





Abstract
Inflammatory bowel disease (IBD) is an intestinal inflammatory condition that affects more than 2 million people in the United States. Although the etiology and pathogenesis of IBD are still largely unknown, dysregulated host/enteric microbial interactions are requisite for the development of IBD. So far, many researchers have tried to identify a precise relationship between IBD and an imbalance of the intestinal microbiota, termed “dysbiosis.” Despite extensive efforts, it is still largely unknown about the interplay among microbes, their hosts, and their environments, and whether dysbiosis is a causal factor or an effect of IBD. Recently, deep-sequencing analyses of the microbiota in patients with IBD patients have been instrumental in characterizing the strong association between dysbiosis and IBD development, although it is still unable to identify specific-associated species level changes in most cases. Based on many recent reports, dysbiosis of the commensal microbiota is implicated in the pathogenesis of several diseases, including IBD, obesity, and allergic disorders, in both human and animal models. In this review article, the authors have focused on explaining the multiple types of dysbiosis, as well as dysbiosis-related diseases and potential treatments to apply this knowledge to understand a possible cause and potentially find therapeutic strategies for IBD as well as the other dysbiosis-related diseases.
The adult human gut contains about 1014 bacterial cells with more than 1000 different bacterial species. There are several major divisions of bacteria found in the normal intestinal microflora, with the most dominant groups being Bacteroidetes and Firmicutes. The gut houses both bacteria that are protective and some bacteria that could be potentially harmful to the host (Fig. 1). In normal conditions of healthy individuals, there is cross talk and cross-regulation between the host and the microbiota that reside in the gut, which creates a homeostatic balance of bacteria so that the gastrointestinal tract remains healthy and free from overgrowth of potentially pathogenic bacteria. The microbiota has a commensal relationship with the host; the bacteria thrive in the rich environment of the gut while the host benefits from multiple functions provided by the bacteria.
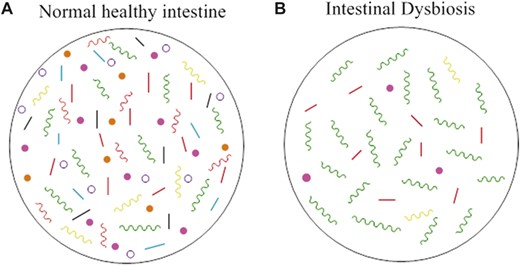
Normal and dysbiotic intestinal microbiota. A, The healthy intestines of normal individuals are colonized by a wide range of bacteria of more than 1000 species. In healthy individuals, these bacteria are in a homeostatic balance between commensal and potentially pathogenic bacteria, and the intestinal tract does not display overgrowth of pathogenic bacteria. The microflora provide the host with protection from foreign microbes, acting as a central line of resistance to colonization by these exogenous bacteria. This protection is known as the “barrier effect,” or colonization resistance.1 Through the mucosal surface of the intestine, the microbiota interacts with the host immune system, providing the host with immune regulatory functions, like priming the mucosal immune system.1,3 The microbiota also possesses various metabolic functions, like breaking down complex carbohydrates and generating short-chain fatty acids, from which the host benefits.1,4 Surprisingly, the gut microbiota is also capable of interacting with distant organs, such as the brain, which has led to studies of the influence of the gut microbiota on mental disorders, like autism, and diseases such as Alzheimer's disease.3 B, When the intestinal bacterial homeostasis is disrupted, dysbiosis occurs. Dysbiosis is defined by an imbalance in bacterial composition, changes in bacterial metabolic activities, or changes in bacterial distribution within the gut. The 3 types of dysbiosis are (1) loss of beneficial bacteria, (2) overgrowth of potentially pathogenic bacteria, and (3) loss of overall bacterial diversity. In most cases, these types of dysbioses occur at the same time. Green colors representing pathogenic bacteria and each different color bacteria representing a different commensal species to show diversity or lack thereof in each case. Dysbiosis has been associated with diseases such as IBD, obesity, type 1 and type 2 diabetes, autism, and certain gastrointestinal cancers.
The homeostatic balance of the intestinal microflora is extremely beneficial to the host; however, if there is a change in the microbial composition that causes a drastic imbalance between the beneficial and potentially pathogenic bacteria, the gut becomes vulnerable to pathogenic insult with gut microbial alterations. This imbalance in the microbial equilibrium is termed “dysbiosis,” which has been further defined as a disturbance to gut microbiota homeostasis due to an imbalance in the flora itself, changes in their functional composition and metabolic activities, or changes in their local distribution.1,2 In general, dysbiosis can be categorized into 3 different types: (1) loss of beneficial organisms, (2) excessive growth of potentially harmful organisms, and (3) loss of overall microbial diversity. It has been found that these 3 types are not mutually exclusive and can occur at the same time, which is most often the case. Dysbiosis has been implicated in a wide range of diseases including inflammatory bowel disease (IBD), obesity, allergic disorders, type 1 diabetes mellitus, autism, obesity, and colorectal cancer (CRC) in both human and animal models. This review will mainly focus on the implication found between dysbiosis and IBD such as Crohn's disease (CD) and ulcerative colitis in addition to the selected dysbiosis-associated diseases.
Dysbiosis-Associated Diseases
Inflammatory Bowel Disease
For many years, researchers have been trying to discover a mono-associated cause of IBD. As a result, there are 3 major pathogens that have been found to be associated the most with IBD. These pathogens are Mycobacterium avium paratuberculosis, which was once thought to be a potential infectious agent associated with the pathogenesis of CD, adherent-invasive Escherichia coli (AIEC), which has been frequently found in patients with IBD with acute/active phases and has been suggested to increase the inflammatory response,3 and Clostridium difficile, which has been detected in patients with ulcerative colitis relapse and remission.5 Although some studies have shown that there may be an increase in comorbidity with these bacteria and IBD, other studies have shown that these results are inconsistent.1,6,7 There is still no direct evidence that any of these bacteria are the sole cause of IBD, and thinking has shifted that IBD may be caused by an imbalance of commensal microflora associated with more complex interactions between the host and the entire intestinal microbiota7 rather than the “one-microbe-one disease” concept. Although imbalance of the microbiota, so-called dysbiosis, seems to be one of the major causes of IBD pathogenesis, further extensive research is needed to provide evidence of this statement because it is still unclear whether the dysbiosis seen in patients with IBD is a risk factor or an effect of the disease.
Intestinal dysbiosis for patients with IBD is shown to have a characteristic pattern of a decrease in commensal bacteria diversity, with a majority of the decrease in Firmicutes and Bacteroides in the intestinal microflora, which are the 2 most abundant groups in the normal flora.8,–10 Based on some research results, dysbiosis in IBD, in particular CD, has also been associated with a relative increase in the Enterobacteriaceae family.1,3,6,8,9 The dysbiosis signature in CD is characterized by 5 bacterial species: an increase in Ruminococcus gnavus, and a decrease in Faecalibacterium prausnitzii, Bifidobacterium adolescentis, Dialister invisus, and an unknown of Clostridium cluster XIVa.11 It has also been reported that unaffected relatives of patients with CD also had an altered intestinal microbiota compared with healthy individuals and also had enhanced mucin degradation with increased epithelial permeability.8,11 Because the intestinal mucosal barrier is the first line of defense against luminal microbiota, this degradation may be an intermediate step toward dysbiosis and CD. With this reasoning, dysbiosis could be a precursor for CD. There are many potential mechanisms being studied that may describe the role of dysbiosis in the pathogenesis of IBD: One of these mechanisms involves the decrease of butyrate-producing bacteria along with an increase in sulfate-reducing bacteria (SRBs), which is frequently seen in dysbiosis of patients with IBD. In these patients, the dysbiosis characterized in the gut has a severe decrease in F. prausnitzii,3,6,8,9 which is a major butyrate-producing bacteria in the intestines. Butyrate is an energy source for intestinal epithelial cells and is needed to help protect the intestinal epithelial barrier from becoming vulnerable to potential pathogens.3 Some studies have also found an increase in SRB.10 SRBs metabolize sulfate into hydrogen sulfide, which is a toxic molecule that can block butyrate utilization and inhibit phagocytosis as well as the killing of bacteria. This hypothesized sequential mechanism for dysbiosis-associated IBD suggests that the dysbiosis characterized by a decrease in butyrate-producing bacteria and increase in SRBs causes a reduced level of butyrate, which causes a decreased expression in epithelial tight junction proteins and therefore increased colonic epithelial permeability, which results in increased bacterial translocation through the intestinal epithelial cells and lamina propria. In genetically predisposed individuals who harbor mutations in the IBD-susceptibility gene(s), killing of bacteria that reaches the lamina propria through the permeable epithelial barrier is impaired by defective phagocytosis, which then leads to excessive Toll-like receptor stimulation, proinflammatory cytokine secretions, and activation of acquired immune responses, all of which increases the intestinal inflammatory responses.12 Another pattern noticed by studying the composition of the microbiota in patients with IBD is the loss of obligate anaerobes such as Bifidobacteria, and the increase of facultative anaerobes like E. coli.13 This pattern has led to the hypothesis that oxygen may be a major factor in causing dysbiosis in patients with IBD. In the healthy intestine, there is normally a low oxygen level and a large obligate anaerobe population. However, in dysbiosis, there is a decrease in Firmicutes, which are obligate anaerobes, and an increase in facultative anaerobes like Enterobacteriaceae. This change in the intestine from obligate to facultative anaerobes suggests that there is a disruption in aanaerobiosis in the intestine, meaning that oxygen and reactive oxygen species may be one of the causal factors of dysbiosis. This finding suggests that an increase in reactive oxygen species would cause a disruption of the anaerobic environment of the gut, which would create a selective advantage to facultative anaerobes or even some aerobes, resulting in overgrowth and causing dysbiosis.13
Investigating the role of dysbiosis as a causal factor of IBD, it has been hypothesized that bile acid (BA) metabolism may also play an important role in the mechanism of dysbiosis-associated IBD. Within the intestinal lumen, bacteria play some role in enzymatic reactions that are responsible for the transformation of BA, the water-soluble, amphipathic end products of cholesterol metabolism, which includes the processes of deconjugation and dehydrogenation. BAs have many metabolic functions such as lipid absorption, maintaining cholesterol homeostasis, and act as anti-inflammatory molecules that are able to decrease proinflammatory cytokine synthesis in monocytes and macrophages. It has been found that patients with IBD exhibit the characteristic dysbiosis pattern and defective BA metabolism in the intestine, particularly during an IBD flare.14 Alterations in the gut microbiota have shown to affect BA metabolism because the microbiota and its associated metabolites mediate some alterations in BA metabolism.15 IBD-associated dysbiosis may alter the capacity for BA modification, which may disrupt the BA-regulated mucosal inflammatory process.16 Firmicutes and Bacteroides, the 2 major groups that are found to be decreased in the microbiota of patients with IBD, are the most potent deconjugating bacteria for BA transformation. With less of these bacteria in the intestine during dysbiosis, there is less conjugation of BAs, leading to increased intestinal inflammation, which could play a crucial role in the worsening of IBD. Therefore, the decrease in BA conjugation could potentially be a reliable marker for intestinal dysbiosis.
Obesity
Obesity is a disease that is on the rise in many countries around the world. According to the Centers for Disease Control and Prevention, in 2014, approximately 78 million adults and 12 million children are obese in the United States. Obesity is a metabolic disorder involving an excess amount of body fat storage that has been considered to be caused by an imbalance of energy, with low energy expenditure and increasing caloric intake. However, recent evidence suggests that obesity is a more complicated disease associated with intestinal dysbiosis in both mice and humans.17
Similar to IBD, a certain microbiota signature seems to be associated with the development of obesity. In obese individuals, there is an overall decrease in bacterial diversity in the intestines.17,18 In a majority of studies in both humans and animal models, obesity seems to be associated with an altered ratio between Bacteroidetes and Firmicutes, which shows a decrease in Bacteroidetes and an increase in Firmicutes.19,–21 This ratio is correlated with body weight and body fat accumulation, demonstrating that the more obese individuals have a more disproportionate ratio of these bacteria. The amount of Bacteroidetes seems to be important in obesity since obese individuals on a caloric-restricted diet with a loss in body weight show that there is an increased ratio of Bacteroidetes species in the intestinal microbiota.22,23 Although the debate about whether dysbiosis is a causal element/factor or just an effect of obesity is still under debate, many experiments involving germ-free (GF) mice suggests that dysbiosis is likely to be a causal factor of obesity. In these experiments, GF mice and wild-type (WT) mice were fed the same high-fat diet (HFD), however only the WT mice developed obesity. When the obese microbiota was transplanted into the GF mice, obesity was then induced.20,24,25 In this same way, it was also found that human obesity could be transferred into lean GF mice via a microbiota transplant.18 Microbiota transplant works in the opposite direction as well, it has been discovered that lean mice microbiota, when transferred into obese mice, can ameliorate symptoms of metabolic syndrome.26 These numerous studies indicate that the intestinal microbiota is closely associated with the development of obesity.
HFDs are shown to alter the microbiota composition, which will be discussed in-depth in the next section. The changes in the microbiota observed in obesity are believed to cause various effects that could promote chronic inflammation-associated obesity. The obese microbiota signature shows an altered ratio of Bacteroidetes and Firmicutes, which contain members that produce short-chain fatty acids (SCFAs). In obese individuals, the levels of SCFAs are significantly decreased compared with lean individuals. The decrease of SCFAs may be due to increased SCFA absorption and the altered composition of intestinal microbiota, which shows a decrease in butyrate-producing bacteria,18 which would cause less SCFA production. SCFAs are believed to inhibit the accumulation of fat in adipose tissue, so a decreased level is thought to contribute to obesity.18
Along with altered SCFA production, the altered obese microbiota has been hypothesized to affect the immune system balance of the host, which could lead to increased bacterial antigen translocation, resulting in chronic inflammation and impaired metabolic functions such as insulin resistance. In both human and animal models, obesity is associated with increased intestinal permeability, which causes decreased intestinal barrier function.17 This decrease in function may cause the passing of molecules like lipopolysaccharides (LPS), which are lipoglycans consisting of a lipid and a polysaccharide and are commonly found in the outer membrane of gram-negative bacteria. LPS functions as an endotoxin by protecting the bacteria from bile salts and lipophilic antibiotics. In addition, LPS can cause endotoxemia, which is the presence of endotoxins in the blood, which could result in septic shock and increased chronic inflammation. LPS is also believed to play a key role in the pathogenesis of the chronic inflammatory state seen in obese individuals. Many studies support the idea that obesity-associated dysbiosis may cause the levels of LPS to increase and cross the “leaky” epithelial barrier into the bloodstream, causing endotoxemia, which leads to the chronic inflammation that drives the obese state.27
Diabetes Mellitus
According to the American Diabetes Association, in 2012, 9.3%, or approximately 29.1 million, Americans had diabetes. This disease is on the rise in the United States, with approximately 1.7 million new diagnoses per year. Diabetes mellitus is a disorder of carbohydrate metabolism that is characterized by inadequate production or utilization of insulin, which is needed to convert sugars and starches into energy for the body to function. There are 2 main types of diabetes: type 1 and type 2. Type 1 diabetes mellitus, or insulin-dependent diabetes mellitus (IDDM), is an autoimmune disease usually diagnosed in children or young adults and is caused by an impairment of insulin production by the beta cells in the pancreas. Type 2 diabetes mellitus, or non–insulin-dependent diabetes mellitus (NIDDM), is normally diagnosed in adults and is characterized by insulin resistance, which is when there is decreased tissue sensitivity to insulin, causing the body to not respond to the insulin that is being produced. NIDDM can be exacerbated by obesity and is also the most common form of diabetes diagnosed. Although IDDM and NIDDM are caused by different mechanisms in the body, studies have found that both types have dysbiosis of the intestinal microbiota, which may contribute to the pathogenesis of the disease.28
IDDM-associated dysbiosis is characterized by a decrease in mucin degrading bacteria, Bifidobacteria, Lactobacillus, and Prevotella, with an increase in Bacteroidetes and Clostridium.29,30 In contrast, NIDDM-associated dysbiosis is characterized by a decrease in Clostridium, an increase in Lactobacillus, and an increase in Bacteroidetes in NIDDM not associated with obesity.31 Both IDDM and NIDDM are associated with a decrease in overall microbial diversity, including a decrease in butyrate-producing bacteria and Firmicutes, as well as a breakdown in intestine epithelial barrier integrity and increased gut permeability.28,31,–33 Increased LPS translocation and endotoxemia are also seen in NIDDM, which, like in obesity, may contribute to the low-grade inflammation that contributes to the development of insulin resistance, which is associated with NIDDM.27
Like in other diseases associated with intestinal dysbiosis, it is unclear if the unbalanced microbiota is a causative factor or an effect of diabetes; however, various human and animal model studies suggest that changes in the microbiota may precede the onset of IDDM. In mice, it has been found that nonobese diabetic (NOD) mice with diabetes already at weaning age have a different microbiota composition than the NOD mice that do not develop diabetes.34 It was also observed that the prevalence of IDDM was dependent on the overall bacterial environment that they were kept in. NOD mice in GF facilities develop disease, whereas mice in specific pathogen-free (SPF) facilities do not.29 When gut microbiota from the SPF housed mice is transferred into the GF housed mice, the diabetes is attenuated.30 The reasoning behind these results is due to reduced expression of MyD88 (myeloid differentiation primary response 88), a universal adaptor protein for almost all Toll-like receptors. MyD88−/− NOD mice in SPF conditions were shown to have a change in the gut microbiota composition and subsequent loss of diabetes mellitus development compared with those in GF conditions. This shows that development of diabetes mellitus is dependent on the commensal bacteria composition in the intestines.30 MyD88 is also vital for bacterial sensing and signaling of the innate immune response, and it is hypothesized that the silencing of this factor may interrupt IDDM development, suggesting that microbial antigens and the innate immune system are linked to the pathogenesis of IDDM.30
Autism Spectrum Disorders
Autism spectrum disorders (ASD), are a group of disorders that include autism and Asperger's syndrome that are characterized by social and communication deficits, repetitive behaviors, and sometimes, cognitive delays.35 The exact cause of ASD is still largely unknown; however, there are strong implications that intestinal dysbiosis may play a role in the pathogenesis of ASD, particularly in autism. This link between ASD and dysbiosis is strengthened by various studies indicating that the gut microbiota and its metabolites seem to affect the central nervous system via the gut–brain axis, which may communicate to the central nervous system through neural, endocrine, and immune pathways to influence brain functions and altered behavior.36 Multiple experiments in mice models have shown that altered gut microbiota can cause the synthesis of neurotoxins, which may interfere with neurodevelopment, causing changes in the brain chemistry and behavior. Subsequently, these dysbiosis-associated neuronal alterations result in causing behavioral changes such as increased anxiety, depression, and cognitive dysfunction, all of which are characteristic features in ASD.35 Experiments using selected antibiotics have shown that probiotics believed to help the normalization of the intestinal microflora also tend to reduce anxiety and improve cognitive functions and behavior,37,38 which shows that the microbiota plays an important role in ASD development.
In children with autism, there is a strong correlation between disease severity and gastrointestinal disorders.39 Common gastrointestinal problems in autistic children include abdominal pain, diarrhea, abdominal bloating, intestinal dysbiosis, and increased intestinal membrane permeability.39 Investigating dysbiosis-associated ASD, multiple studies found that there was a decrease in Firmicutes, and beneficial bacteria such as Bifidobacteria and Prevotella, with an increase in Bacteroidetes and potentially pathogenic bacteria such as Proteobacteria and Clostridiales.38 In many studies, the increase in Clostridiales bacteria, presumably some selected clusters of Clostridia, seems to play an important role in the development of autism. A strongly proposed mechanism in which Clostridiales may help propagate autism pathogenesis is the production of propionic acid, one of the members of SCFAs, which can cross the physiological barrier between the gut and the blood and permeate into the blood–brain barrier, where it can alter functions in several parts of the brain, causing cognitive impairments and symptoms normally attributed to autism.40
Cancer
Intestinal dysbiosis has also been linked to CRC, which is the third most common cancer and second leading cause of cancer death (both sexes combined) in the United States according to the American Cancer Society. CRC has several risk factors, including IBD, obesity, diabetes, and a diet consisting of high fat and protein, all of which also are linked to intestinal dysbiosis. This suggests that dysbiosis may also play a key role in CRC pathogenesis.
A general dysbiosis pattern has been found in patients in CRC that involves a decrease of butyrate-producing bacteria along with an increase in the proportion of several potentially pathogenic bacteria. In various literature sources, it was found that there is a decrease in Proteobacteria, Bifidobacteria, Prevotella, and reduced SCFA production rates, whereas there is an increase in Firmicutes, Bacteroidetes, Enterobacteriaceae, and Fusobacteria.41 Various studies have also shown that 2 specific species of bacteria, Akkermansia muciniphila and Fusobacterium nucleatum, are increased in CRC tissues.42 Both these bacterial species are associated with strong local inflammatory responses, which may be involved in inflammation-associated diseases and would be representative of a high risk of CRC. F. nucleatum is also associated with increased amounts of CRC tumors and lymph node metastasis.42 It was also found that the composition and luminal numbers of dominant microbial species seen in CRC-associated dysbiosis differs depending on disease severity and tumor stage/status. Significant differences were seen in both mucosal and fecal microbial compositions between patients in CRC with polyps and those with tumors, with the most significant change being Enterobacteriaceae, which was increased in the mucosa of patients in CRC with tumors compared with those patients with polyps, and Bacteroidetes, which was increased in CRC tissues with tumors than those without tumors.43
There is strong preclinical experimental evidence indicating that dysbiosis plays a causal effect in CRC pathogenesis and tumorigenesis in CRC-susceptible patients. When gut microbiota from mice with tumors was transplanted into C57BL/6 mice in a GF facility, the rate of tumorigenesis in the colon increased significantly.44 The manipulation of the intestinal microbiota with antibiotic treatment also demonstrated the importance of the microbiota in tumor status, showing a decrease in number and size of tumors in CRC-susceptible mice when antibiotics were administered.41,44 Experiments in MyD88-deficient mice showed a reduction of tumor progression and neoplastic development,41 and suggest that MyD88 may transmit a carcinogenic signal that could be one of the major causal factors in CRC tumorigenesis.
The experiments involving the GF mice as well as the antibiotic experiments show that changes in the gut microbiota during dysbiosis are associated with tumorigenesis and strongly seem to contribute to this process. Various studies investigating the role of dysbiosis in CRC development have suggested multiple mechanisms of how this may occur. One such mechanism involves the absence of SCFAs, which are significantly reduced in the fecal microbiota during the course of CRC development.41 When butyrate was given as a supplement to HFD-fed genetically susceptible K-rasG12Dint mice, the incidence of tumors significantly decreased,41 suggesting that this SCFA is important in protection against tumorigenesis. Another suggested mechanism involves the overproduction of α-defensins, which are small, cysteine-rich, cationic proteins associated with innate immunity that are important for protection against pathogens, including many gram-negative and gram-positive bacteria, fungi, and enveloped viruses, as well as involved in cell signaling and shaping of the intestinal microbiota. In sporadic patients with CRC with adenomatous polyps, the overproduction of α-defensins may cause dysbiosis, because there is a correlation involving a decrease in beneficial mucosa-adherent bacteria, including Bifidobacterium infantis, along with an increase in α-defensin production in these patients.45 This dysbiosis may lead to a decrease in regulating colonic epithelial proliferation, which is one of the precarcinogenic factors, indicating a potential role for dysbiosis in CRC development. The altered microbiota metabolism present in dysbiosis is also thought to release toxic metabolites, which may be associated with neoplastic changes in intestinal epithelial cells. Intestinal dysbiosis is believed to contribute to exacerbating chronic inflammation, which is thought to be the leading cause of colitis-associated CRC development.46 It has been suggested that an increased immune response toward the antigens of the dysbiotic gut microbiota may lead to this chronic, potentially CRC-causing inflammation.47
Diet and the Microbiome
Diet is one of the most important influences on homeostasis of the gut microbiota, and is important from birth, as indicated by studies showing different gut microbial compositions in breast-fed versus formula-fed infants.48 Specific foods and dietary components have been investigated to determine the type of effects they have on the whole organism. For years, many researchers across the world have found an effect between diet and dysbiosis-associated diseases such as obesity, diabetes, and allergic disorders; however, recent research shows that diet may also impact diseases like IBD and certain cancer types, such as CRC.
When comparing the “Western” diet, which mainly consists of animal protein and high in sugar and saturated fat, with an agrarian diet that mainly consists of low animal protein and saturated fat and high carbohydrates and simple sugars, the composition of the intestinal microbiota showed drastic differences: When analyzing the microbiota of people consuming the Western diet, there was an increase in Firmicutes and Proteobacteria as well as some Bacteroides, whereas people consuming the agrarian diet had increased amounts of Actinobacteria, mostly Prevotella.49 In mice, it was found that a high-fat/sugar diet results in dysbiosis in those with altered host barrier function.3 In people consuming the agrarian diet, there was an increase in SCFA levels and reduced SCFAs in people consuming the Western diet.49 SCFAs, or volatile fatty acids, are synthesized in the colon during the process of dietary fiber fermentation. The most common SCFAs include acetic, propionic, and butyric acids. SCFAs, in particular, butyric acid, are thought to be protective against the establishment of potentially pathogenic intestinal bacteria, which indicates that SCFAs may be a regulatory factor in the origin of dysbiosis. In summary, diets high in fiber are shown to have a higher gut microbial diversity, mostly of commensal bacteria, which limits the colonization of potentially pathogenic bacteria, which have been associated with various diseases such as IBD and CRC.
Recent studies have shown that vitamin D is a probable chemoprotectant, and has been shown to decrease the risk of IBD and colitis. In mice with dextran sulfate sodium-induced colitis, administration of vitamin D was shown to cause a better recovery of weight as well as less bloody diarrhea.50 Vitamin D promotes epithelial cell resistance to injury and suppresses inflammatory responses, which is important in patients with IBD.50 Vitamin D is also believed to be a factor in regulating gut bacterial homeostasis. Studies in mice of both vitamin D and vitamin D receptor deficiency have shown composition changes in the intestinal microbiota, with a decrease in Lactobacilli and Firmicutes and an increase in Clostridium and Bacteroidetes in the fecal stool compared with WT mice.51,–54 Lactobacilli are gram-positive, fermentative bacteria that produce lactic acid, which has anti-inflammatory and antitumorigenic effects by balancing intestinal homeostasis.51 Vitamin D deficiency, with a decrease in lactic acid producing bacteria and an increase in potentially pathogenic bacteria, may increase the risk of chronic inflammation and the following colitis-associated CRC development. Vitamin D has also shown, in mice, to protect against pathogenic bacterial colonization by maintaining the intestinal epithelial barrier function.52,53 In vitamin D–deficient mice, intestinal epithelial barrier dysfunction occurs, which allows pathogenic bacteria such as adherent-invasive E. coli to colonize the gut, causing dysbiosis and allowing for more dysbiosis-associated inflammation to occur, which increases the risk of CD.52,54
High consumption of whole grains and dietary fiber show strong inverse correlations with CRC risk, along with reducing inflammation, and reducing the risk for other diseases such as cardiovascular disease, diabetes, advanced CRC, and obesity-related diseases.55 In 2011, the World Cancer Research Fund/American Institute for Cancer Research concluded that fiber was “probable” to decrease colorectal neoplasia, with fiber being inversely proportional to CRC.56 Whole grains are rich in phenolic compounds, the most abundant being ferulic acid (FA). The fiber in whole-grain foods delivers the FAs to the gut, where they are slowly and continuously released by the gut microbiota, which also convert the FAs into circulating dihydroferulic acid.55 FA and dihydroferulic acid have been considered to be protective against neoplastic changes in colonic epithelial cells,57 as well as an increase in immune response and have been shown to possibly decrease the risk of obesity-related diseases when whole grains are consumed long-term. The increased FA may directly have an antimicrobial effect toward potentially pathogenic bacteria.55 Along with these acids, whole-grain consumption has been shown to decrease tumor necrosis factor α production along with increasing Bacteroides and Lactobacillus, which helps to ameliorate subclinical inflammation.55
A decrease in fiber is associated with a higher risk of advanced CRC.58 Patients with advanced CRC were shown to have decreased butyrate-producing bacteria in the gut microbiota, which causes less SCFA fermentation in the gut, which may lead to the formation of advanced CRC. In contrast, increased fiber intake is shown to have an increase in butyrate-producing bacteria and an overall balanced gut microbiota compared with individuals with decreased fiber, indicating fiber's important role in decreasing CRC risk and its role in maintaining gut microbial homeostasis.56,58 Butyrate, one of the most important SCFAs, has been shown to have an antitumorigenic effect by inhibiting proliferation of neoplastic cells, restricting tumor angiogenesis, and inducing apoptosis.59 Diets high in whole grains and fiber may influence the gut microbiota at the phylum level, specifically causing a decrease in Bacteroides when consumed long term and increasing Bifidobacteria and Roseburia, both of which are butyrate-producing bacteria suggested to influence immunity and inflammatory functions in animal models. In studies investigating the roles of high-protein/low-carb versus high-protein/moderate-carb diets, the presence of fiber and nondigestible carbohydrates contribute to the prevention of CRC development.57 This is believed to be in part due to increased SCFA fermentation, which can affect the colon by promoting mucus secretion and modulating the inflammatory immune response. These studies also showed that high-protein/moderate-carb diets modulated potentially carcinogenic N-nitroso compound (NOC) formation, which was previously believed to only be affected by the increased consumption of meat and protein; however, these studies show that the magnitude of the NOC effects depend on the amount of carbohydrates ingested; with increased carbohydrate intake, there is less NOC formation and therefore a decreased risk of CRC formation. Some studies have shown that there may be sex differences in the effect of dietary fiber on decreasing colorectal neoplasia risk, showing that men may benefit from the effect of fiber more than women. It is suggested that in women, fiber decreased estrogen, which may be protective against neoplastic changes of epithelial cells.60 Another hypothesis as to why there may be sex differences on the effects of fiber is that women are more prone to developing pure right-sided (proximal) polyps than men. If fiber is more effective in preventing left-sided (distal) polyps, this could explain why these differences occurred.60,61
An increase in fruit and vegetable fiber consumption has been shown to be chemoprotective and is associated with increased gut microbial diversity and decreased CRC risk.56 People with a diet high in vegetables have been shown to be less likely to develop IBD as well as advanced CRC risk.58,59 Most research shows that vegetables are inversely proportional to cancer risk; however, some studies show that only cooked leafy vegetables have this effect, but not all vegetables.62 There is also a sex difference seen in the consumption of fruit and vegetables, with these 2 food groups being inversely proportional to CRC risk in men but not women.62 The consumption of red wine has also been studied to show that its consumption can significantly modify the gut microbial diversity in humans, showing increased probiotic bacteria such as Prevotella, Proteobacteria, and Bifidobacteria and decreased potentially pathogenic bacteria like Clostridium,63 which is the result of the polyphenols present in the wine. Polyphenols are present in many foods such as wine, tea, fruit, vegetables, and chocolate and usually arrive at the colon intact, where they can have regulatory functions and antimicrobial properties that modify the gut microbiota.63 Along with decreasing cancer risk, polyphenols are also associated with lowering blood pressure and cholesterol levels, as well as decreasing the risk for cardiovascular disorders including cardiovascular disease, stroke, and aortic aneurysm.
Genetics and Dysbiosis
Along with environmental factors such as diet, genetic components are also strongly associated with the development of dysbiosis. This is most prevalent in dysbiosis that is associated with CD. Using genome-wide association studies, there are now more than 160 IBD-associated loci; however, this review only focuses on variants of interleukin (IL)-10/IL-10R, which have been associated with early-onset IBD, as well as variants of NOD2 and ATG16L1, the 2 most studied IBD susceptibility genes and their associated proteins that have been found to increase the risk of CD through altered interaction with the gut microbiota.
Nucleotide Oligomerization Domain 2
The first identified CD susceptibility gene, nucleotide oligomerization domain 2 (NOD2), also known as NOD2/CARD15 (CAspase Recruitment Domain-containing protein 15) is located on human chromosome 16q12 and synthesizes NOD2 protein, which acts as a type of pathogen-associated molecular pattern and specifically recognizes muramyl dipeptide, which is a component of the bacterial peptidoglycan cell wall present in both gram-positive and gram-negative bacteria. NOD2 is expressed in many types of cells including dendritic cells, macrophages, Paneth cells, as well as intestinal epithelial cells, and epithelial cells in the lungs and oral cavity.64 NOD2 protein is important in innate immunity and microbial regulation, where it is involved in pathogen recognition and defense against these organisms.65 Muramyl dipeptide binds and activates NOD2, which then is localized to the plasma membrane and initiates signaling cascades by recruiting receptor-interacting protein 2, followed by activating the mitogen-activated protein kinase and nuclear factor kappa B cascades, resulting in the release of proinflammatory molecules to help kill the pathogenic bacteria.66 NOD2 is also needed for expression of defensins by specialized epithelial cells called Paneth cells, which are located in small intestinal crypts.65 Furthermore, NOD2 is important in inducing autophagy in epithelial cells by guiding ATG16L1 (autophagy-related 16-like 1) protein to the plasma membrane to induce autophagosome formation66 (Fig. 2). Functioning NOD2 is essential for both gram-positive and gram-negative bacterial regulation and helps to prevent pathogenic bacteria colonization and dysbiosis.
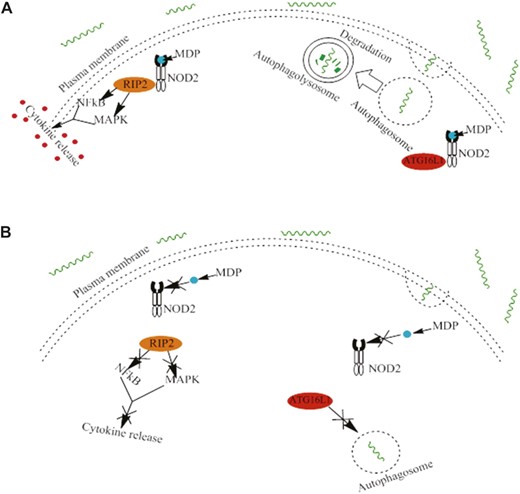
Normal and mutated NOD2 functions. A, Normal functions of NOD2. The normal NOD2 variant is activated by muramyl dipeptide, which is a component of bacterial cell walls. Activated NOD2 recruits receptor-interacting protein 2 (RIP2), which then activates the mitogen-activated protein kinase (MAPK) and nuclear factor kappa B (NF-κB) cascades. This results in the release of proinflammatory molecules to help kill pathogenic bacteria (left). Activated NOD2 also guides autophagy-related 16-like 1 (ATG16L1) protein from the cytoplasm to the plasma membrane to initiate autophagosome formation (right). B, Mutated variants of NOD2 cause impairments in sensing and recognizing muramyl dipeptide. Without this activation, RIP2 and the resulting cytokine release does not occur (left). Without active NOD2, ATG16L1 is not guided to the plasma membrane and remains in the cytosol, which impairs autophagosome formation and results in impaired killing of invading bacteria (right). NOD2 variants and their impaired cellular functions may lead to dysbiosis within the intestinal epithelium, and are associated with earlier onset of ileal CD.
There are 3 NOD2 gene variants that are associated with increased risk for CD susceptibility: 2 missense mutations (R702W and G908R) and 1 frameshift mutation (L1007fsinsC).66 These variations alter the leucine-rich repeat (LRR) domain or the neighboring region, which causes a significant decrease or complete loss of NOD2 function.67 NOD2 is important in regulating the gut microbiota in the intestine by regulating colonization and suppressing opportunistic pathogens. NOD2 WT mice have shown to clear pathogens like Helicobacter hepaticus quickly; however, in NOD2 knockout mice, the clearance of this pathogen was delayed and the number of H. hepaticus in the terminal ileum was significantly higher than in the WT mice.68 Without NOD2, sensing and recognition of bacterial muramyl dipeptide is impaired and these bacteria cannot be neutralized, so they continue to grow in the gut, causing dysbiosis. This dysbiosis alters the host–microbial interactions in the ileal mucosa, leading to an increased inflammatory response that may lead to CD. In humans, NOD2 mutations, in particular the frameshift NOD2 mutation, are strongly associated with decreased α-defensin expression in Paneth cells, which would impair its antibacterial function, increasing the risk of infection, and resulting in more CD-associated inflammation.64 NOD2 mutations can also impair autophagy: The frameshift NOD2 variant (L1007fsinsC) keeps the ATG16L1 protein in the cytoplasmic compartment and never localizes to the plasma membrane for initiation of autophagosome formation.66 People who are homozygous for 1 of the 3 major NOD2 variants (R702W, G908R, and L1007fsinsC) have a 20-fold increase in CD risk; however, less than 20% of patients with CD are homozygous for a NOD2 variant,69 which shows that NOD2 variants are not the sole causal factors of CD. However, these 3 NOD2 variations are associated with the occurrence of fibrostenotic CD of the small intestine.70
NOD2 and the commensal bacteria have a regulatory relationship with each other. In GF mice, expression of NOD2 in the terminal ileum was lower compared with SPF mice.71 After the GF mice were reconstituted with commensal bacteria, the expression levels of NOD2 in the ileum significantly increased, which indicates that commensal bacteria positively regulate NOD2, whereas NOD2 function negatively regulates the commensal flora.71 Without this relationship, the homeostasis in the gut, or dysbiosis, breaks down. NOD2 knockout(−/−) mice have shown to have dysbiosis in the terminal ileum, with an increase in commensal bacteria along with an increase in susceptibility to pathogenic bacteria. This is because of the inability to regulate the commensal bacteria and prevent pathogenic bacteria colonization. In NOD2−/− mice, an altered gut microbiota was seen in the terminal ileal mucosa, mostly consisting of an increase of Bacteroides and Firmicutes compared with NOD2 WT mice, which had barely detectable amounts of these bacteria genera.68,72 These data also correlates to what was seen in humans containing NOD2 deficiencies.60 Mice with CD-associated NOD2 variants have also been shown to have increased numbers of Enterobacteriaceae.73 There is also an allele-dosage effect that is seen with NOD2 deficiency, with a significant correlation between the number of NOD2 risk alleles and the increased amount of Enterobacteriaceae present.73 Patients with CD who have NOD2 mutations have shown an ineffective recognition of M. avium paratuberculosis, which is a pathogenic bacteria that has been found in a significant minority of patients and may be associated with granuloma formation.74
ATG16L1
The ATG16L1 gene encodes for the ATG16L1 protein, which is essential for autophagy and autophagosome formation.66 ATG16L1 is expressed in the intestinal epithelium, antigen-presenting cells, T cells, and B cells.65 It is normally found in the cytosol until it is recruited by NOD2 and guided to the plasma membrane, where it recruits 1A/1B-light chain 3 (LC3) for autophagosome formation initiation.66 ATG16L1 has also been indicated to play a role in both mice and humans by regulating granule secretion in Paneth cells, which can affect the gut microbiota composition.75
A single-nucleotide polymorphism in the ATG16L1 gene that changes a threonine to alanine (T300A) causes a decrease in or lack of ATG16L1 protein expression.66 In both humans and mice homozygous for the risk allele, there were no morphology abnormalities in the ileum, but abnormalities in Paneth cell granule secretion were displayed.75 With ATG16L1 depletion, Paneth cells had significantly decreased numbers of antibacterial granules that it normally secretes, which causes impairment in microbiota regulation in the gut and can lead to CD-associated dysbiosis. A decrease or lack of ATG16L1 also causes a defect in the role NOD2 plays in autophagosome formation, which is ATG16L1 dependent for this process. Without ATG16L1, a lack of bacterial-infected cytoplasmic autophagy occurs, giving the invading bacteria an opportunity to overcolonize and result in dysbiosis in the gut epithelia that can lead to inflammation and an increased risk of developing CD.
Like NOD2 polymorphisms, ATG16L1 variants are associated with impaired bacterial clearance through autophagy against intracellular bacteria and also have an allele-dosage effect on the intestinal microbiota.76 ATG16L1 deficiency has been shown to be associated with an increase in susceptibility to Helicobacter pylori strain s1m1, which leads to chronic inflammation that may create a procarcinogenic environment.77 It was also seen that in ATG16L1-deficient cells, there was a significant increase in the adherent-invasive E. coli LF82 strain, which was isolated from the ileocecal lesions with active inflammation in CD patients.78 Other work has shown that the decrease in autophagy of ileal bacteria in ATG16L1-deficient cells is not pathogen specific.76,78 Of note, ATG16L1 deficiency is associated with an increased invasion of opportunistic pathogens, some of which that have been tested are Mycobacterium tuberculosis and Streptococcus pyrogenes.78 The increased amounts of intracellular pathogenic bacteria causes an imbalance in the gut microbiota, this dysbiosis then can lead to chronic inflammation, which increases the risk of developing diseases such as IBD and can become a carcinogenic environment.
Interleukin-10/Interleukin-10R
IL-10 is an anti-inflammatory cytokine that is secreted mostly by monocytes/macrophages, but can also be produced by lymphocytes such as Th2 cells and some regulatory T cells/B cells. IL-10 expression is important for mucosal immune homeostasis within the gastrointestinal tract of both mice and humans, where it initiates a cascade to ultimately restrict excessive immune responses and intestinal inflammation. This cascade is initiated when IL-10 binds to IL-10R (IL-10 receptor), which is a protein that comprises 2 alpha (IL-10RA) and 2 beta (IL-10RB) subunits. When IL-10 binds to these receptors, activation of JAK1 and TYK2 leads to the phosphorylation of STAT3, which then moves into the nucleus and downregulates proinflammatory gene expression.79 This cascade is important for restricting excessive Th17 (T helper 17) cells in the mucosa, of which an excessive expansion has been associated with CD.79,80 IL-10 also restricts the secretion of proinflammatory cytokines, such as tumor necrosis factor α and IL-12, which contribute to harmful inflammation in the intestines.81
Genetic mutations in the IL-10 and IL-10R genes result in loss-of-functional mutations in IL-10 and IL-10R proteins, respectively, and have been associated with early-onset IBD (EO-IBD), which can affect children ranging from as young as a few months old to 18 years. Along with IBD and colonic inflammation, patients with EO-IBD may also develop perianal disease, respiratory diseases, arthritis, and may have abscesses, fistulae, and ulcers of the intestinal mucosa.82,83 There have been a wide variety of mutations of the IL-10R protein subunits reported, ranging from amino acid substitutions, deletions, point mutations, and frameshift mutations resulting in a premature stop codon.79,83 These mutations cause a defect in the IL-10-mediated anti-inflammatory signaling cascade in both IL-10R−/− mice and humans, which resulted in severe intestinal inflammation and increased proinflammatory cytokines, most notably an increase in tumor necrosis factor α, from peripheral blood mononuclear cells.81,84 The lack of an anti-inflammatory response and the subsequent increase in proinflammatory cytokines is especially severe in the intestine, where the presence of commensal bacteria leads to a large unregulated immune response, which causes a hyperinflammatory state and leads to the development of EO-IBD.79,–81,83 The loss of the IL-10 signal cascade on innate cells also can impair the cross talk with T cells, which leads to further mucosal immune imbalance and contributes more to intestinal inflammation.80
Patients with EO-IBD usually do not respond to immunosuppressive therapies such as corticosteroids and thalidomide81,82; however, because the defects in patients with IL-10/IL-10R pathway is in hematopoietic lineage cells, allogenic hematopoietic stem cell transplantation has been the choice option for curative treatment. From both familial as well as unrelated donors, hematopoietic stem cell transplantation has so far shown to be successful, with patients with EO-IBD going into remission.81,82 This treatment option seems to be promising; however, one review indicated that the primary graft rejection rate was high, and suggests that patients with EO-IBD have a regimen of T-cell replete haplo-identical bone marrow transplantation with posttransplant cyclophosphamide to reduce the rate of rejection.83
Symbiotic Intervention
The significant progress in recent years in understanding host-microbial cross talk, microbiome profiles/signatures, and molecular techniques to study gut microbiota have facilitated the design and administration of interventions to reverse the microbiota-mediated ill effects during disease state. Classical interventions include compounds such as diet (as described in above section) and antibiotics that have direct effects on the gut microbiome homoeostasis. Alternatively, one can also introduce exogenous bacteria, directly or indirectly, to manipulate and alter endogenous gut bacterial flora in the host. These may come in the form of probiotics, fecal microbiota transplantation, and environmental changes in bacterial flora that create a symbiotic intervention with their own advantages and disadvantages (Fig. 3). They can have profound effects on the host and the gut microbial community such as (1) facilitating biofilm formation by enhancing microbial coaggregation, (2) production of bacteriocins that have selective antimicrobial effects, (3) host immunity stimulation, and (4) enhancement of gut barrier function though host epithelial cells.
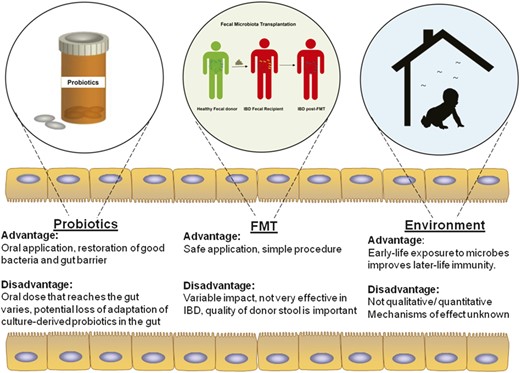
Advantages and disadvantages of different symbiotic interventions of gut microbial flora. Exogenous bacteria can influence the dysbiotic gut to achieve restoration of a healthy flora. These exogenous bacteria can be introduced in the form of probiotics or fecal microbiota transplantation that has their own advantages and disadvantages. Microbes present in the environment can also alter the endogenous gut microbiome composition and confer disease susceptibility or protection.
Probiotics
Probiotics are live, mostly gram-positive, bacteria (e.g., Bifidobacterium spp., Lactobacillus spp., Lactococcus spp., Pediococcus spp. and other nonpathogenic strains of E. coli). They generally promote intestinal barrier integrity, prevent bacterial translocation in the gut and reduce inflammatory response. However, it was suggested that the effects of probiotics may be transient, because it was shown that administration of Lactobacillus plantarum can result in an increase in the amount of Lactobacillus in the feces over time, but not in the intestinal biopsy.85 Nevertheless, although transient, in vivo human response showed that the effects of L. plantarum can be dramatic in the host to establish immune tolerance by changing the intestinal mucosa gene expression profile, mainly involving a NF-kB-dependent pathway.86 In addition to changes in host gene transcriptome, fecal pyrosequencing of healthy individuals given probiotics (3 probiotic strains: Lactobacillus rhamnosus CNCM I-4036, Lactobacillus paracasei CNCM I-4034, and Bifidobacterium breve CNCM I-4035) was reported to alter general gut flora abundance under healthy conditions.87
In disease context, probiotics in the form of fermented milk can induce weight gain in individuals with severe acute malnutrition by modulating the endogenous gut microbiota.88 This will be beneficial in developing countries with high risk of malnutrition, especially in children. However, administration of other certain strains of probiotics can have antiobesity effects. For example, L. plantarum and L. gasseri consumption will lead to weight loss through the suppression of several hormonal pathways.88 This means that different probiotics strains can confer different effects and thus the type of bacteria to be supplemented have to be carefully characterized and chosen for the desired effect. With that, Shen et al89 recently demonstrated a proof of concept, in which a defined consortium of 8 bacteria (altered Schadler flora) can reduce hyperammonemia-associated neurotoxicity and encephalopathy. These altered Schadler flora possess minimal urease effect, and colonization of this consortium promoted the establishment of a new community of bacteria that promotes long-term reduction in urease activity and ammonia production. This results in reduced ammonia and carbon dioxide production originating from the metabolism of host-derived urea by bacterial-derived urease. Similarly, in intestinal inflammation, a 5-bacterial strain milk product was reported to improve the degree of colitis in T-bet−/− x Rag2−/− mice by changing endogenous gut bacterial composition.90 Recent follow-up studies by the same group subsequently identified Lactococcus lactis I-1631 alone is sufficient to ameliorate colitis as tested in 3 preclinical mouse models of colitis.91 Colonization of L. lactis in the gut is not necessary to improve colitis, but rather host lysozyme-mediated lysis during colitis leads to the release of superoxide dismutase by L. lactis that reduces oxidative stress during inflammation. This suggests that bacteria can provide benefits to the host but will require certain host-derived factors to exert these beneficial effects.
The disadvantage of probiotics is a more quantitative. Oral doses of probiotics are significantly much less (typically 3- to 4-folds) than those present in the colon and the amount that will eventually reach the colon will further reduce on passing through the harsh conditions of the stomach and small bowel. In addition, most strains of probiotics used are derived and originated from the gut (e.g., Lactobacilli, Bifidobacteria and Escherichia). These strains may have lost their adaptation to the gut environment during ex vivo cultivation.
Fecal Microbiota Transplantation
Fecal microbiota transplantation (FMT; aka fecal bacteriotherapy or fecal infusion) is an emerging method to treat patients with dysbiosis. The method uses the principle of engrafting the microbiota from healthy donors into a patient recipient to re-introduce or reestablish a stable environment that influences both the endogenous microbes, as well as the host. The relationship between the donors and patient does not seem to affect the outcome.92,93 FMT was originally performed through fecal enemas, subsequently also through nasoduodenal tubes (1991) and colonoscopy administration (2000). Recent reports have demonstrated the development of capsulated frozen inoculum that can be administered orally with no apparent side effects and thus avoids unnecessary invasive gastrointestinal procedures (2012).94
Notably, the best example of how FMT can ameliorate disease is in the case of C. difficile infection (CDI), with a cure rate exceeding 90% to 95% worldwide.95 Patients with CDI usually will remain on antimicrobials until 2 to 3 days before FMT. Donor stool, commonly processed and used within 8 hours of passage, is suspended in nonbacteriostatic saline and filtered to remove large particulate matter. Post-FMT results in the expansion and restoration of bacteria derived from the donor in the patient within 2 weeks to 1 month.96 Currently, FMT is being considered for other diseases such as insulin resistance, and IBD.26,97 Patients with IBD receiving FMT may develop fever and temporary reduction in C-reactive protein.98 Temporal characterization of FMT in patients with IBD showed a shift toward donor phylotypes in the recipient, including Faecalibacterium prausnitzii, Rosebura faecis, and Bacteroides ovatus, which produce anti-inflammatory and/or short-chain fatty acids. This effect may kick in 3 days after transplantation and may remain for up to 12 weeks.98 Such time-resolved analysis of microbial profile post-FMT can thus be used to determine colonization efficacy and for monitoring FMT success rate.
However, the success rate of FMT on IBD is much less than those compared with CDI, with 62% to 71% of patients with IBD achieving resolution or reduction of symptoms.97 The data collected for FMT on IBD are usually performed on small cohorts and thus also lack standardized procedures, including donor selection and sample preparation. Another concern for FMT on IBD is the route of administration, which will depend on the genre of dysbiosis. For instance, Bacteroidetes may be destroyed by gastric acid and thus using the lower route of administration will be preferred when performing FMT. Conversely, transition through the upper gastrointestinal tract is required for certain spore-forming Firmicutes to be effective functionally.99 Other general concerns about FMT include safety, cost, ethical acceptance, and risks including the possible transmission of infectious agents.
Environmental Interventions
The environment has a profound impact on the gut microbiota in mediating general homeostasis that begins right after birth. The microbiome composition/profile of babies that were delivered by caesarean section was different from those babies born vaginally; the former are first exposed to bacteria from the hospital environment, whereas the latter are colonized at first by maternal fecal and vaginal bacteria.100 Caesarean-born babies have been shown to lack Bifidobacteria species that are important to postnatal immunity development compared with vaginally delivered babies who show predominance of these species.101
Postnatally, the “hygiene hypothesis” subsequently governs the development and changes of the gut homeostasis. It is based on the need for developing infants to be exposed to plenty of both pathogenic and commensal microorganisms for their immune systems to develop and function properly. This is evidently supported by many studies using animal models demonstrating how lack of microbial exposure during early life affects not just immunity development, but also controls the maturation and function of other organs including the central nervous system. Early-life exposure to microbes has persistent effects on invariant natural killer T cells and response to diseases such as IBD and asthma.102 In piglets, the environmental microbial diversity affects the numbers of CD4+, CD4+CD25+ effector T cells, and CD4+CD25+ Foxp3+ regulatory T cells, as well as the serum immunoglobulin G antibody response.103 The contribution of environmental microbes on disease development is also evident in many spontaneous models of IBD, such as in the T-cell receptor α chain (TCR-α)−/− mice that do not develop disease in GF conditions, but rather in SPF and CV environments.102 In addition to controlling immunity response, environmental microbes also affect host cell development and function. Recently, Erny et al104 reported that GF mice displayed global defects in microglia and morphology and disturb cellular networks. They determined that the microbial production of SCFA and microbiota fermentation products are present in SPF mice, but not GF, and are key determinants of this cause, because reintroduction of these components into GF mice can rescue the phenotype.
Later in life, the type of lifestyle can also promote environmental-associated dysbiosis. For example, intestinal microbiota of both humans and mice has been shown to exhibit diurnal oscillation that can be influenced by feeding rhythms or induction of jet lag. Jet lag induces dysbiosis in both human and mice and results in glucose intolerance and obesity.105 Transfer of the aberrant flora through FMT to GF mice will result in the same phenotype. Together, all these examples show that changing the environment microbial status can either promote disease or confer protection. Improving our understanding toward all these factors, as well as the establishments of major microbiome research groups such as the National Institutes of Health (NIH)-supported Human Microbiome Project in the United States and the MetaHIT (Metagenomics of the Human Intestinal Tract) Project established by the International Human Microbiome Consortium in Europe, will help better facilitate intervention design against dysbiosis.
Conclusions
This review shows that dysbiosis is a complicated disorder in the intestinal microbiota, i.e., strongly believed to play a role in the pathogenesis of IBD and other disorders like CRC and allergic disorders; however, future work must be done to confirm this hypothesis. Future researchers must also be aware of the various factors, such as genetics, diet, and environmental factors, which impact the formation of gut dysbiosis. Based on this knowledge, along with the continuing work of identifying the gut microbiota present in humans, future researchers should be able to come closer in successfully intervening against dysbiosis and its associated diseases.
Acknowledgments
The authors are grateful to Dr. Rita Kumari for her helpful discussions and advice.
References
Author notes
Reprints: Emiko Mizoguchi, MD, PhD, Department of Medicine, Gastrointestinal Unit, Massachusetts General Hospital, GRJ 825D, 55 Fruit Street, Boston, MA 02114 (e-mail: emizoguchi@mgh.harvard.edu).
Supported by the National Institute of Health (R01-DK80070, DK91247, AI81807), and grants from the Broad Medical Foundation and American Gastroenterological Association Foundation to E. Mizoguchi.
The authors have no conflict of interest to disclose.


{kind=link}
{kind=link}
{kind=link}