, Weinmann A, Centner C, Piendl A, Lohse AW, Galle PR et al. (2009). Hepatocellular carcinoma in patients with autoimmune hepatitis. World J Gastroenterol 15: 578–582.
Thenappan A, Li Y, Kitisin K, Rashid A, Shetty K, Johnson L et al. (2010). Role of transforming growth factor beta signaling and expansion of progenitor cells in regenerating liver. Hepatology 51: 1373–1382.
Thorgeirsson SS, Grisham JW . (2002). Molecular pathogenesis of human hepatocellular carcinoma. Nat Genet 31: 339–346.
Umemura T, Ichijo T, Yoshizawa K, Tanaka E, Kiyosawa K . (2009). Epidemiology of hepatocellular carcinoma in Japan. J Gastroenterol 44 (Suppl 19): 102–107.
Ura S, Honda M, Yamashita T, Ueda T, Takatori H, Nishino R et al. (2009). Differential microRNA expression between hepatitis B and hepatitis C leading disease progression to hepatocellular carcinoma. Hepatology 49: 1098–1112.
Varnholt H, Drebber U, Schulze F, Wedemeyer I, Schirmacher P, Dienes HP et al. (2008). MicroRNA gene expression profile of hepatitis C virus-associated hepatocellular carcinoma. Hepatology 47: 1223–1232.
Wakita T, Pietschmann T, Kato T, Date T, Miyamoto M, Zhao Z et al. (2005). Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat Med 11: 791–796.
Walters KA, Syder AJ, Lederer SL, Diamond DL, Paeper B, Rice CM et al. (2009). Genomic analysis reveals a potential role for cell cycle perturbation in HCV-mediated apoptosis of cultured hepatocytes. PLoS Pathogens 5: e1000269.
Wang AG, Lee DS, Moon HB, Kim JM, Cho KH, Choi SH et al. (2009a). Non-structural 5A protein of hepatitis C virus induces a range of liver pathology in transgenic mice. J Pathol 219: 253–262.
Wang N, Liang Y, Devaraj S, Wang J, Lemon SM, Li K . (2009b). Toll-like receptor 3 mediates establishment of an antiviral state against hepatitis C virus in hepatoma cells. J Virol 83: 9824–9834.
Waris G, Livolsi A, Imbert V, Peyron JF, Siddiqui A . (2003). Hepatitis C virus NS5A and subgenomic replicon activate NF-kappaB via tyrosine phosphorylation of IkappaBalpha and its degradation by calpain protease. J Biol Chem 278: 40778–40787.
Waris G, Tardif KD, Siddiqui A . (2002). Endoplasmic reticulum (ER) stress: hepatitis C virus induces an ER-nucleus signal transduction pathway and activates NF-kappaB and STAT-3. Biochem Pharmacol 64: 1425–1430.
Waris G, Turkson J, Hassanein T, Siddiqui A . (2005). Hepatitis C virus (HCV) constitutively activates STAT-3 via oxidative stress: role of STAT-3 in HCV replication. J Virol 79: 1569–1580.
Whittaker S, Marais R, Zhu AX . (2010). The role of signaling pathways in the development and treatment of hepatocellular carcinoma. Oncogene 29: 4989–5005.
Whyte P, Buchkovich KJ, Horowitz JM, Friend SH, Raybuck M, Weinberg RA et al. (1988). Association between an oncogene and an anti-oncogene: the adenovirus E1A proteins bind to the retinoblastoma gene product. Nature 334: 124–129.
Yeh MM, Daniel HD, Torbenson M . (2010). Hepatitis C-associated hepatocellular carcinomas in non-cirrhotic livers. Mod Pathol 23: 276–283.
Yeoman AD, Al-Chalabi T, Karani JB, Quaglia A, Devlin J, Mieli-Vergani G et al. (2008). Evaluation of risk factors in the development of hepatocellular carcinoma in autoimmune hepatitis: implications for follow-up and screening. Hepatology 48: 863–870.
Yi M, Villanueva RA, Thomas DL, Wakita T, Lemon SM . (2006). Production of infectious genotype 1a hepatitis C virus (Hutchinson strain) in cultured human hepatoma cells. Proc Natl Acad Sci USA 103: 2310–2315.
Yoshizawa H . (2002). Hepatocellular carcinoma associated with hepatitis C virus infection in Japan: projection to other countries in the foreseeable future. Oncology 62 (Suppl 1): 8–17.
You LR, Chen CM, Yeh TS, Tsai TY, Mai RT, Lin CH et al. (1999). Hepatitis C virus core protein interacts with cellular putative RNA helicase. J Virol 73: 2841–2853.
Zhu N, Khoshnan A, Schneider R, Matsumoto M, Dennert G, Ware C et al. (1998). Hepatitis C virus core protein binds to the cytoplasmic domain of tumor necrosis factor (TNF) receptor 1 and enhances TNF-induced apoptosis. J Virol 72: 3691–3697.
Acknowledgements
This work was supported in part by the University Cancer Research Fund and grants from the National Institutes of Health: P20-CA127004 and R21-AI081058.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
McGivern, D., Lemon, S. Virus-specific mechanisms of carcinogenesis in hepatitis C virus associated liver cancer. Oncogene 30, 1969–1983 (2011). https://doi.org/10.1038/onc.2010.594
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/onc.2010.594
Keywords
This article is cited by
An overview of mouse models of hepatocellular carcinoma
Infectious Agents and Cancer (2023)
Association of MTHFR and TYMS gene polymorphisms with the susceptibility to HCC in Egyptian HCV cirrhotic patients
Clinical and Experimental Medicine (2022)
Effects of hepatitis C virus core protein and nonstructural protein 4B on the Wnt/β-catenin pathway
BMC Microbiology (2017)
Immunotherapy against cancer-related viruses
Cell Research (2017)
Hepatitis C virus drives the pathogenesis of hepatocellular carcinoma: from immune evasion to carcinogenesis
Clinical & Translational Immunology (2016)
