


Abstract
Bromodomain and extraterminal inhibitors (BETi) are promising cancer therapies, yet prominent side effects of BETi at effective doses have been reported in phase I clinical trials. Here, we screened a panel of small molecules targeting epigenetic modulators against human metastatic melanoma cells. Cells were pretreated with or without ascorbate (vitamin C), which promotes DNA demethylation and subsequently changes the sensitivity to drugs. Top hits were structurally unrelated BETi, including JQ1, I-BET151, CPI-203, and BI-2536. Ascorbate enhanced the efficacy of BETi by decreasing acetylation of histone H4, but not H3, while exerting no effect on the expression of BRD proteins. Histone acetyltransferase 1 (HAT1), which catalyzes H4K5ac and H4K12ac, was downregulated by ascorbate mainly via the TET-mediated DNA hydroxymethylation pathway. Loss of H4ac, especially H4K5ac and H4K12ac, disrupted the interaction between BRD4 and H4 by which ascorbate and BETi blocked the binding of BRD4 to acetylated histones. Cotreatment with ascorbate and JQ1 induced apoptosis and inhibited proliferation of cultured melanoma cells. Ascorbate deficiency as modeled in Gulo−/− mice diminished the treatment outcome of JQ1 for melanoma tumorgraft. In contrast, ascorbate supplementation lowered the effective dose of JQ1 needed to successfully inhibit melanoma tumors in mice. On the basis of our findings, future clinical trials with BETi should consider ascorbate levels in patients. Furthermore, ascorbate supplementation might help reduce the severe side effects that arise from BETi therapy by reducing the dosage necessary for treatment.
Significance: This study shows that ascorbate can enhance the efficacy of BET inhibitors, providing a possible clinical solution to challenges arising in phase I trials from the dose-dependent side effects of this class of epigenetic therapy. Cancer Res; 78(2); 572–83. ©2017 AACR.
Introduction
Aberrant epigenetic alterations along with genetic mutations contribute to melanoma. Histone acetylation is one major epigenetic mark that regulates transcription. Bromodomain-containing proteins bind acetylated lysines of histones and play a key role in acetylation-dependent assembly of transcriptional regulator complexes (1). On the basis of structural domains, bromodomain-containing proteins are further divided into bromodomain and extraterminal (BET) subfamily and non-BET subfamilies (2). The BET subfamily includes bromodomain-containing 2 (BRD2), BRD3, BRD4, and testis-specific BRDT. Abnormal histone acetylation and aberrant BET protein binding have been linked to various diseases, particularly the development of cancers including cutaneous melanoma (3). BET proteins promote the expression of certain oncogenes such as MYC, resulting in cell proliferation and tumorigenesis (4). Recent work found that BRD4 expression is much higher in primary and metastatic melanoma relative to nevi (5). It is known that the initiation and progression of various cancers depends on the continued expression of BRD4 (6). Indeed, decreasing BRD4 expression by siRNA dramatically reduced melanoma growth in vitro and in vivo (5), suggesting that BRD4 is a therapeutic target of melanoma.
BET proteins, such as BRD4, contain two bromodomains (BD1 and BD2), which are druggable targets (7, 8). JQ1 (thieno-triazolo-1,4-diazepine) is the first published small molecule that binds competitively to bromodomains (7, 9). A majority of these BET inhibitors (BETi) block both BD1 and BD2, whereas others may specifically inhibit BD1 or BD2 (10). Of the known BETi, most can effectively block BRD4. Preclinical studies have shown that BETi are promising melanoma therapies (11). For instance, BETi including CPI-203 and I-BET151 induce apoptosis and cell-cycle arrest in cultured melanoma cells (12, 13). BETi I-BET151 and Histone deacetylase (HDAC) inhibitor LBH589 synergistically promote apoptosis in melanoma cells (14). Furthermore, BETi JQ1 and BRAF inhibitor vemurafenib also act synergistically against BRAF-mutant melanoma (15, 16). The suppressing effect of BETi on gene transcription is not limited to MYC, but can affect other cancer-related genes (17). Overall, published preclinical studies indicate that BETi are effective for melanoma treatment.
BETi, like other pharmacotherapies, exert potential toxicities at the effective doses. From a few completed phase I trials, prominent side effects such as gastrointestinal toxicity, thrombocytopenia, anemia, neutropenia, diarrhea, fatigue, and nausea have been reported (18–20). Most of these side effects are dose-dependent. If by any means melanoma or other cancer cells could be sensitized to BETi, lower doses could then conceivably be used to achieve an increased therapeutic index, which could translate to a reduced side effect profile.
One recent breakthrough in the field of epigenetics is the identification of an active DNA demethylation pathway in which a group of enzymes, termed ten-eleven translocation (TET) methylcytosine dioxygenases, were found to convert 5-methylcytosine (5mC) to 5-hydroxymethylcytosine (5hmC; refs. 21, 22), and further to 5-formylcytosine (5fC) and 5-carboxylcytosine (5caC) that are replaced by unmodified C to complete DNA demethylation (23, 24). In addition to being a DNA demethylation intermediate, 5hmC also is an epigenetic mark with unique regulatory capacities (25). TETs belong to the Fe(II) and 2-oxoglutarate (2OG)-dependent dioxygenase family. We and others have shown that vitamin C, which exists predominantly as the ascorbate anion under physiological pH, acts as an additional cofactor for TETs by reducing the redox-inactive Fe(III)/Fe(IV) to Fe(II; refs. 26–30). Thus, ascorbate has an impact on DNA demethylation by promoting the availability of redox-active Fe(II) to TETs.
The uptake rate of ascorbate by melanoma cells is only approximately 50% of the uptake rate by healthy melanocytes (31) due to the lower expression of sodium dependent vitamin C transporter 2 (SVCT2; ref. 32), suggesting a need to compensate for the potential ascorbate deficiency within melanoma cells. Furthermore, conventional drug discovery and development involves cell-based in vitro screening. However, ascorbate is often absent from the media for culturing various cells, including melanoma cells (33). In light of the function of ascorbate in regulating DNA demethylation, it appears that the potential role of ascorbate in modulating cellular functions and drug responses of cancer cells has been overlooked. The goal of this work is to determine whether ascorbate, by regulating DNA demethylation and subsequently the transcriptome, changes the sensitivity of melanoma to BETi and to elucidate the molecular mechanism by which ascorbate enhances the efficacy of BETi.
Materials and Methods
Cell culture and treatment
Adult human melanocytes (NHEM), derived from a healthy human subject, were purchased from Lonza. Human melanoma cell lines (A2058, SK-MEL28, SK-MEL2, C8161, and 1205Lu) were purchased from the ATCC in a period between the year of 2014 to 2017 without further authentication. As described previously (34), frozen cells were newly thawed from low (3–10) passages. Mycoplasma was tested using PlasmoTest Mycoplasma detection kits (Invitrogen) and only negative cells were included in the experiments. Melanoma cells were maintained under a 5% CO2 atmosphere in RPMI medium (Life technologies) supplemented with 10% heat-inactivated FBS, 100 U/mL penicillin and 100 μg/mL of streptomycin. Melanocytes were cultured in 254 medium supplemented with Human melanocyte growth supplements (Thermo Fisher Scientific). Cells were seeded for 24 hours, and subsequently treated with sodium ascorbate (Sigma-Aldrich). Media were changed daily to avoid the accumulation of unabsorbed ascorbate. All cells tested negative for Mycoplasma by PCR.
Compound screening
For primary screen, cells were pretreated with or without 50 μmol/L sodium ascorbate for 72 hours in flasks. Then, 500 cells per well were seeded in a 384-well plate. Compounds were added in duplicates to the testing plates at a final concentration of 1 μmol/L. After plates were incubated for 72 hours, CellTiter-Glo reagents (Promega) were added to each well. Viability was measured on Envision Multi-Label Reader (PerkinElmer) as a percentage of response relative to both cells treated with DMSO alone (0% response) and cells treated with 10 μmol/L Velcade (100% response). Compounds with both low response (≤ mean + 3 SD cutoff) in cells not pretreated with ascorbate and high response (≥ mean + 3 SD cutoff) in cells pretreated with ascorbate were considered hits. The Z-factor for all the plates was greater than 0.9, demonstrating the robustness of the assay.
EC50 measurements
A2058, 1205Lu, C8161, SK-MEL 2, and SK-MEL 28 cells were pretreated with or without 50 μmol/L sodium ascorbate for 72 hours. On a 384-well drug plate, a 3-fold serial dilution of BETi including BI-2536, CPI-203, BET-151 (Selleck chemicals) or JQ1 (kindly provided by the Bradner laboratory, Dana-Farber Cancer Institute, Harvard University) were prepared with a starting concentration of 10 μmol/L. On the day of treatment, BETi were simultaneously transferred to the cell plates with a 384-pipettor head using a FLIPR tetra instrument (molecular devices). Cell viability was measured by CellTiter-GLo assay. Each concentration of BETi was plotted against percent cell survival. EC50 values were calculated from 4-parameter fitted curves by solving for the X-intercept value at the 50% inhibition level of the Y-intercept value.
Apoptosis assay
Melanoma cells were seeded in 24-well plates and pretreated with 50 μmol/L sodium ascorbate for 72 hours. Three pretreated wells were then further treated with either 0.25 μmol/L JQ1 or 0.5 μmol/L JQ1 each for 48 hours. Another two groups of non-pretreated cells were subjected to JQ1 treatment (0.25 or 0.5 μmol/L each) in triplicate. Apoptotic cell number was determined by Fluorescein-based TUNEL (Sigma-Aldrich) assay following the manufacturer's protocol.
MTT assay
Melanoma cells were pretreated with sodium ascorbate (0, 10, 50, and 100 μmol/L) for 72 hours followed by JQ1 treatment (0, 0.1, 0.25, 0.5, and 1 μmol/L) for 48 hours. Cell growth rate was measured by TACS MTT Cell Proliferation Assay (Trevigen) following the manufacturer's instructions.
siRNA
siRNA sequences directed against human BRD2, BRD3, BRD4, and non-targeting scrambled siRNA were purchased from Thermo Fisher Scientific. Cells were plated in growth medium without antibiotics at 30% to 50% confluence. Transfection of siRNA was performed using Lipofectamine 2000 (Invitrogen). Media were changed 6 hours after transfection to eliminate the toxic effects of transfecting reagents. After maintaining for 72 hours, cells were harvested for RNA and protein extraction. Individual Accell siRNA targeted against human TET1, TET2, and TET3 were designed and synthesized by Dharmacon and transfected following the manufacturer's instruction.
Quantitative RT-PCR
Total RNA was extracted from cells using the RNeasy Mini Kit (Qiagen). cDNA was synthesized from 1 μg of total RNA with the Super Script III First-Strand Synthesis System (Invitrogen). Real-time Quantitative PCR was performed with 100 ng of diluted cDNA, TaqMan Fast Advanced Master Mix (Applied Bio systems) and TaqMan gene-expression assay targeted towards either BRD 2, 3, 4 or GAPDH as internal control (Applied Biosystems) on an Applied Biosystems 7900HT. Relative gene expression was determined using the 2−ΔΔCT method. RT-PCR of HAT1 gene was performed using the same methods with the exception of using PowerUp SYBR Green Master Mix (Applied Biosystems). Primers were designed to span introns with the following sequences; HAT1 (forward primer: 5′-TCTAAAGTTGATGAGAACTTTGACT GT-3′, reverse primer: 5′-TTGTCTAATTTTGCCCTCAACA-3′;) GAPDH (forward primer: 5′-TGGACCTGACCTGCCGTCTA-3′, reverse primer: 5′-CCCTGTTGCTGTAGCCAAATTC-3′).
Subcellular fractionation and immunoblotting
Cells were lysed with RIPA buffer (50 mmol/L Tris-HCl, 150 mmol/L NaCl, 0.1% SDS, 0.5% sodium deoxycholate, 1% NP40) in the presence of protease and phosphatase inhibitors. For subcellular fractionation, 2 × 108 cells were collected for the extraction and both NE-PER nuclear and cytoplasmic extraction kits (Thermo Fisher Scientific) were used. Before SDS-PAGE, cell lysates were resuspended in SDS sample buffer (60 mmol/L Tris–HCl, 1% SDS, 10% glycerol, 0.05% bromophenol blue, pH 6.8, with 2% β-mercaptoethanol). Samples were subjected to 10% SDS-PAGE (Bio-Rad) and transferred to PVDF membranes (Bio-Rad). Transfer efficiency was determined by Ponceau S staining (Sigma-Aldrich). PVDF membranes were incubated with blocking solution (TBS containing 0.1% Tween 20 and 5% BSA) and were probed with specific antibodies (anti-PanH4ac (rabbit): # 39925 Active motif, Carlsbad, CA; anti-pan H4ac (mouse): 61337 #anti-PanH3ac: # 61638 Active motif; anti-H3 total: #61648 Active motif; anti-H4K12ac: # 39165 Active motif; anti-K5ac: # 39699 Active motif; anti-K8ac: # 2594S Cell Signaling Technology; anti-K16ac # 13534S Cell Signaling Technology; anti-GAPDH: Santa Cruz Biotechnology; anti-BRD4: Abcam). Protein bands were detected using the Chemiluminescence Kit (Millipore). ImageJ was used to quantify immunoblot results.
Immunofluorescence
Melanoma cells were seeded in 12-well culture dish with coverslips for 24 hours before treatment. After treatment, coverslips were washed with cold PBS. The cells were fixed for 10 minutes at room temperature with 4% paraformaldehyde, permeabilized for 5 minutes with 0.2% Triton X-100 PBS, and blocked for 30 minutes with 5% BSA. Cells were then incubated with the primary antibodies at 1:50 dilution overnight at 4°C, followed by the secondary antibodies at 1:250 dilution for another hour at room temperature. To stain the nucleus, cells were incubated with 40 μg/ml 4′,6-diamidino-2-phenylindole (DAPI) for 20 minutes at room temperature. The coverslips were then mounted on glass slides and examined at room temperature with a Zeiss LSM 710 confocal microscope.
Image acquisition and analysis
All fluorescence imaging was acquired using a Zeiss LSM 710 confocal microscope and captured into a 512 × 512 frame size by averaging four times at a bit depth of 16. Fluorescence intensity was quantified using Fiji (ImageJ). Average intensity values were measured from every cell within the image field from a minimum of three ×40 images per condition. The intensity values from individual cells were plotted and statistically analyzed by one-way ANOVA with the Tukey post hoc test using GraphPad Prism 7 from GraphPad Software.
Chromatin immunoprecipitation
Cells were crosslinked with 1% formaldehyde solution in PBS for 10 minutes. Afterwards, freshly prepared 0.125 mol/L Glycine was added followed by gently shaking the cells at room temperature to stop the crosslinking process. Cells were collected by centrifugation and resuspended in ChIP buffer (100 mmol/L NaCl; 50 mmol/L Tris-HCL, pH 8.1; 5mmol/L EDTA, pH 8.0; 0.5% SDS; 5% triton X-100, 1 × protease inhibitor). Cells were then subjected to sonication in a Bioruptor pico machine on a 30 seconds on/off cycle for 5 minutes. Sonication efficiency was confirmed by observing 500 bp DNA fragments. Protein concentrations were quantified by the BCA Protein Assay Kit (Thermo Fisher Scientific). One-hundred micrograms of protein from each sample were incubated with either 5 μg of IgG or 3 μg of mouse monoclonal pan H4ac primary antibody overnight by rotating at 14 rpm at 4°C. After incubation with Protein A/G sepharose beads (Thermo Fisher Scientific), samples were centrifuged to collect the immunocomplex. After pellets being washed with ChIP buffer, samples were boiled at 95°C for 5 minutes with 1 × laemmli buffer (Bio-Rad) and subjected to Immunoblot.
Animal studies
The Institutional Animal Care and Use Committee at the University of Miami has approved this study. C57Bl/6J Gulo−/− mice were maintained with an ascorbate sufficient water diet (0.33g/L) and C57Bl/6J (Wild type) were maintained with no additional ascorbate in the diet. One month before experiment, ascorbate was removed from the diet of Gulo−/− mice to induce ascorbate deficiency. Mice were then engrafted on the neck by injecting 2 × 105 mouse melanoma cells, B16-F10, to produce experimental tumor. For human melanoma cell xenograft experiments, four groups of Nu/J mice (# 002019, The Jackson Laboratory) were used. Two groups of Nu/J mice were maintained with high dose of ascorbate supplemented water (3.3 g/L) and the other two with regular water. Once palpable tumors were visible, C57Bl/6J mice (WT and Gulo−/−) were randomly distributed into groups of five within themselves and one subset of five from both groups received JQ1 (50 mg/Kg body weight/d) via intraperitoneal injection. Upon appearance of tumors, Nu/J mice were randomly sorted into two groups that received JQ1 (50 mg/kg body weight/d) or no treatment. Mice within these groups were either supplemented with ascorbate or provided regular water only. Tumor volume and body weight were measured with caliper and scale. After extraction, tumors were washed in PBS, weighed, and then fixed for further experiments.
Statistical analysis
All observations in this study were analyzed in triplicate or more and each experiment was repeated three times. GraphPad Prism was also used to generate and analyze data. Dose–response data were analyzed by ANOVA followed by Tukey post hoc comparison of all the means to determine significance. Values represent the mean ± SEM of three independent experiments. To compare two groups, Student t test was used and P < 0.05 was considered as statistically significant.
Results
Ascorbate sensitizes melanoma cells to BETi
We screened a panel of small molecules (n = 164; Supplementary Table S2), which target various epigenetic modulators such as DNA methyltransferases and HDACs, against human metastatic melanoma A2058 cells pretreated with or without ascorbate (50 μmol/L, the median plasma level in healthy humans) for 72 hours. To avoid the interference of pH levels by protons derived from ascorbic acid, all experiments were conducted using sodium ascorbate. Overall, ascorbate treatment increased the activity of approximately 50% and suppressed the activity of approximately 19% of the tested compounds while the rest remained largely unchanged. Strikingly, the top hit compounds were a number of structurally different BETi including JQ1, I-BET151, CPI-203, and BI-2536, whose activity in suppressing A2058 melanoma cell proliferation was increased by ascorbate (Fig. 1A).

Ascorbate treatment enhances the efficacy of BETi. A, Representative data of the compound screen (n = 164) targeting epigenetic modulators in A2058 cells. The responses of JQ1, CPI-203, BI-2536, and I-BET151 were drastically increased by pretreatment with 50 μmol/L ascorbate. B, The survival rate of A2058 cells was decreased after BRD4 knockdown by siRNA compared with scramble siRNA (P = 0.0008). Ascorbate (50 μmol/L) further decreased the survival rate (P = 0.0002). C–E, Dose–response curve of BETi. Ascorbate (50 μmol/L) increased the response of A2058 cells to JQ1, CPI-203, BI-2536, and I-BET151, whereas GSH (50 μmol/L) had no effect (n = 3). All data are mean ± SEM.
Ascorbate treatment enhances the efficacy of BETi. A, Representative data of the compound screen (n = 164) targeting epigenetic modulators in A2058 cells. The responses of JQ1, CPI-203, BI-2536, and I-BET151 were drastically increased by pretreatment with 50 μmol/L ascorbate. B, The survival rate of A2058 cells was decreased after BRD4 knockdown by siRNA compared with scramble siRNA (P = 0.0008). Ascorbate (50 μmol/L) further decreased the survival rate (P = 0.0002). C–E, Dose–response curve of BETi. Ascorbate (50 μmol/L) increased the response of A2058 cells to JQ1, CPI-203, BI-2536, and I-BET151, whereas GSH (50 μmol/L) had no effect (n = 3). All data are mean ± SEM.
This study was then focused on BETi in melanoma. We first measured the transcription of BRD proteins in melanoma cell lines by quantitative RT-PCR (qRT-PCR). The mRNA level of BRD2 and BRD4, but not BRD3, was significantly higher in A2058 melanoma cells compared with healthy melanocytes (Supplementary Fig. S1). This is consistent with the previous report that BRD4 is overexpressed in melanoma (5). BETi block the binding of BRD4 with acetylated lysine residues in histones, which could be alternatively mimicked by decreasing the expression of BRD4. We then used siRNA to knock down the expression of BRD4 in A2058 melanoma cells. The effect of siRNA on BRD4 expression was confirmed by qRT-PCR and immunoblot (Supplementary Fig. S2). BRD4 siRNA suppressed cell survival, while scrambled siRNA control had no obvious effect. Ascorbate treatment (50 μmol/L) further enhanced the effect of BRD4 siRNA on inhibiting melanoma cell survival (Fig. 1B). These results suggest that ascorbate potentiates the inhibition on the bromodomain protein complex by either BETi or BRD4 siRNA in A2058 melanoma cells.
Cells were then treated with a 10-point/3-fold serial dilution of BETi including JQ1, I-BET151, CPI-203, and BI-2536 with a starting concentration of 10 μmol/L to verify the effect of ascorbate. Five melanoma cell lines were used including 4 lines (1205Lu, C8161, SK-MEL-28, A2058) that contain BRAF mutations and one line (SK-MEL-2) that harbors an NRAS mutation. Ascorbate (50 μmol/L) pretreatment for 72 hours reduced the half-maximal effective concentration (EC50) value of these BETi by an average of more than 4-fold (Fig. 1C–F; Supplementary Table S3) in all 5 melanoma cell lines tested. In contrast, glutathione (GSH) exerted no obvious effect, suggesting that the action of ascorbate on BETi is independent of its property as a general reducer.
Ascorbate suppresses the acetylation of H4, but not H3, in melanoma cells
Two potential mechanisms could underline the enhancing effect of ascorbate on BETi. One possibility might be a reduced expression of BRD proteins and the other could be downregulated histone acetylation, both of which can result in less BRD protein binding to acetylated histones. We first examined our existing RNA-seq data (32), which indicate that BRD transcripts remain at comparable levels in the A2058 melanoma cells treated with or without ascorbate. To verify this, we measured the transcripts of BRDs in A2058 melanoma cells by qRT-PCR. The results confirmed that there was no obvious change at the mRNA level of BRDs including BRD4 (Supplementary Fig. S3), suggesting that ascorbate does not change BRD4 expression.
We then turned our attention to histone acetylation. The level of H3 acetylation (H3ac) and H4 acetylation (H4ac) was evaluated by immunoblot of whole-cell lysate using antibodies against pan-acetylated histones. The immunoblot densitometric data showed ascorbate treatment decreased H4, but not H3, total acetylation in both A2058 and 1205Lu melanoma cells (Fig. 2A–C).

Ascorbate treatment reduces H4ac but not H3ac in melanoma cells. A, Immunoblot of H3ac and H4ac in A2058 and 1205Lu cells treated with or without ascorbate (50 μmol/L) for 72 hours. Total H3 and H4 were used for loading controls. B, Densitometry analysis of H3ac normalized by total H3. No significant change in H3ac was observed after ascorbate treatment. C, Densitometry analysis of H4ac normalized by total H4. The levels of H4ac were reduced by ascorbate treatment in A2058 and 1205Lu cells. D and F, Immunofluorescence and quantification of H3ac in A2058 and 1205Lu cells treated with or without ascorbate (50 μmol/L) for 72 hours. No significant change in H3ac was observed in A2058 and 1205Lu cells. G and I, Immunofluorescence and quantification of H4ac in A2058 and 1205Lu cells treated with or without ascorbate (50 μmol/L) for 72 hours. H4ac was decreased by ascorbate treatment in A2058 and 1205Lu cells. All data are mean ± SEM.
Ascorbate treatment reduces H4ac but not H3ac in melanoma cells. A, Immunoblot of H3ac and H4ac in A2058 and 1205Lu cells treated with or without ascorbate (50 μmol/L) for 72 hours. Total H3 and H4 were used for loading controls. B, Densitometry analysis of H3ac normalized by total H3. No significant change in H3ac was observed after ascorbate treatment. C, Densitometry analysis of H4ac normalized by total H4. The levels of H4ac were reduced by ascorbate treatment in A2058 and 1205Lu cells. D and F, Immunofluorescence and quantification of H3ac in A2058 and 1205Lu cells treated with or without ascorbate (50 μmol/L) for 72 hours. No significant change in H3ac was observed in A2058 and 1205Lu cells. G and I, Immunofluorescence and quantification of H4ac in A2058 and 1205Lu cells treated with or without ascorbate (50 μmol/L) for 72 hours. H4ac was decreased by ascorbate treatment in A2058 and 1205Lu cells. All data are mean ± SEM.
Immunoblot of cell lysates can be used to detect the overall H3ac and H4ac in the cell but cannot be used to separate the newly synthesized histones from the histones incorporated in the chromatin. We used immunofluorescence to visualize H3ac and H4ac. The results showed that both H3ac and H4ac immunofluorescence mainly localized within the nucleus. Immunofluorescence confirmed no change in H3ac expression by ascorbate treatment in A2058 and 1205Lu melanoma cells (Fig. 2D). Conversely, ascorbate treatment decreased H4ac immunofluorescence in both cell lines (Fig. 2E). Immunofluorescence quantification of nuclear H3ac or H4ac further confirmed these findings (Fig. 2E–I). These results suggest that ascorbate treatment downregulates H4ac, which could, in principle, underline the enhanced effect of BETi.
Ascorbate inhibits the expression of HAT1
To understand the potential mechanism by which ascorbate downregulates H4ac, we investigated our published RNA-seq data (32). By querying all HATs and HDACs, we found that HAT1 transcripts were decreased by 100 μmol/L ascorbic acid in our previous study. To be consistent with the ascorbate used in the drug screen experiment, we examined the impact of sodium ascorbate, instead of ascorbic acid, on HAT1 expression. First, the level of HAT1 mRNA was reduced in both A2058 and 1205Lu melanoma cells by ascorbate (50 μmol/L) treatment for 72 hours (Fig. 3A). Second, immunoblot and quantification confirmed that ascorbate treatment downregulated HAT1 protein levels in the two melanoma cell lines (Fig. 3B and C). Furthermore, immunofluorescence also showed a decrease of HAT1 signal in these cell lines treated with ascorbate (Fig. 3D). Immunofluorescence quantification confirmed the decrease of HAT1 signal after ascorbate treatment (Fig. 3E and F). Taken together, results of qRT-PCR, immunoblot, and immunofluorescence suggest that ascorbate downregulates the expression of HAT1 in melanoma cells.
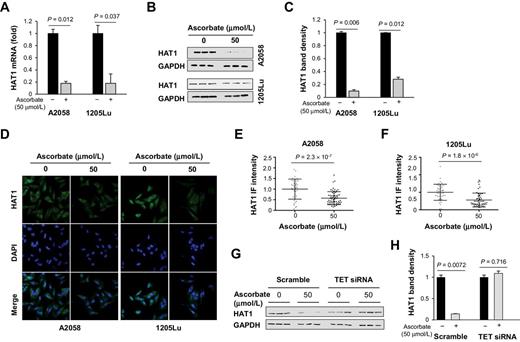
Downregulation of HAT1 by ascorbate treatment. A, qRT-PCR of HAT1. HAT1 mRNA were reduced by ascorbate (50 μmol/L) treatment for 72 hours in A2058 cells (P = 0.012) and in 1205Lu cells (P = 0.037). B, Immunoblot of HAT1 in A2058 and 1205Lu cells. C, Densitometry analysis of HAT1 normalized by GAPDH shows that HAT1 was decreased by ascorbate treatment in A2058 cells and 1205Lu cells. D, Immunofluorescence of HAT1 in A2058 and 1205Lu cells treated with or without ascorbate (50 μmol/L) for 72 hours. E and F, Quantification of HAT1 immunofluorescence. The level of HAT1 protein was decreased by ascorbate treatment in A2058 and 1205Lu cells. G-H, HAT1 protein remained unchanged by ascorbate treatment in TETs knockdown A2058 cells as shown by immunoblot and quantification.
Downregulation of HAT1 by ascorbate treatment. A, qRT-PCR of HAT1. HAT1 mRNA were reduced by ascorbate (50 μmol/L) treatment for 72 hours in A2058 cells (P = 0.012) and in 1205Lu cells (P = 0.037). B, Immunoblot of HAT1 in A2058 and 1205Lu cells. C, Densitometry analysis of HAT1 normalized by GAPDH shows that HAT1 was decreased by ascorbate treatment in A2058 cells and 1205Lu cells. D, Immunofluorescence of HAT1 in A2058 and 1205Lu cells treated with or without ascorbate (50 μmol/L) for 72 hours. E and F, Quantification of HAT1 immunofluorescence. The level of HAT1 protein was decreased by ascorbate treatment in A2058 and 1205Lu cells. G-H, HAT1 protein remained unchanged by ascorbate treatment in TETs knockdown A2058 cells as shown by immunoblot and quantification.
In an attempt to understand whether ascorbate is involved in HAT1 expression through TET-mediated DNA hydroxymethylation, we conducted hMeDIP-PCR, a method that enriches hydroxymethylated DNA by anti–5hmC-based immunoprecipitation for PCR assays. Primers were designed to cover the HAT1 gene, including two CpG rich regions in the promoter and all 11 exons. Out of a total 13 amplicons, 7 amplicons, including ones at CpG region 2 and exons 2, 4, 5, 6, 7, and 11 showed stronger real-time PCR signals after ascorbate treatment, indicating higher 5hmC levels in those regions (Supplementary Fig. S4). The elevated 5hmC could potentially underpin the decreased expression of HAT1. Indeed, after knocking down the expression of TETs by siRNA, HAT1 protein remained largely unchanged after ascorbate (50 μmol/L) treatment. Whereas the control group (transfected with scramble siRNA) showed a significant reduction in HAT1 after the same ascorbate treatment (Fig. 3G and H). These results suggest that the suppression of HAT1 by ascorbate is likely through the TETs-mediated DNA demethylation pathway.
Ascorbate decreases the acetylation of H4K5 and H4K12 in melanoma cells
HAT1 is regarded as the enzyme that acetylates the NH2-terminal tail of newly synthesized H4 at lysine positions 5 and 12, which could eventually be incorporated into chromatin (35). A decreased expression of HAT1 would change the acetylation of H4 at the two positions. We therefore tested whether ascorbate treatment changes H4K5 acetylation (H4K5ac) and H4K12 acetylation (H4K12ac). Treatment with ascorbate (50 μmol/L) for 72 hours indeed decreased H4K5 acetylation (H4K5ac) and H4K12 acetylation (H4K12ac) in A2058 melanoma cells shown by the immunoblot of whole-cell lysates (Fig. 4A–C). The suppression of H4K5ac and H4K12ac by ascorbate was more dramatic in 1205Lu cells (Fig. 4A–C). Immunofluorescence also showed decreased H4K5ac and H4K12ac in both A2058 and 1205Lu melanoma cells after ascorbate treatment (Fig. 4D–I). Immunofluorescence quantification of nuclear H4K5ac and H4K12ac confirmed these effects. To further examine the acetylation of newly synthesized H4, immunoblot was performed on fractionated cytosol and nuclei samples in A2058 cells. The results showed that the levels of H4ac in both nuclear and cytoplasmic fractions were inhibited by ascorbate (Supplementary Fig. S5). Comparatively, the cytosolic bands decreased more drastically than the nuclear bands by ascorbate. Furthermore, acetylation of H4 at two other residues (K8 and K16), which are not reported targets of HAT1, remained largely unchanged after ascorbate treatment (Supplementary Fig. S6). Overall, these results suggest that ascorbate treatment inhibits H4K5ac and H4K12ac by suppressing HAT1 expression.
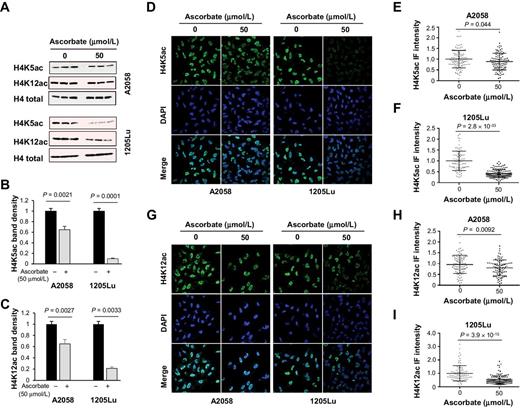
H4K5ac and H4K12ac are decreased by ascorbate treatment. A, Immunoblot of H4K5ac and H4K12ac in A2058 and 1205Lu cells treated with or without ascorbate (50 μmol/L) for 72 hours. B and C, Densitometry analysis of H4K5ac and H4K12ac normalized by total H4. H4K5ac and H4K12ac were decreased by ascorbate treatment in A2058 and 1205Lu cells. D–F, Immunofluorescence and quantification of H4K5ac in A2058 and 1205Lu cells treated with or without ascorbate (50 μmol/L) for 72 hours. H4K5ac was decreased by ascorbate treatment in A2058 and 1205Lu cells. G and I, Immunofluorescence and quantification of H4K12ac in A2058 and 1205Lu cells treated with or without ascorbate (50 μmol/L) for 72 hours. H4K12ac was decreased by ascorbate treatment in A2058 and 1205Lu cells. All data are mean ± SEM.
H4K5ac and H4K12ac are decreased by ascorbate treatment. A, Immunoblot of H4K5ac and H4K12ac in A2058 and 1205Lu cells treated with or without ascorbate (50 μmol/L) for 72 hours. B and C, Densitometry analysis of H4K5ac and H4K12ac normalized by total H4. H4K5ac and H4K12ac were decreased by ascorbate treatment in A2058 and 1205Lu cells. D–F, Immunofluorescence and quantification of H4K5ac in A2058 and 1205Lu cells treated with or without ascorbate (50 μmol/L) for 72 hours. H4K5ac was decreased by ascorbate treatment in A2058 and 1205Lu cells. G and I, Immunofluorescence and quantification of H4K12ac in A2058 and 1205Lu cells treated with or without ascorbate (50 μmol/L) for 72 hours. H4K12ac was decreased by ascorbate treatment in A2058 and 1205Lu cells. All data are mean ± SEM.
Inhibition of BRD4 binding to the acetylated histones
By inhibiting H4ac, specifically H4K5ac and H4K12ac, ascorbate alone could decrease the binding of BRD4 to histones, which may underpin the enhanced efficacy of BETi by ascorbate as shown by the compound screen of melanoma cells (Fig. 1). To test whether ascorbate in combination with BETi further decreases the binding of BRD4 with H4ac, we first used immunofluorescence to detect the colocalization of BRD4 and H4ac. As shown in Fig. 5A, there was substantial BRD4 with H4ac colocalization signal in A2058 melanoma cells without any treatment. Treatment of A2058 melanoma cells with JQ1 (0.5 μmol/L) decreased the binding of BRD4 with H4ac. Ascorbate (50 μmol/L) treatment downregulated H4ac and reduced its colocalization with BRD4. Combination treatment with ascorbate and JQ1 further diminished the binding of BRD4 with H4ac in A2058 and 1205Lu cells (Fig. 5A and B). The inhibitory effects of ascorbate and JQ1 on the binding of BRD4 to H4ac was further verified by quantification of colocalization signals (Fig. 5C and D). We then used chromatin immunoprecipitation (ChIP) to further verify the effect of ascorbate on the binding of BRD4 to H4ac. Cross-linked samples were immunoprecipitated by mouse monoclonal anti-H4ac antibody and immunoblotted with anti-BRD4 antibody. In both A2058 and 1205Lu melanoma cells, ascorbate (50 μmol/L) treatment decreased the ChIP bands by over approximately 50% (Fig. 5E and F). These results suggest that ascorbate enhances the effect of JQ1 in blocking the binding of BRD4 to the acetylated H4.

The binding of BRD4 to acetylated H4 is interrupted by ascorbate treatment. A and B, Confocal microscopy images of H4ac, BRD4, and their colocalization in A2058 and 1205Lu cells. C and D, Quantification of H4ac and BRD4 colocalization in A2058 and 1205Lu cells. The colocalization coefficient of H4ac and BRD4 was reduced by JQ1. Ascorbate treatment alone reduced the signal of H4ac and subsequently the H4ac-BRD4 colocalization. The colocalization coefficient of H4ac and BRD4 was further decreased by cotreatment with ascorbate and JQ1. E, ChIP assay of H4ac and BRD4 binding in A2058 cells and 1205Lu cells. F, Densitometry analysis of BRD4 in the ChIP assay. The binding between H4ac and BRD4 was reduced by ascorbate treatment in A2058 cells and 1205Lu cells. All data are mean ± SEM (n = 3).
The binding of BRD4 to acetylated H4 is interrupted by ascorbate treatment. A and B, Confocal microscopy images of H4ac, BRD4, and their colocalization in A2058 and 1205Lu cells. C and D, Quantification of H4ac and BRD4 colocalization in A2058 and 1205Lu cells. The colocalization coefficient of H4ac and BRD4 was reduced by JQ1. Ascorbate treatment alone reduced the signal of H4ac and subsequently the H4ac-BRD4 colocalization. The colocalization coefficient of H4ac and BRD4 was further decreased by cotreatment with ascorbate and JQ1. E, ChIP assay of H4ac and BRD4 binding in A2058 cells and 1205Lu cells. F, Densitometry analysis of BRD4 in the ChIP assay. The binding between H4ac and BRD4 was reduced by ascorbate treatment in A2058 cells and 1205Lu cells. All data are mean ± SEM (n = 3).
Ascorbate enhances the efficacy of JQ1 in treating melanoma cells
On the basis of the result that ascorbate and BETi combination blocked the binding of BRD4 to acetylated H4, it is reasonable to predict that ascorbate sensitizes melanoma cells to BETi treatment. We first examined the effect of combinatorial treatment with ascorbate and JQ1 on apoptosis. Ascorbate (50 μmol/L) alone did not induce apoptosis measured by TUNEL assay in A2058 melanoma cells. JQ1 (0.25 and 0.5 μmol/L) dose-dependently promoted apoptosis of these cells. Comparatively, JQ1 in combination with ascorbate further enhanced apoptosis of A2058 melanoma cells compared to JQ1 treatment alone (Fig. 6A). This enhancement of JQ1-induced apoptosis after ascorbate treatment was further verified in another two melanoma lines, 1205Lu and C6181 (Fig. 6B and C).

Reduction of melanoma cell malignancy by ascorbate and JQ1. A–C, Induction of apoptosis in A2058, 1205Lu, and C6181 cells by combination treatment of ascorbate and JQ1. Ascorbate alone at 50 μmol/L did not have obvious consequence on apoptosis but enhanced the effect of JQ1 at 0.25 and 0.5 μmol/L to induce apoptosis. D–F, Cell viability after combination treatment of ascorbate and JQ1. Ascorbate at 50 and 100 μmol/L reinforced the inhibitory effect of JQ1 (0.1, 0.25, 0.5, and 1 μmol/L) on the viability of A2058, 1205Lu, and C6181 cells; *, P < 0.001; All data are mean ± SEM.
Reduction of melanoma cell malignancy by ascorbate and JQ1. A–C, Induction of apoptosis in A2058, 1205Lu, and C6181 cells by combination treatment of ascorbate and JQ1. Ascorbate alone at 50 μmol/L did not have obvious consequence on apoptosis but enhanced the effect of JQ1 at 0.25 and 0.5 μmol/L to induce apoptosis. D–F, Cell viability after combination treatment of ascorbate and JQ1. Ascorbate at 50 and 100 μmol/L reinforced the inhibitory effect of JQ1 (0.1, 0.25, 0.5, and 1 μmol/L) on the viability of A2058, 1205Lu, and C6181 cells; *, P < 0.001; All data are mean ± SEM.
We then examined the combined treatment of ascorbate and JQ1 on melanoma cell proliferation. Ascorbate at concentrations less than 50 μmol/L did not obviously affect A2058 melanoma cell proliferation, whereas 100 μmol/L decreased cell proliferation as measured by MTT assay. JQ1 (0.1–1 μmol/L) hampered melanoma cell proliferation in a dose-dependent manner. In addition, ascorbate (10–100 μmol/L) also dose-dependently enhanced the inhibition of JQ1 on cell proliferation (Fig. 6D). The inhibitory effects on cell growth by JQ1 and ascorbate was then validated in another two melanoma lines 1205Lu and SK-MEL-2 (Fig. 6E and F). By diet and oral delivery, the concentration of ascorbate in the plasma of humans can reach 100 μmol/L (36). Here ascorbate at 100 μmol/L exerted a stronger effect on the inhibition of cell growth caused by JQ1.
The relationship between ascorbate and BETi in the combination treatment was then analyzed by isobologram and combination index. The results indicated that the effective concentration in combination at EC50, which fall below the individual EC50 line on isobologram, corresponded to combination index (CI) values of 0.789 and 0.696. Furthermore, at EC70, the combination dose also fell below the EC70 line with calculated CI values at 0.448 and 0.869 respectively. These results suggest a possible synergistic interaction between ascorbate and BETi in treating melanoma cells (Supplementary Fig. S7).
Ascorbate sensitizes melanoma xenograft to JQ1 treatment
Most mammals such as mice synthesize ascorbate de novo in the liver. In contrast, humans as well as other high primates, guinea pigs, and fruit bats no longer can synthesize ascorbate due to a mutant and nonfunctional gulonolactone oxidase (Gulo), the enzyme catalyzing the last step of ascorbate biosynthesis. For these animals, ascorbate is an essential vitamin that needs to be supplied via diet and dietary supplements. We first examined the syngenic murine melanoma formation in immune sufficient Gulo−/− and Gulo+/+ (wild type) mice. Gulo−/− mice, like humans, cannot synthesize ascorbate and were supplemented with ascorbate (0.033 g/L) in the drinking water, which prevents scurvy in adult Gulo−/− mice but does not allow the ascorbate levels in the plasma to reach that of Gulo+/+ mice (37). Similar to human melanoma cells, ascorbate (50 μmol/L) enhanced the efficacy of BETi by decreasing EC50 values in murine B16-F10 melanoma cells (Supplementary Fig. S8). The effect of ascorbate on BETi was also mediated by a reduction in H4ac as shown by immunoblot (Supplementary Fig. S8). After these verifications, B16-F10 cells were injected subcutaneously into Gulo−/− and Gulo+/+ mice. JQ1 (50 mg/kg body weight) was intraperitoneally injected daily for 14 days. Without JQ1 treatment, the tumor size was 48% bigger in Gulo−/− mice than in Gulo+/+ mice. Treatment with JQ1 decreased the size of tumors in Gulo+/+ mice by 68% compared with ascorbate deficient Gulo−/− mice (Fig. 7A and B). These results suggest that insufficient ascorbate antagonizes the treatment effect of JQ1.

Inhibition of melanoma tumorgraft by ascorbate and JQ1. A, Photograph of murine B16-F10 melanoma syngenic tumorgraft from Gulo−/− and Gulo+/+ (wild-type) mice treated with or without JQ1. B, Quantification of B16-F10 melanoma tumor weights. Without JQ1 treatment, tumors in ascorbate sufficient mice are smaller (P = 0.001) compared with ascorbate-deficient mice. In ascorbate-sufficient mice, tumors were drastically reduced by JQ1 (50 mg/kg body weight; P = 0.01). However, in ascorbate-deficient mice, tumors were not significantly reduced by the same dose of JQ1 treatment (P = 0.85). C, Photograph of human A2058 melanoma xenografts treated with or without JQ1 from nude mice supplemented with or without ascorbate. D, Quantification of A2058 melanoma xenograft weights. Without ascorbate supplements, xenografts remained at similar sizes after JQ1 (50 mg/kg body weight) injection (P = 0.20). However, xenografts were reduced in mice treated with JQ1 when supplemented with ascorbate compared with the mice supplemented with ascorbate but no JQ1 treatment (P = 0.01), or in comparison to the mice treated only with JQ1 (P = 0.02).
Inhibition of melanoma tumorgraft by ascorbate and JQ1. A, Photograph of murine B16-F10 melanoma syngenic tumorgraft from Gulo−/− and Gulo+/+ (wild-type) mice treated with or without JQ1. B, Quantification of B16-F10 melanoma tumor weights. Without JQ1 treatment, tumors in ascorbate sufficient mice are smaller (P = 0.001) compared with ascorbate-deficient mice. In ascorbate-sufficient mice, tumors were drastically reduced by JQ1 (50 mg/kg body weight; P = 0.01). However, in ascorbate-deficient mice, tumors were not significantly reduced by the same dose of JQ1 treatment (P = 0.85). C, Photograph of human A2058 melanoma xenografts treated with or without JQ1 from nude mice supplemented with or without ascorbate. D, Quantification of A2058 melanoma xenograft weights. Without ascorbate supplements, xenografts remained at similar sizes after JQ1 (50 mg/kg body weight) injection (P = 0.20). However, xenografts were reduced in mice treated with JQ1 when supplemented with ascorbate compared with the mice supplemented with ascorbate but no JQ1 treatment (P = 0.01), or in comparison to the mice treated only with JQ1 (P = 0.02).
Human melanoma xenograft was then analyzed in immune-deficient nude mice, which produce endogenous ascorbate with a plasma concentration around 50 μmol/L (38). The cell-based experiments clearly suggested that 100 μmol/L ascorbate treatment, rather than 50 μmol/L, is the optimal dose for enhancing the effect of JQ1. To reach approximately 100 μmol/L ascorbate in the plasma, additional ascorbate (3.3 g/L) was provided in the drinking water as previously described (39). Without JQ1 treatment, ascorbate (3.3 g/L) alone moderately decreased (∼34%) the xenograft size. JQ1 (50 mg/kg body weight) inhibited xenograft growth in nude mice but the melanoma xenograft was further diminished (∼60%) by the combined treatment with ascorbate and JQ1 (Fig. 7C and D). Taken together, these results suggest that ascorbate supplements enhance, whereas ascorbate deficiency as modeled in Gulo−/− mice diminishes, the treatment outcome of JQ1 for melanoma.
Discussion
There is strong enthusiasm for the anti-cancer properties of BETi in both academia and the pharmaceutical industry. Currently, there are about 20 clinical trials at early stages evaluating various BETi in treating malignancies including solid cancers (2). However, concerns about the safety and uncertain outcomes of these clinical trials have been raised, mainly due to the rudimentary understanding of BRD biology and the lack of selective inhibitors for individual BRD proteins (40). Indeed, dose-dependent side effects in patients have been reported from a few of completed phase I clinical trials. These side effects include gastrointestinal toxicity and fatigue (18, 19), thrombocytopenia, anemia, neutropenia, diarrhea, fatigue, and nausea (20). For the next set of clinical trials, the difficult decision to lower doses of existing BETi to minimize side effects might consequently compromise the treatment outcome.
This preclinical study provides a translational solution to help overcome the dilemma of existing BETi clinical trials. We show that ascorbate sensitizes melanoma cells to BETi by decreasing H4k5ac and H4k12ac, therefore, allowing fewer binding sites for BRD proteins. Importantly, ascorbate is a readily available, well-tolerated, and an essential micronutrient for humans, which can be conveniently delivered in oral form. Thus, ascorbate could be considered as a cotreatment in BETi clinical trials.
The average concentration of ascorbate in the plasma of healthy humans is at approximately 50 μmol/L range (36). We thus chose 50 μmol/L ascorbate to pretreat melanoma cells for drug screen and subsequent mechanistic analyses in cell-based experiments. Previously, we showed that the expression of SVCT2, through which ascorbate enters and accumulates within the cell, was decreased in melanoma cells, especially the lines derived from metastatic stage tumors (32). This is consistent with the report that ascorbate uptake rate by melanoma cells is only approximately 50% of the uptake rate by healthy melanocytes (31). These studies suggest there is a shortage of intracellular ascorbate in melanoma, which needs to be compensated by higher levels of ascorbate. As presented in this study, JQ1 failed to suppress tumor growth in ascorbate deficient Gulo−/− mice. Cell-based analysis further showed that ascorbate at 100 μmol/L was more effective than at 50 μmol/L in suppressing melanoma cell growth and inducing apoptosis. Ascorbate at 100 μmol/L in the plasma can be conveniently reached by diet and dietary supplement in mice as well as in humans. When additional ascorbate was provided, JQ1 showed much stronger inhibition of xenograft growth in nude mice.
The mechanism by which ascorbate sensitizes melanoma cells to BETi is mainly through the suppression of HAT1 expression. Our earlier work showed that ascorbate is a cofactor for TETs to convert 5mC to 5hmC (26, 27), which has been confirmed by later reports (28–30). This finding revealed that ascorbate plays a role in promoting active DNA demethylation. We showed that ascorbate treatment partially reestablished 5hmC content in melanoma cells and caused a shift in the transcriptome (30). HAT1 expression is downregulated by ascorbate treatment, which is correlated with 5hmC increase in its promoter and gene body. It is thus likely that ascorbate promotes DNA demethylation in the HAT1 gene region, which further suppresses the transcription of HAT1.
Emerging evidence shows that HAT1 can play a critical role in chromatin remodeling (41). HAT1 acetylates the soluble H4 at lysine positions 5 and 12 in the cytoplasm. The acetylated H4 is eventually incorporated into chromatin (35). By inhibiting HAT1 expression, ascorbate decreases H4K5ac and H4K12ac, thus reducing the binding sites for BRD proteins. Possible approaches to block the function of BRDs, especially BRD4 in melanoma, may include (i) attenuating BRDs; (ii) decreasing histone acetylation; and (iii) reducing BRDs expression. Ascorbate in combination with BETi could effectively execute the first two approaches for melanoma treatment. The synergy between ascorbate and JQ1 in inhibiting BRD4 binding to the acetylated H4 was confirmed by ChIP and confocal imaging in two melanoma cell lines.
Decreased H4K5ac and H4K12ac would affect gene-expression profiles in melanoma cells. It is thus plausible that the transcriptomic changes, as we have shown before (32), in response to ascorbate treatment could be caused by enhanced DNA demethylation and reduced H4K5ac and H4K12ac. A reanalysis of RNA-seq revealed that many genes involved in cell proliferation and apoptosis, such as Clusterin, were changed by ascorbate (34). These genes might contribute to the decreased malignancy phenotypes in vitro and in vivo by ascorbate.
In this study, we focused on melanoma due to the finding that BRD4 is overexpressed in human melanoma samples (5). It remains to be seen whether ascorbate has a similar effect on other types of cancer. Ascorbate pretreatment decreased the EC50 value of some structurally different BETi to about 10-fold in cultured human melanoma cell lines and one murine line. We provided evidence to show, in principle, that insufficient ascorbate diminishes the treatment effect of JQ1 and higher ascorbate improves the treatment outcome of JQ1 for melanoma.
In conclusion, these data suggest that ascorbate epigenetically sensitizes melanoma cells to BETi treatment by suppressing HAT1 expression, further reducing H4K5ac and H4K12ac. BETi clinical trials should consider ascorbate levels in patients in the study design. The potential effect of ascorbate cotreatment in reducing BETi doses could further be extended by investigating BETi therapeutic index and side effects.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Authors' Contributions
Conception and design: S.P. Brothers, C. Wahlestedt, G. Wang
Development of methodology: S. Mustafi, C.-H. Volmar, S.P. Brothers, C. Wahlestedt
Acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.): S. Mustafi, V. Camarena, C.-H. Volmar, Z.-J. Liu, C. Wahlestedt, G. Wang
Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): S. Mustafi, C.-H. Volmar, T.C. Huff, D.W. Sant, S.P. Brothers, C. Wahlestedt, G. Wang
Writing, review, and/or revision of the manuscript: S. Mustafi, V. Camarena, C.-H. Volmar, T.C. Huff, D.W. Sant, S.P. Brothers, Z.-J. Liu, C. Wahlestedt, G. Wang
Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): C.-H. Volmar, Z.-J. Liu, G. Wang
Study supervision: S.P. Brothers, C. Wahlestedt, G. Wang
Acknowledgments
The authors sincerely thank the Bradner laboratory and Dr. Jun Qi at Dana-Farber Cancer Institute, Harvard University for providing JQ1 used in this research. This work was supported by NIH grants R21CA191668 and R01NS089525 (to G. Wang), R01MH110441 and R01NS092671 (to S. P. Brothers), and R01DA035055 and R01AA023781 (to C. Wahlestedt).
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.