
Abstract
The observation that altered metabolism is the fundamental cause of cancer was made by Otto Warburg nearly a century ago. However, the subsequent identification of oncogenes and tumor suppressor genes has displaced Warburg's theory pointing towards genetic aberrations as the underlining cause of cancer. Nevertheless, in the last decade, cancer-associated mutations have been identified in genes coding for tricarboxylic acid cycle (TCA cycle, also known as Krebs cycle) and closely related enzymes that have essential roles in cellular metabolism. These observations have revived interest in Warburg's hypothesis and prompted a flurry of functional studies in the hope of gaining mechanistic insight into the links between mitochondrial dysfunction, metabolic alterations, and cancer. In this review, we discuss the potential pro-oncogenic signaling role of some TCA cycle metabolites and their derivatives (oncometabolites). In particular, we focus on their effects on dioxygenases, a family of oxygen and α-ketoglutarate-dependent enzymes that control, among other things, the levels and activity of the hypoxia-inducible transcription factors and the activity of DNA and histone demethylases.
Similar content being viewed by others


Introduction
At the beginning of the 20th century, Otto Warburg observed that cancer tissues have high rates of glycolysis even in the presence of oxygen, a metabolic phenotype that he labeled “aerobic glycolysis” ([1] and reviewed in [2]). Warburg attributed these metabolic changes to defects in mitochondrial respiration and ATP production (oxidative phosphorylation). The aerobic glycolysis of cancer cells has been widely investigated throughout the years, and it is now considered as one of the metabolic hallmarks of cancer transformation and has been exploited to develop novel diagnostic and therapeutic tools [3]. Nevertheless, in subsequent decades after Warburg's pioneering findings, belief in his theory waned, partly due to the identification of mutations in cancer predisposition genes including oncogenes (pro-tumorigenic) and tumor suppressor genes (anti-tumorigenic). Furthermore, convincing evidence that mitochondrial dysfunction is the actual cause of the metabolic switch in cancer and of tumorigenesis in general was scarce [2]. However, in the last decade, the identification of loss- or gain-of-function mutations in key metabolic enzymes that have a causal role in tumorigenesis has awakened interest in Warburg's hypothesis [4, 5].
Genetic evidence for the involvement of metabolic enzymes in tumorigenesis
SDH mutations in hereditary paragangliomas and pheochromocytomas and other sporadic tumors
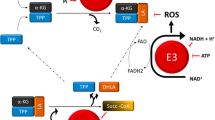
Mitochondrial succinate–coenzyme Q oxidoreductase, which catalyzes the conversion of succinate to fumarate in the TCA cycle (Fig. 1a) while simultaneously transferring electrons from succinate to coenzyme Q (complex II of the respiratory chain), is comprised of four subunits: succinate dehydrogenase (SDH) SDHA, SDHB, SDHC, and SDHD all of which are nuclear genes encoding mitochondrial enzymes. A decade ago, heterozygous germline mutations in SDHB, SDHC, and SDHD subunits were identified as the causal tumor suppressor genes in hereditary paragangliomas and pheochromocytomas (hPGL), a rare hereditary cancer predisposition syndrome of the chromaffin tissue arising in the adrenal medulla pheochromocytoma (PCC) or derived from the parasympathetic tissue of the head and neck paraganglioma (PGL) [6–8]. Also, more recently, mutations in SDHA and the SDH assembly factor SDHAF2 (formerly known as SDH5 and required for flavination of SDH) have been described in hPGL [9–11]. Though primarily associated with hPGL, SDHB mutation carriers have additional increased susceptibility to renal cell cancers (RCC) [12–14]. In all cases, the loss-of-function germline mutations are followed by a somatic “second hit” of the second allele (usually deletion) in the tumor cells [15]. Furthermore, somatic mutations in several SDH genes are increasingly appreciated in sporadic PGL, PCC, and RCC [16]. Recently, a role for SDH mutations in gastrointestinal stromal tumors (GISTs) was also proposed. In particular, it was found that mutations in SDHB, SDHC, and SDHD are correlated with the rare development of a combination of hPGL and GIST, defined as Carney–Stratakis syndrome, and with the nonfamilial Carney triad, characterized by the presence of extra-adrenal paragangliomas, GIST, and pulmonary chondromas (reviewed in [17]).

The biochemistry and pathophysiology of oncometabolites accumulation in cancer. a Fumarate and succinate accumulate in the mitochondria and in the cytosol of cells expressing loss-of-function mutants of SDH or FH. 2-Hydroxyglutarate is accumulated as a consequence of neomorphic mutations in IDH1 in the cytosol and IDH2 in the mitochondria. b Biochemical effects of the accumulated oncometabolites in the cell. The effects are color coded: red for succinate, blue for fumarate, and green for 2HG. The accumulation of succinate impairs the enzymatic activity of several aKG-dependent dioxygenases: JMJd3, which regulates chromatin structure; PHD3, which is involved in promoting neuronal apoptosis in response to NGF withdrawal; and PHD2, which primarily regulates HIFα stability. Similarly, fumarate inhibits PHD2 enzymatic activity causing HIF stabilization. 2HG accumulation impairs DNA demethylation via the inhibition of the aKG-dependent dioxygenase TET2 and affects hematopoietic cells differentiation
FH mutations in hereditary leiomyomatosis and renal cell cancer
Fumarate hydratase (FH) catalyzes the reversible conversion of fumarate to malate in the TCA cycle (Fig. 1a). Loss-of-function germline mutations in FH predispose to hereditary leiomyomatosis and renal cell cancer (HLRCC), inherited leiomyomas (generally benign tumors of the smooth muscle), and renal (type II papillary and collecting duct) carcinoma [18, 19]. There is evidence to suggest that FH mutations may also be involved in the pathogenesis of breast, bladder, and testicular (Leydig cell) cancers [20, 21]. Similar to SDH in hPGL, enzymatic activity of FH is absent in HLRCC tumors and loss of the wild-type allele is observed in the majority of tumors [19].
IDH mutations in glioma and AML
Recently, mutations in isocitrate dehydrogenase (IDH) have been identified in gliomas and acute myelogenous leukemia (AML) [22–26]. IDH1 is one of three human IDH enzymes that catalyze the oxidative decarboxylation of isocitrate to α-ketoglutarate (aKG, also known as 2-oxoglutarate). IDH1 mutations are somatic and unlike SDH and FH mutations in hPGL and HLRCC respectively, no loss of heterozygosity has been demonstrated (i.e., all tumors retained one wild-type allele). Furthermore, the majority of IDH1 mutations in gliomas are strictly confined to a single residue, R132, whereas mutations in SDH and FH occur throughout the gene and though predominantly missense, changes also manifest as truncating, insertion, and deletion mutations. Sequence analysis of gliomas that are wild type for IDH1 R132 revealed a subset of tumors harboring mutations in the homologous exon of IDH2 and at the equivalent residue R172 [26]. Therefore, from a genetic point of view, IDH1/IDH2 mutations in glioma and AML appear dominant, oncogenic gain-of-function mutations. Indeed, a recent study demonstrated that these specific point mutations in IDH change the activity of the enzyme which no longer produces α-ketoglutarate but rather uses (reduces) α-ketoglutarate to a less familiar metabolite, 2-hydroxyglutarate (2HG) [27] (see below and Fig. 1a).
Biochemical and biological consequences of mutations in the cancer-associated metabolic enzymes
SDH and FH mutations in cancer: a tale of PHDs and HIFs
The observation that tumors derived from SDH or FH mutations are genetically and histologically characterized by a strong hypoxic signature and are significantly more vascularized [28–30] suggested a causal link between TCA cycle dysfunctions and the activation of the hypoxia-inducible transcription factors (HIFs), master regulators of the response to low oxygen. Importantly, HIF activation can also explain some of the metabolic alterations observed in these tumors and it may play a supportive role in the tumorigenic process. In fact, HIF is known to orchestrate the metabolic and genetic reprogramming required to sustain tumor cell growth, vascularization, and proliferation [31, 32]. The molecular link between TCA cycle dysfunction and HIF activation was initially proposed by Selak and colleagues who demonstrated that the accumulation of succinate in SDH-deficient cells causes the inhibition of the HIF prolyl 4-hydroxylases (PHDs), important regulators of the stability of the α subunit of HIF [33]. In normoxic conditions, PHDs hydroxylate two proline residues on the oxygen-dependent degradation domain of HIFα, targeting it to the ubiquitin–proteasome degradation machinery (Fig. 1b). This hydroxylation requires oxygen and α-ketoglutarate and produces carbon dioxide and succinate. For that reason, the accumulated succinate in SDH-deficient cells impairs PHDs activity and leads to HIFα stabilization under normoxic conditions, a phenomenon that has been defined as pseudohypoxia [15]. Importantly, the metabolic inhibition of PHDs is not unique to succinate as fumarate, which is accumulated in HLRCC cells, was also demonstrated to be a potent inhibitor of PHDs [34]. The accumulation of succinate and fumarate in the relevant cancer tissues as well as the accompanied PHD inhibition and pseudohypoxia was confirmed both in HLRCC leiomyomas and PGL tissues by Pollard et al. [35]. The biochemical characterization of the inhibition of PHDs by succinate and fumarate, as opposed to other TCA cycle metabolites, has been reported [36]. Furthermore, other biochemical studies showed that PHD activity is competitively inhibited by succinate or fumarate and, therefore, the ratio between α-ketoglutarate and succinate (or fumarate) rather than the absolute concentrations of these metabolites dictates PHD activity. In line with this, MacKenzie et al. used cell-permeable esters of α-ketoglutarate to reactivate PHDs enzymatic activity and thus alleviated pseudohypoxia caused by the accumulation of succinate or fumarate [37, 38].
An alternative model that links SDH and FH deficiency with PHD inactivation and HIF stabilization was proposed: Relying on a previously characterized role of respiratory chain-derived reactive oxygen species (ROS) as signals for HIF stabilization under hypoxia [39, 40], Guzy and colleagues showed that cells expressing mutant SDHB, but not mutant SDHA, are characterized by significant mitochondrial ROS production required, together with succinate, for a complete inactivation of PHDs [41]. Interestingly, these results fit better with earlier observations when SDHA was the only subunit of SDH that appeared not to be associated with hPGL. However, as discussed above, SDHA was recently added to the list of tumor suppressors in hPGL [11]. Nevertheless, it is still plausible that ROS accumulation in some SDH-mutated tumors may have an additive modifying role that affects the type and severity of the tumor (SDHB mutations are associated with RCC and more aggressive PCC). Of note, ROS accumulation was observed also in FH-deficient cells though the underlining mechanism appeared to be different. In fact, Sudarshan et al. demonstrated that in an FH-deficient cell line, defects in oxidative phosphorylation cause upregulation of glycolysis that initiates ROS generation by NADPH oxidase [42]. Unfortunately, conflicting findings do not allow for defining a clear biological picture of the role of ROS in these tumors. For instance, the findings of Selak and colleagues could not support the need of ROS for HIFα stabilization in SDH-deficient cells [43]. In addition, the reduction of fumarate levels, without the recovery of mitochondrial function obtained by reconstituting a cytosolic-confined FH into FH-deficient cells, was found to be sufficient for reactivating PHDs and for HIFα degradation [44].
Recently, the relevance of succinate as a regulator of HIF stability was extended also to hypoxia. Puisségur and colleagues found that the HIF-dependent expression of microRNA-210 (miR-210) targets several subunits of the respiratory chain and, importantly, SDHD, which causes accumulation of succinate [45]. This amplification loop suggests that the stabilization of HIFα under hypoxia could be modulated not only by the concentration of oxygen but also by the concentration of succinate allowing a further control of PHDs activity.
Is HIF the only relevant target?
While the key function of PHDs is to hydroxylate and destabilize HIFα, it is likely that other substrates of PHDs are playing roles in the response to hypoxia. Therefore, when PHDs are inhibited by succinate or fumarate, the inhibition of hydroxylation of these PHD substrates may contribute to tumorigenesis regardless of HIF activity. Two studies proposed a novel, HIF-independent role of PHD3 in neuronal apoptosis [46, 47]. These investigators found that apoptosis of neuronal or neuroendocrine-derived cells (like PGL and PCC) at early stages of development requires PHD3 activity (Fig. 1b). In this scenario, the accumulation of succinate due to SDH deficiency would impair PHD3-dependent apoptosis, therefore setting the stage for their neoplastic transformation. These findings also help to explain why SDH mutations predispose patients to PGL and PCC. However, direct PHD3 targets that may mediate the apoptotic response have not been identified. A similar PHD3-dependent developmental defect in FH-deficient cells has not been reported. In contrast, a genome-wide transcriptomic analysis revealed that FH-deficient leiomyomas are characterized by a significant downregulation of the serum response factor (SRF) and its target genes FOS and JUNB [48]. This genetic signature suggests that the cause of leiomyoma formation might be a defective SRF-dependent smooth muscle differentiation. However, whether PHDs could have a role in SRF-mediated differentiation is not known.
So far, succinate and fumarate have been depicted as mitochondria-to-cytosol signaling molecules that activate the HIF pathway via the inactivation of PHDs. However, the activity of other enzymes could be affected by the accumulation of TCA cycle metabolites. In particular, all members of the α-ketoglutarate-dependent dioxygenase family [49] may be inhibited by succinate or fumarate since similar to PHDs, their enzymatic activity requires α-ketoglutarate and they produce succinate. Among the members of this family, the jumonji domain- containing (JMJC) histone demethylases have been recently investigated. These enzymes remove the methyl marks on the arginine and lysine residues of histones after performing an α-ketoglutarate- and oxygen-dependent hydroxylation. It was shown that succinate accumulation in SDH-deficient cells impairs the activity of the histone demethylase JMJD3, leading to changes in the methylation mark of histone H3 on arginine 27 (Fig. 1b) [50]. Similarly, in a yeast model of paraganglioma, another histone demethylase, Jhd1, was found to be inhibited both in vitro and in vivo by succinate accumulation; importantly, this effect was reverted by the addition of exogenous, cell-permeable α-ketoglutarate[51].
Histone methylation is an important epigenetic modification which has been demonstrated to regulate gene expression by modulating chromatin structure and the binding of transcription factors [52]. Interestingly, it was recently shown that HIF1 promotes the transcription of two JMJ domain containing proteins, JMJD1A and JMJD2B [53, 54], possibly to facilitate the binding of HIF to the promoter regions of other HIF target genes. These findings might suggest that HIF transcriptional activity requires chromatin remodeling via the activity of these α-ketoglutarate-dependent enzymes. Importantly, unlike PHDs, these dioxygenases retain their activity even at low oxygen levels [55]. It is therefore likely that the presence of inhibiting concentrations of succinate or fumarate may modulate transcriptional activity of HIF and hence, may be responsible for transcriptional differences between hypoxic and pseudohypoxic conditions. Therefore, it is tempting to speculate that succinate and fumarate could act not only as mitochondria-to-cytosol but also as mitochondria-to-nucleus signaling units with the power to regulate gene expression by regulating chromatin structure.
Extracellular roles of succinate and fumarate: ways to help tumorigenesis?
Beyond the inhibition of α-ketoglutarate-dependent dioxygenases, a potentially additional role in the pathophysiology of TCA cycle metabolites was recently identified. In 2004, it was found that two orphan G protein-coupled receptor, namely GPR99 and GPR91, members of the purinergic P2 receptors family, were not activated by nucleotides, as initially proposed, but were actually responsive to extracellular α-ketoglutarate and succinate, respectively. These studies suggest that α-ketoglutarate and succinate can have a hormone-like role; they can be secreted into the bloodstream and convey specific signals to distal cells and tissues. In particular, it was shown that the stimulation of the GPR91 receptor by succinate triggers the secretion of renin from the kidney and leads to increased blood pressure [56]. More recent reports have shown that several other organs such as liver, adipose tissue, and the retina could sense succinate accumulation via the membrane receptor GRP91 (see [57] for a review). Interestingly, in ischemic retina, succinate activates GPR91 in retinal ganglion cells thus promoting vascular endothelial growth factor secretion and local neovascularization which is HIF- independent [58]. This important observation may suggest that in addition to the PHD-HIF pathway, some of the metabolic and pro-angiogenic effects of succinate in hPGL may actually depend on, or synergize with, the paracrine activation of the succinate receptor. Intriguingly, fumarate was found to have a role in regulating blood pressure and its accumulation in the blood correlates with hypertension in a salt-sensitive rat model [59]. It is tempting to speculate that the hormone-like physiological roles of succinate and potentially of fumarate may complement their intracellular metabolic signaling effects and further alter tumor microenvironment to support tumorigenesis.
IDH: a tumor suppressor or an oncogene?
In 2008, a genome-wide screening revealed somatic mutations of the TCA cycle enzyme IDH1 in low grade gliomas and secondary glioblastomas [24]. Just a few months after this seminal discovery, a large parallel DNA sequencing study found IDH1 mutations in AML patients [60]. Interestingly, subsequent studies demonstrated that also IDH2 is mutated in AML and glioma patients [26, 61, 62]. IDH1 and IDH2 are closely related NADP-linked enzymes, while IDH3 is NAD-linked (Fig. 1a). IDH2 and IDH3 are mitochondrial enzymes that function in the TCA cycle while IDH1 is located in the cytosol and peroxisomes, where it supplies NADPH-reducing equivalents for biosynthetic and other reactions [5]. It should be stressed that the canonical NAD-dependent reaction in the TCA cycle is a specific task of IDH3 which did not appear to be mutated in these cancers. As discussed above, the pattern of mutations (always at the same amino acid residue) and the heterozygosity state of the tumors (no loss of the wild-type allele) suggested an oncogenic, rather than a tumor suppressive role for IDH mutants. An unbiased metabolomic analysis of mutant IDH1-overexpressing cells found a striking intracellular accumulation of a poorly characterized metabolite, 2HG. In order to explain this finding, Dang et al. demonstrated that the mutant IDH1 acquires a neomorphic catalytic activity that allows a NADPH-dependent reduction of α-ketoglutarate into 2HG [27]. Importantly, this metabolite is significantly accumulated in glioma cells and in the blood of AML patients and therefore, despite its unclear role in tumorigenesis, the definition “oncometabolite” was coined by Dang et al. to portray the potential oncogenic contribution of 2HG (see [5] for discussion).
Other than the obvious genetic and biochemical differences between the loss of SDH and FH functions to the gain of IDH functions in tumors, several biological differences suggested that IDH mutations in cancer significantly differ from the genuine TCA cycle dysfunctions. First, as anticipated above, IDH1 and IDH2 appear to be mostly involved in the regulation of the NADP/NADPH ratio in the mitochondria and the cytosol [5] rather than in NADH production, a primary role of the TCA cycle which is executed by IDH3. Of note, the limited roles of IDH1 and IDH2 in the TCA cycle were substantiated by the absence of significant changes in the levels of TCA cycle metabolites in IDH1/IDH2-mutated cells [27, 62]. In addition, despite the fact that 2HG was proposed to act, similarly to succinate and fumarate, as an inhibitor of PHDs [63, 64], the presence of a HIF signature in IDH mutant cells and in gliomas is still debated. For the above reasons, while there may be some overlapping roles of 2HG, succinate, and fumarate, the underlining biochemical features which contribute to tumorigenesis in IDH and FH/SDH mutant cells seem to differ. A recent study in AML demonstrated genetic redundancy (mutual exclusiveness) between IDH mutations and TET2 deletions [65]. Importantly, TET2 is a recently discovered DNA demethylase which is also an α-ketoglutarate-dependent dioxygenase [66]. Indeed, Figueroa et al. demonstrated that 2HG can inhibit the DNA methylation status of TET2-expressing cells (Fig. 1b).
Summary
So far, alterations in three enzymatic reactions have been reported to possess genetically causal links to cancer formation: (1) Germline mutations in FH are associated with HLRCC (leiomyomas and RCC) and potentially with other tumors. (2) Both germline and somatic mutations in any of the four subunits of SDH (SDHA, SDHB, SDHC, SDHD) or the SDH assembly factor SDHAF2 are associated with PGL, PCC, and/or RCC. (3) Somatic mutations in either IDH1 or IDH2 are associated with gliomas or AML. While the FH and SDH mutations are typically loss-of-function mutations and the genes involved behave genetically like tumor suppressors, the IDH mutations lead to a gain of a new NADPH-dependent α-ketoglutarate-reductase activity which generates 2HG. Considering the fact that one wild-type IDH allele is retained in tumors with IDH mutations, and no significant changes in α-ketoglutarate or isocitrate levels were observed in these tumors, it is safe to propose that IDH1/IDH2 mutations are oncogenic gain-of-function mutations.
In all three types of these genetic-metabolic events, it appears that the underlying mechanism of tumorigenesis involved the accumulation of metabolites that convey oncogenic signals (oncometabolites). Although part of this oncogenic activity might be attributed to hormone-like effects of these molecules, strong evidence indicates that the principal oncometabolic activities of succinate, fumarate and 2HG are related to the inhibition of the α-ketoglutarate-dependent dioxygenases, alas with different specificities to different enzymes and therefore, with different biochemical and biological consequences. More generally, these observations reveal a dynamic and bidirectional interaction between the metabolic status of the cell and its genetic profile and propose that small metabolites may be novel and unexplored signaling units.
References
Warburg O (1956) On the origin of cancer cells. Science 123:309–314
Frezza C, Gottlieb E (2009) Mitochondria in cancer: not just innocent bystanders. Semin Cancer Biol 19:4–11
Tennant DA, Duran RV, Gottlieb E (2010) Targeting metabolic transformation for cancer therapy. Nat Rev Cancer 10:267–277
Kaelin WG Jr (2009) SDH5 mutations and familial paraganglioma: somewhere Warburg is smiling. Cancer Cell 16:180–182
Frezza C, Tennant DA, Gottlieb E (2010) IDH1 mutations in gliomas: when an enzyme loses its grip. Cancer Cell 17:7–9
Baysal BE, Ferrell RE, Willett-Brozick JE, Lawrence EC, Myssiorek D, Bosch A, van der Mey A, Taschner PE, Rubinstein WS, Myers EN, Richard CW 3rd, Cornelisse CJ, Devilee P, Devlin B (2000) Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science 287:848–851
Niemann S, Muller U (2000) Mutations in SDHC cause autosomal dominant paraganglioma, type 3. Nat Genet 26:268–270
Astuti D, Latif F, Dallol A, Dahia PL, Douglas F, George E, Skoldberg F, Husebye ES, Eng C, Maher ER (2001) Gene mutations in the succinate dehydrogenase subunit SDHB cause susceptibility to familial pheochromocytoma and to familial paraganglioma. Am J Hum Genet 69:49–54
Hao HX, Khalimonchuk O, Schraders M, Dephoure N, Bayley JP, Kunst H, Devilee P, Cremers CW, Schiffman JD, Bentz BG, Gygi SP, Winge DR, Kremer H, Rutter J (2009) SDH5, a gene required for flavination of succinate dehydrogenase, is mutated in paraganglioma. Science 325:1139–1142
Bayley JP, Kunst HP, Cascon A, Sampietro ML, Gaal J, Korpershoek E, Hinojar-Gutierrez A, Timmers HJ, Hoefsloot LH, Hermsen MA, Suarez C, Hussain AK, Vriends AH, Hes FJ, Jansen JC, Tops CM, Corssmit EP, de Knijff P, Lenders JW, Cremers CW, Devilee P, Dinjens WN, de Krijger RR, Robledo M (2010) SDHAF2 mutations in familial and sporadic paraganglioma and phaeochromocytoma. Lancet Oncol 11:366–372
Burnichon N, Briere JJ, Libe R, Vescovo L, Riviere J, Tissier F, Jouanno E, Jeunemaitre X, Benit P, Tzagoloff A, Rustin P, Bertherat J, Favier J, Gimenez-Roqueplo AP (2010) SDHA is a tumor suppressor gene causing paraganglioma. Hum Mol Genet 19:3011–3020
Housley SL, Lindsay RS, Young B, McConachie M, Mechan D, Baty D, Christie L, Rahilly M, Qureshi K, Fleming S (2010) Renal carcinoma with giant mitochondria associated with germ-line mutation and somatic loss of the succinate dehydrogenase B gene. Histopathology 56:405–408
Ricketts C, Woodward ER, Killick P, Morris MR, Astuti D, Latif F, Maher ER (2008) Germline SDHB mutations and familial renal cell carcinoma. J Natl Cancer Inst 100:1260–1262
Henderson A, Douglas F, Perros P, Morgan C, Maher ER (2009) SDHB-associated renal oncocytoma suggests a broadening of the renal phenotype in hereditary paragangliomatosis. Fam Can 8:257–260
Gottlieb E, Tomlinson IP (2005) Mitochondrial tumour suppressors: a genetic and biochemical update. Nat Rev Cancer 5:857–866
van Nederveen FH, Korpershoek E, Lenders JW, de Krijger RR, Dinjens WN (2007) Somatic SDHB mutation in an extraadrenal pheochromocytoma. N Engl J Med 357:306–308
Stratakis CA, Carney JA (2009) The triad of paragangliomas, gastric stromal tumours and pulmonary chondromas (Carney triad), and the dyad of paragangliomas and gastric stromal sarcomas (Carney–Stratakis syndrome): molecular genetics and clinical implications. J Intern Med 266:43–52
Launonen V, Vierimaa O, Kiuru M, Isola J, Roth S, Pukkala E, Sistonen P, Herva R, Aaltonen LA (2001) Inherited susceptibility to uterine leiomyomas and renal cell cancer. Proc Natl Acad Sci USA 98:3387–3392
Tomlinson IP, Alam NA, Rowan AJ, Barclay E, Jaeger EE, Kelsell D, Leigh I, Gorman P, Lamlum H, Rahman S, Roylance RR, Olpin S, Bevan S, Barker K, Hearle N, Houlston RS, Kiuru M, Lehtonen R, Karhu A, Vilkki S, Laiho P, Eklund C, Vierimaa O, Aittomaki K, Hietala M, Sistonen P, Paetau A, Salovaara R, Herva R, Launonen V, Aaltonen LA (2002) Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat Genet 30:406–410
Carvajal-Carmona LG, Alam NA, Pollard PJ, Jones AM, Barclay E, Wortham N, Pignatelli M, Freeman A, Pomplun S, Ellis I, Poulsom R, El-Bahrawy MA, Berney DM, Tomlinson IP (2006) Adult leydig cell tumors of the testis caused by germline fumarate hydratase mutations. J Clin Endocrinol Metab 91:3071–3075
Lehtonen HJ, Kiuru M, Ylisaukko-Oja SK, Salovaara R, Herva R, Koivisto PA, Vierimaa O, Aittomaki K, Pukkala E, Launonen V, Aaltonen LA (2006) Increased risk of cancer in patients with fumarate hydratase germline mutation. J Med Genet 43:523–526
Marcucci G, Maharry K, Wu YZ, Radmacher MD, Mrozek K, Margeson D, Holland KB, Whitman SP, Becker H, Schwind S, Metzeler KH, Powell BL, Carter TH, Kolitz JE, Wetzler M, Carroll AJ, Baer MR, Caligiuri MA, Larson RA, Bloomfield CD (2010) IDH1 and IDH2 gene mutations identify novel molecular subsets within de novo cytogenetically normal acute myeloid leukemia: a Cancer and Leukemia Group B study. J Clin Oncol 28:2348–2355
Balss J, Meyer J, Mueller W, Korshunov A, Hartmann C, von Deimling A (2008) Analysis of the IDH1 codon 132 mutation in brain tumors. Acta Neuropathol 116:597–602
Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, Olivi A, McLendon R, Rasheed BA, Keir S, Nikolskaya T, Nikolsky Y, Busam DA, Tekleab H, Diaz LA Jr, Hartigan J, Smith DR, Strausberg RL, Marie SK, Shinjo SM, Yan H, Riggins GJ, Bigner DD, Karchin R, Papadopoulos N, Parmigiani G, Vogelstein B, Velculescu VE, Kinzler KW (2008) An integrated genomic analysis of human glioblastoma multiforme. Science 321:1807–1812
Bleeker FE, Lamba S, Leenstra S, Troost D, Hulsebos T, Vandertop WP, Frattini M, Molinari F, Knowles M, Cerrato A, Rodolfo M, Scarpa A, Felicioni L, Buttitta F, Malatesta S, Marchetti A, Bardelli A (2009) IDH1 mutations at residue p.R132 (IDH1 R132) occur frequently in high-grade gliomas but not in other solid tumors. Hum Mutat 30:7–11
Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, Kos I, Batinic-Haberle I, Jones S, Riggins GJ, Friedman H, Friedman A, Reardon D, Herndon J, Kinzler KW, Velculescu VE, Vogelstein B, Bigner DD (2009) IDH1 and IDH2 mutations in gliomas. N Engl J Med 360:765–773
Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, Fantin VR, Jang HG, Jin S, Keenan MC, Marks KM, Prins RM, Ward PS, Yen KE, Liau LM, Rabinowitz JD, Cantley LC, Thompson CB, Vander Heiden MG, Su SM (2009) Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 462:739–744
Gimenez-Roqueplo AP, Favier J, Rustin P, Mourad JJ, Plouin PF, Corvol P, Rotig A, Jeunemaitre X (2001) The R22X mutation of the SDHD gene in hereditary paraganglioma abolishes the enzymatic activity of complex II in the mitochondrial respiratory chain and activates the hypoxia pathway. Am J Hum Genet 69:1186–1197
Dahia PL, Ross KN, Wright ME, Hayashida CY, Santagata S, Barontini M, Kung AL, Sanso G, Powers JF, Tischler AS, Hodin R, Heitritter S, Moore F, Dluhy R, Sosa JA, Ocal IT, Benn DE, Marsh DJ, Robinson BG, Schneider K, Garber J, Arum SM, Korbonits M, Grossman A, Pigny P, Toledo SP, Nose V, Li C, Stiles CD (2005) A HIF1alpha regulatory loop links hypoxia and mitochondrial signals in pheochromocytomas. PLoS Genet 1:72–80
Vanharanta S, Pollard PJ, Lehtonen HJ, Laiho P, Sjoberg J, Leminen A, Aittomaki K, Arola J, Kruhoffer M, Orntoft TF, Tomlinson IP, Kiuru M, Arango D, Aaltonen LA (2006) Distinct expression profile in fumarate-hydratase-deficient uterine fibroids. Hum Mol Genet 15:97–103
Cervera AM, Apostolova N, Crespo FL, Mata M, McCreath KJ (2008) Cells silenced for SDHB expression display characteristic features of the tumor phenotype. Cancer Res 68:4058–4067
Ashrafian H, O'Flaherty L, Adam J, Steeples V, Chung YL, East P, Vanharanta S, Lehtonen H, Nye E, Hatipoglu E, Miranda M, Howarth K, Shukla D, Troy H, Griffiths J, Spencer-Dene B, Yusuf M, Volpi E, Maxwell PH, Stamp G, Poulsom R, Pugh CW, Costa B, Bardella C, Di Renzo MF, Kotlikoff MI, Launonen V, Aaltonen L, El-Bahrawy M, Tomlinson I, Pollard PJ (2010) Expression profiling in progressive stages of fumarate-hydratase deficiency: the contribution of metabolic changes to tumorigenesis. Cancer Res 70:9153–9165
Selak MA, Armour SM, MacKenzie ED, Boulahbel H, Watson DG, Mansfield KD, Pan Y, Simon MC, Thompson CB, Gottlieb E (2005) Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell 7:77–85
Isaacs JS, Jung YJ, Mole DR, Lee S, Torres-Cabala C, Chung YL, Merino M, Trepel J, Zbar B, Toro J, Ratcliffe PJ, Linehan WM, Neckers L (2005) HIF overexpression correlates with biallelic loss of fumarate hydratase in renal cancer: novel role of fumarate in regulation of HIF stability. Cancer Cell 8:143–153
Pollard PJ, Briere JJ, Alam NA, Barwell J, Barclay E, Wortham NC, Hunt T, Mitchell M, Olpin S, Moat SJ, Hargreaves IP, Heales SJ, Chung YL, Griffiths JR, Dalgleish A, McGrath JA, Gleeson MJ, Hodgson SV, Poulsom R, Rustin P, Tomlinson IP (2005) Accumulation of Krebs cycle intermediates and over-expression of HIF1alpha in tumours which result from germline FH and SDH mutations. Hum Mol Genet 14:2231–2239
Hewitson KS, Lienard BM, McDonough MA, Clifton IJ, Butler D, Soares AS, Oldham NJ, McNeill LA, Schofield CJ (2007) Structural and mechanistic studies on the inhibition of the hypoxia-inducible transcription factor hydroxylases by tricarboxylic acid cycle intermediates. J Biol Chem 282:3293–3301
MacKenzie ED, Selak MA, Tennant DA, Payne LJ, Crosby S, Frederiksen CM, Watson DG, Gottlieb E (2007) Cell-permeating alpha-ketoglutarate derivatives alleviate pseudohypoxia in succinate dehydrogenase-deficient cells. Mol Cell Biol 27:3282–3289
Porcelli AM, Ghelli A, Ceccarelli C, Lang M, Cenacchi G, Capristo M, Pennisi LF, Morra I, Ciccarelli E, Melcarne A, Bartoletti-Stella A, Salfi N, Tallini G, Martinuzzi A, Carelli V, Attimonelli M, Rugolo M, Romeo G, Gasparre G (2010) The genetic and metabolic signature of oncocytic transformation implicates HIF1alpha destabilization. Hum Mol Genet 19:1019–1032
Guzy RD, Hoyos B, Robin E, Chen H, Liu L, Mansfield KD, Simon MC, Hammerling U, Schumacker PT (2005) Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab 1:401–408
Mansfield KD, Guzy RD, Pan Y, Young RM, Cash TP, Schumacker PT, Simon MC (2005) Mitochondrial dysfunction resulting from loss of cytochrome c impairs cellular oxygen sensing and hypoxic HIF-alpha activation. Cell Metab 1:393–399
Guzy RD, Sharma B, Bell E, Chandel NS, Schumacker PT (2008) Loss of the SdhB, but not the SdhA, subunit of complex II triggers ROS-dependent HIF activation and tumorigenesis. Mol Cell Biol 28:718–731
Sudarshan S, Sourbier C, Kong HS, Block K, Valera Romero VA, Yang Y, Galindo C, Mollapour M, Scroggins B, Goode N, Lee MJ, Gourlay CW, Trepel J, Linehan WM, Neckers L (2009) Fumarate hydratase deficiency in renal cancer induces glycolytic addiction and hypoxia-inducible transcription factor 1alpha stabilization by glucose-dependent generation of reactive oxygen species. Mol Cell Biol 29:4080–4090
Selak MA, Duran RV, Gottlieb E (2006) Redox stress is not essential for the pseudo-hypoxic phenotype of succinate dehydrogenase deficient cells. Biochim Biophys Acta 1757:567–572
O'Flaherty L, Adam J, Heather LC, Zhdanov AV, Chung YL, Miranda MX, Croft J, Olpin S, Clarke K, Pugh CW, Griffiths J, Papkovsky D, Ashrafian H, Ratcliffe PJ, Pollard PJ (2010) Dysregulation of hypoxia pathways in fumarate hydratase-deficient cells is independent of defective mitochondrial metabolism. Hum Mol Genet 19:3844–3851
Puissegur MP, Mazure NM, Bertero T, Pradelli L, Grosso S, Robbe-Sermesant K, Maurin T, Lebrigand K, Cardinaud B, Hofman V, Fourre S, Magnone V, Ricci JE, Pouyssegur J, Gounon P, Hofman P, Barbry P, Mari B (2011) miR-210 is overexpressed in late stages of lung cancer and mediates mitochondrial alterations associated with modulation of HIF-1 activity. Cell Death Differ (in press)
Lee S, Nakamura E, Yang H, Wei W, Linggi MS, Sajan MP, Farese RV, Freeman RS, Carter BD, Kaelin WG Jr, Schlisio S (2005) Neuronal apoptosis linked to EglN3 prolyl hydroxylase and familial pheochromocytoma genes: developmental culling and cancer. Cancer Cell 8:155–167
Schlisio S, Kenchappa RS, Vredeveld LC, George RE, Stewart R, Greulich H, Shahriari K, Nguyen NV, Pigny P, Dahia PL, Pomeroy SL, Maris JM, Look AT, Meyerson M, Peeper DS, Carter BD, Kaelin WG Jr (2008) The kinesin KIF1Bbeta acts downstream from EglN3 to induce apoptosis and is a potential 1p36 tumor suppressor. Genes Dev 22:884–893
Raimundo N, Vanharanta S, Aaltonen LA, Hovatta I, Suomalainen A (2009) Downregulation of SRF-FOS-JUNB pathway in fumarate hydratase deficiency and in uterine leiomyomas. Oncogene 28:1261–1273
Schofield CJ, Ratcliffe PJ (2004) Oxygen sensing by HIF hydroxylases. Nat Rev Mol Cell Biol 5:343–354
Cervera AM, Bayley JP, Devilee P, McCreath KJ (2009) Inhibition of succinate dehydrogenase dysregulates histone modification in mammalian cells. Mol Cancer 8:89
Smith EH, Janknecht R, Maher LJ 3rd (2007) Succinate inhibition of alpha-ketoglutarate-dependent enzymes in a yeast model of paraganglioma. Hum Mol Genet 16:3136–3148
Teperino R, Schoonjans K, Auwerx J (2010) Histone methyl transferases and demethylases; can they link metabolism and transcription? Cell Metab 12:321–327
Pollard PJ, Loenarz C, Mole DR, McDonough MA, Gleadle JM, Schofield CJ, Ratcliffe PJ (2008) Regulation of Jumonji-domain-containing histone demethylases by hypoxia-inducible factor (HIF)-1alpha. Biochem J 416:387–394
Beyer S, Kristensen MM, Jensen KS, Johansen JV, Staller P (2008) The histone demethylases JMJD1A and JMJD2B are transcriptional targets of hypoxia-inducible factor HIF. J Biol Chem 283:36542–36552
Yang J, Jubb AM, Pike L, Buffa FM, Turley H, Baban D, Leek R, Gatter KC, Ragoussis J, Harris AL (2010) The histone demethylase JMJD2B is regulated by estrogen receptor alpha and hypoxia, and is a key mediator of estrogen induced growth. Cancer Res 70:6456–6466
He W, Miao FJ, Lin DC, Schwandner RT, Wang Z, Gao J, Chen JL, Tian H, Ling L (2004) Citric acid cycle intermediates as ligands for orphan G-protein-coupled receptors. Nature 429:188–193
Peti-Peterdi J (2010) High glucose and renin release: the role of succinate and GPR91. Kidney Int 78:1214–1217
Sapieha P, Sirinyan M, Hamel D, Zaniolo K, Joyal JS, Cho JH, Honore JC, Kermorvant-Duchemin E, Varma DR, Tremblay S, Leduc M, Rihakova L, Hardy P, Klein WH, Mu X, Mamer O, Lachapelle P, Di Polo A, Beausejour C, Andelfinger G, Mitchell G, Sennlaub F, Chemtob S (2008) The succinate receptor GPR91 in neurons has a major role in retinal angiogenesis. Nat Med 14:1067–1076
Tian Z, Liu Y, Usa K, Mladinov D, Fang Y, Ding X, Greene AS, Cowley AW Jr, Liang M (2009) Novel role of fumarate metabolism in dahl-salt sensitive hypertension. Hypertension 54:255–260
Mardis ER, Ding L, Dooling DJ, Larson DE, McLellan MD, Chen K, Koboldt DC, Fulton RS, Delehaunty KD, McGrath SD, Fulton LA, Locke DP, Magrini VJ, Abbott RM, Vickery TL, Reed JS, Robinson JS, Wylie T, Smith SM, Carmichael L, Eldred JM, Harris CC, Walker J, Peck JB, Du F, Dukes AF, Sanderson GE, Brummett AM, Clark E, McMichael JF, Meyer RJ, Schindler JK, Pohl CS, Wallis JW, Shi X, Lin L, Schmidt H, Tang Y, Haipek C, Wiechert ME, Ivy JV, Kalicki J, Elliott G, Ries RE, Payton JE, Westervelt P, Tomasson MH, Watson MA, Baty J, Heath S, Shannon WD, Nagarajan R, Link DC, Walter MJ, Graubert TA, DiPersio JF, Wilson RK, Ley TJ (2009) Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med 361:1058–1066
Gross S, Cairns RA, Minden MD, Driggers EM, Bittinger MA, Jang HG, Sasaki M, Jin S, Schenkein DP, Su SM, Dang L, Fantin VR, Mak TW (2010) Cancer-associated metabolite 2-hydroxyglutarate accumulates in acute myelogenous leukemia with isocitrate dehydrogenase 1 and 2 mutations. J Exp Med 207:339–344
Ward PS, Patel J, Wise DR, Abdel-Wahab O, Bennett BD, Coller HA, Cross JR, Fantin VR, Hedvat CV, Perl AE, Rabinowitz JD, Carroll M, Su SM, Sharp KA, Levine RL, Thompson CB (2010) The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 17:225–234
Zhao S, Lin Y, Xu W, Jiang W, Zha Z, Wang P, Yu W, Li Z, Gong L, Peng Y, Ding J, Lei Q, Guan KL, Xiong Y (2009) Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1alpha. Science 324:261–265
Pollard PJ, Ratcliffe PJ (2009) Cancer. Puzzling patterns of predisposition. Science 324:192–194
Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, Li Y, Bhagwat N, Vasanthakumar A, Fernandez HF, Tallman MS, Sun Z, Wolniak K, Peeters JK, Liu W, Choe SE, Fantin VR, Paietta E, Lowenberg B, Licht JD, Godley LA, Delwel R, Valk PJ, Thompson CB, Levine RL, Melnick A (2010) Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 18:553–567
Ko M, Huang Y, Jankowska AM, Pape UJ, Tahiliani M, Bandukwala HS, An J, Lamperti ED, Koh KP, Ganetzky R, Liu XS, Aravind L, Agarwal S, Maciejewski JP, Rao A (2010) Impaired hydroxylation of 5-methylcytosine in myeloid cancers with mutant TET2. Nature 468:839–843
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Frezza, C., Pollard, P.J. & Gottlieb, E. Inborn and acquired metabolic defects in cancer. J Mol Med 89, 213–220 (2011). https://doi.org/10.1007/s00109-011-0728-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-011-0728-4