




Abstract
Lung cancer is the leading cause of cancer deaths worldwide and current therapies fail to treat this disease in majority of cases. Antrodia camphorata is a medicinal mushroom being widely used as food dietary supplement for cancer prevention. The sesquiterpene lactone antrocin is the most potent among >100 secondary metabolites isolated from A. camphorata . However, the molecular mechanisms of antrocin-mediated anticancer effects remain unclear. In this study, we found that antrocin inhibited cell proliferation in two non-small-cell lung cancer cells, namely H441 (wild-type epidermal growth factor receptor, IC 50 = 0.75 μM) and H1975 (gefitnib-resistant mutant T790M, IC 50 = 0.83 μM). Antrocin dose dependently suppressed colony formation and induced apoptosis as evidenced by activated caspase-3 and increased Bax/Bcl2 ratio. Gene profiling studies indicated that antrocin downregulated Janus kinase/signal transducer and activator of transcription (JAK/STAT) signaling pathway. We further demonstrated that antrocin suppressed both constitutively activated and interleukin 6-induced STAT3 phosphorylation and its subsequent nuclear translocation. Such inhibition is found to be achieved through the suppression of JAK2 and interaction between STAT3 and extracellular signal-regulated kinase. Additionally, antrocin increased microRNA let-7c expression and suppressed STAT signaling. The combination of antrocin and JAK2/STAT3 gene silencing significantly increased apoptosis in H441 cells. Such dual interruption of JAK2 and STAT3 pathways also induced downregulation of antiapoptotic protein mcl-1 and increased caspase-3 expression. In vivo intraperitoneal administration of antrocin significantly suppressed the growth of lung cancer tumor xenografts. Our results indicate that antrocin may be a potential therapeutic agent for human lung cancer cells through constitutive inhibition of JAK2/STAT3 pathway.
Introduction
Lung cancer continues to be the leading form of cancer death in both men and women in the United States with an estimated 166 280 deaths attributed to lung cancer in 2008 ( 1 ). Given the inherent insensitivity to cytotoxic agents, identifying molecules that drive lung cancer growth, survival and metastasis is critical as is the development of novel therapeutics. The Janus kinase/signal transducer and activator of transcription (JAK/STAT) signaling components are potential targets in lung cancer and other cancers ( 2 ). These proteins are activated by upstream tyrosine kinase signaling and control genes that regulate cancer hallmarks. Persistent activation of JAK/STAT signaling contributes to the malignancy of tumors by promoting tumor cell proliferation and survival, angiogenesis and immune evasion ( 3–6 ). A number of recent studies have found activated JAK/STAT in lung cancer cell lines and tissues, thus suggesting a functional role for this target in subsets of lung cancer ( 7–10 ).
Recently, small-molecule inhibitors of JAK/STAT signaling have been identified for development of cancer therapeutics. Discovery of the JAK2 V617F mutation in myeloproliferative disease has prompted development of selective JAK2 inhibitors for treatment of hematologic disorders ( 11–13 ). In addition to its clear role in hematologic malignancies, the JAK/STAT pathway is also activated in many solid tumors, including head and neck squamous cell carcinoma, non-small-cell lung cancer (NSCLC), and small-cell lung cancer (SCLC). It is important to define the role of this pathway in these solid tumors, as better therapies are desperately needed for these malignancies, and new therapeutic agents that target both JAK and STAT are being developed. Indeed, JAK kinase inhibitors are already being tested in clinical trials ( 14 , 15 ). Furthermore, PD180970 has been reported to inhibit tyrosine phosphorylation of JAK1 and this off-target effect of the compound could contribute to its effects on STAT3 ( 16 ). In addition to JAK molecules, the epidermal growth factor receptor (EGFR) is also a candidate tyrosine kinase responsible for STAT3 activation. Interestingly, small-molecule inhibitors of JAK/STAT3 signaling have been reported to suppress cancer cell growth, both in vitro and in vivo ( 17 , 18 ).
Antrocin is a sesquiterpene lactone isolated from a medicinal mushroom Antrodia camphorata . Antrodia camphorata has been widely used as food dietary supplement for cancer prevention and hepatoprotection in several Asian and European countries ( 19 ). The chemistry of this medicinal mushroom has been extensively studied leading to the identification of >100 secondary metabolites. Among these, particular attention has been directed to antrocin. Presently, this compound is reputed as the most potent that contribute A. camphorata pharmacological efficacy ( 19 ). In our previous study, antrocin has been reported as a potent antagonist in various cancer cells including breast, lung, liver and colon, being highest in metastatic breast cancer MDA-MB-231 cells ( 20 ). Additionally, we also demonstrated that antrocin possesses potent anticancer property through promoting apoptosis in MDA-MB-231 cells ( 20 ). However, whether antrocin could inactivate other survival signaling pathways to achieve its antitumor effect remains unclear.
In this study, we aimed to investigate the effect of antrocin on non-small lung cancer cells and identified that at least one of its action mechanisms through the inhibition of JAK/STAT3 signaling pathway. Furthermore, to better support in vivo studies, we also assessed the effects of antrocin in animal tumor models.
Materials and methods
Reagents, cell lines and culture
Antrocin used in our experiments was synthesized following the procedures described recently ( 21 ). Human lung adenocarcinoma cell lines with increasing invasive ability (CL1-0 and CL1-5) were generous gifts from Dr Pan-Chyr Yang, School of Medicine, National Taiwan University. Lung cancer cell lines (i.e. A549, H1975, H441, PC9 and BEAS-2B) were obtained from the ATCC. The cells were grown in RPMI 1640 culture medium or Dulbecco’s modified Eagle’s medium (GIBCO-Life Technologies, Gaithersburg, MD), supplemented with 1.5g/l of NaHCO 3 , and 10% fetal bovine serum (GIBCO-Life Technologies). Propidium iodide (PI), dimethyl sulfoxide and sulforhodamine B (SRB) were purchased from Sigma–Aldrich (St Louis, MO). The Annexin-V/FITC Apoptosis Kits were obtained from R&D Systems (Minneapolis, MN). Antibodies to STAT3 (Tyr705), p-TKY, p-JAK1 (Try1022), p-JAK2 (Try1007), p-Akt (Ser473), Akt and MAPK (Thr202/Tyr204), poly-(ADP) ribose polymerase, cleaved poly-(ADP) ribose polymerase, gp130, cleaved caspase-3 (Asp175), β-actin and horseradish-peroxidase-linked rabbit IgG were purchased from Cell Signaling Technology (Beverly, MA). Antibodies to B-cell lymphoma 2 (Bcl-2), survivin, Bax and p53 were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). A TRIzol RNA isolation kit was obtained from Life Technologies (Rockville, MD), and primers for reverse transcription–polymerase chain reaction (RT–PCR), deoxynucleoside triphosphate, reverse transcriptase and Taq polymerase were obtained from Gibco BRL (Cergy Pontoise, France). All other chemicals were of the highest pure grade available.
Cell growth and cell death/apoptosis assays
The effect of antrocin on cell number was estimated by the mitochondrial-metabolism-based SRB colorimetric methods in 96-well plate format. Apoptosis was evaluated by staining with 4′,6-diamidino-2-phenylindole. DNA fragmentation analysis and cell-cycle distribution were carried out with PI staining according to the method as we reported previously ( 20 ) and flow cytometry using a FACSCalibur flow cytometer (Becton Dickinson, San Jose, CA).
Anchorage-independent growth
H441 cells were harvested and seeded in six-well plates coated with 1% agarose. Anchorage-independent growth was assessed after incubation for 20 days in culture media without or with antrocin (5 and 10 μM), which was replaced every 4 days. Plates were stained with 0.005% crystal violet, and the colonies were counted manually under a microscope and photographed.
Microarray expression profiling and data analysis
H441 cells were seeded at 5 × 10 6 cells/10cm plate overnight at <60% confluency and subsequently treated with either vehicle control (0.2% dimethyl sulfoxide) or antrocin (5 µM) for 12h. Total RNA from cell cultures of H441 was isolated using a Trireagent according to the manufacturer’s protocols. The Illumina TotalPrep RNA Amplification Kit was used for generating biotinylated, amplified RNA for hybridization with Illumina arrays. Complementary RNAs were hybridized to the HumanWG-6 BeadChip array, which allows to assay >48 000 transcripts (Whole Genome). The images were analyzed using the Genome studio software. Differential gene expression analysis between control and antrocin-treated samples was subsequently conducted using the Illumina customer model, which applies multiple testing corrections, that determines the false discovery rate (FDR). Data were normalized and selected by detecting P -value < 0.05, fold >1.5. Gene network and pathway analysis was performed using Ingenuity Pathways Analysis ( http://www.ingenuity.com ).
RNA extraction and RT–PCR
Cellular RNA was extracted with a TRIzol RNA isolation kit as described in the manufacturer’s manual. RNA concentration and purity were determined based on measurement of the absorbance at 260 and 280nm. After the addition of RNase inhibitor (20U), the total RNA was stored at −70°C. The sense and antisense primer sequences used were as follows: Bcl-2: 5′-TATAAGCTGTCGCAGAGGGGCTA-3′ and 5′-GTACTCAGTCATCCAC AGGGCGAT-3′; Survivin: 5′-AGATGACGACCCCATGCAAA-3′ and 5′-CGC ACTTTCTCCGCAGTTTC-3′; Cyclin D1: 5′-GCCCTCGGTGTCCTACTTC -3′ and 5′-AGGAAGCGGTCCAGGTAGTT-3′; Mcl-1: 5′-GAGACCTTACG ACGGGTT-3′ and 5′-TTTGATGTCCAGTTTCCG-3′; glyceraldehyde 3-phosphate dehydrogenase: 5′-TCCAAAATCAAGTGGGGCGA-3′ and 5′-AAAT GAGCCCCAGCCTTCTC-3′. From each sample, 250ng of RNA was reverse transcribed, using 200U of SuperScript II™ RNase-H reverse transcriptase, 20U of RNase inhibitor, 0.6mM deoxynucleoside triphosphates and 0.5mg/ml oligo (dT) 12–18. PCR was performed with Platinum Taq polymerase under the following conditions: 35 cycles of 94°C for 1min, 56°C (Bcl-2, CyclinD1, Mcl-1 and survivin) or 63°C (glyceraldehyde 3-phosphate dehydrogenase) for 1min, 72°C for 2min followed by 10min at 72°C. A 10 μl aliquot from each PCR was separated in a 1.8% agarose gel containing 0.2mg/ml ethidium bromide.
Immunofluorescence analysis
For immunofluorescence analysis, cells were plated in six-well chamber slides (Nunc™, Thermo Fisher Scientific) for 24h before treatment with 5 μM antrocin for 48h. Afterwards, cells were fixed in 2% paraformaldehyde for 10min at room temperature, permeabilized with 0.1% Triton X-100 in 0.01M phosphate-buffered saline (PBS), pH 7.4 containing 0.2% bovine serum albumin, air dried and rehydrated in PBS. Cells were then incubated with a rabbit polyclonal antibody against p-STAT-3 and diluted 1:500 in PBS containing 3% normal goat serum for 2h at room temperature. Negative controls were performed by omitting the primary antibody. After washing twice in PBS for 10min, an anti-rabbit IgG fluorescein isothiocyanate-conjugated secondary antibody (Jackson Immunoresearch) that was diluted 1:500 in PBS was added. The cells were let to sit for a period of 1h at room temperature. Cells were then washed in PBS and mounted in Vectashield mounting medium with 4′,6-diamidino-2-phenylindole to counter stain DNA. Cells were observed using a Zeiss Axiophot (Carl Zeiss) fluorescence microscope. Microphotographs were acquired using AxioCam MRc digital video camera and Axiovision Zeiss software (Carl Zeiss).
Preparation of cell lysates and nuclear fractions
The cell lysates and nuclear fractions were prepared using the Nuclear Extraction Kit (Panomics). Briefly, harvested cells (1 × 10 6 cells/6 cm plate) were washed twice with 5ml cold 1× PBS. A 0.5ml aliquot of Buffer A working reagent (a combination of 0.5ml 1× Buffer A, 5 μl dithiothreitol, 5 μl protease inhibitor cocktail and 20 μl 10% IGEPAL) was added to each plate. The plate was transferred to an ice bucket on a rocking platform at 150 r.p.m. for 10min. Each sample was centrifuged at 14 000 g for 3min at 4°C. The supernatant (cytosolic fraction) was removed and the pellet kept on ice. A 75 ml aliquot of Buffer B working reagent [a combination of 0.5ml 1× Buffer B, 5 μl dithiothreitol, and 5 μl (protease inhibitor cocktail)] was added to each pellet and vortexed at the highest setting for 10 s. The Eppendorf tube was placed horizontally in an ice bucket that was transferred to a rocking platform at 150 r.p.m. for 2h. After centrifugation at 14 000 g for 5min at 4°C, the supernatant (nuclear extract) was transferred to a new Eppendorf tube for the measurement of the protein concentration of each sample and was stored at −80°C.
Western blotting
Samples (10 μg) of total cell lysates were size fractionated electrophoretically by a 10% polyacrylamide sodium dodecyl sulfate–polyacrylamide gel electrophoresis and transferred onto a polyvinylidene difluoride membrane using the BioRad Mini Protean electrotransfer system. The blots were subsequently incubated with 5% skim milk in phosphate-buffered saline with Tween-20 for 1h to block non-specific binding and was probed overnight at 4°C with the antibodies against total and phosphorylated STAT3 (Tyr705), p-TKY, p-JAK1 (Try1022), p-JAK2 (Try 1007), p-Akt (Ser473), Akt, gp130 and MAPK (Thr202/Tyr204); Bcl-2, Bax, survivin, poly-(ADP) ribose polymerase, p53, caspase-3 and β-actin. The membranes were sequentially detected with an appropriate peroxidase-conjugated secondary antibody incubated at room temperature for 1h. Intensive PBS washing was performed after each incubation. After the final PBS washing, signals were developed using the enhanced chemiluminescence detection system and Kodak X-OMAT Blue Autoradiography Film.
Small interfering RNA knockdown of STAT3 and JAK2 expression
H441 cells were transfected with STAT3 and JAK2 small interfering RNA (siRNA) duplexes or non-specific control siRNA duplexes (Upstate Biotechnology, NY) using Lipofectamine 2000 (Invitrogen). Immunoblot analyses showed that STAT3 and JAK2 levels remained low but detectable, and expression of β-actin was unaffected by siRNA treatment.
Ectopic expression of microRNA let-7c
The miR-let-7c expression in cells was increased by transfection with precursor (SC400005, Origene). The cells were plated in culture dishes or 24/96-well plates for 24h and then transfected with let-7c precursor with Lipofectamine 2000 (Invitrogen) for 24h. After transfection, the cells were subjected to further assays or for RNA/protein extraction.
Non-invasive bioluminescence evaluation of antrocin-mediated tumor suppression
H441 cells were modified to express dual optical reporter system, L2G fusion construct (firefly luciferase 2 and enhanced green fluorescent protein) for in vivo tracking purposes. L2G system was a generous gift from Dr Gambhir, Stanford University, CA, USA. Stable L2G-expressing H441 cells were generated according to described protocol ( 22 ). All procedures involving animals were carried out according to Taipei Medical University guidelines for the handling and use of laboratory animals. Additionally, experimental protocols were approved by the ‘Ethics Committee for Animal Experimentation’ of the Taipei Medical University (Approved protocol Number LAC-99–0304). H441-L2G cells (5.5 × 10 5 cells/100 µl PBS) were systemically injected into non-obese diabetic/severe combined immunodeficiency mice (female, 4–6 weeks of age) via the later tail vein. Mice with approximately equal bioluminescence intensity were separated into three groups (N ≥ 4 in each group), one being vehicle control and the other treated with antrocin (5 mg/kg/day and 10mg/kg/day). d -Luciferin (Promega) was injected intraperitoneally at a dose of 150mg/kg, followed by inhaled isoflurane anesthesia and bioluminescence imaging (IVIS 200, Caliper Life Sciences, Hopkinton, MA). Animals were imaged weekly and data were analyzed using the Living Images software package (Caliper Life Sciences). Data are represented as the average fold change in bioluminescence intensity = total bioluminescence/total bioluminescence (initial bioluminescence from week 1). The error bars indicate standard error of the average. All animals were humanely killed after experiments. A Kaplan–Meier survival curve was plotted to estimate the survival of antrocin-treated and control animals.
Replications and statistical evaluations
Each result was replicated in at least two independent experiments, and many were done multiple times. A two-way between-subjects analysis of variance was conducted to evaluate the effect of Let-7c and antrocin on STAT3. Following with a significant analysis of variance, a post hoc comparison using the Bonferroni adjustment was applied. The results indicate that there was a significant difference among groups ( P < 0.05)
Results
Antrocin inhibits the proliferation of human NSCLC cells
Previously we have demonstrated that antrocin inhibited the cell proliferation in various cancer cells ( 20 ). In this study, we examined the antrocin effect on a panel of NSCLC cell proliferation using SRB assay. As shown in Figure 1A , treatment of these NSCLC cell lines with various concentrations of antrocin for 48h resulted in a significant reduction in cell viability. It was evident from the data that different NSCLC cell lines differed in their sensitivity to antrocin. Importantly, under the same conditions, antrocin-medicated growth inhibition was significantly lower in normal human bronchial epithelial cells (BEAS-2B) compared with NSCLC cell lines. To further determine antrocin-induced antiproliferation effect was associated with the induction of apoptosis, two representative cell lines, H1975 and H441, were treated with various concentrations of antrocin. As shown in Figure 1B , antrocin treatment for 48h resulted in an increase in the number of cells in early (lower right) and late (upper left) stage of apoptosis in a dose-dependent fashion. These results provided evidence that antrocin induced apoptosis in human NSCLC cells. To determine whether cell proliferation inhibition with antrocin affects anchorage-independent growth, H1975 and H441 cells exhibiting high sensitivity to antrocin were tested. Figure 1C shows that treatment with 5 or 10 μM antrocin results in a dose-dependent decrease in colony formation in the H1975 cells (36–78% inhibition; P < 0.001) and H441 cells (49–85% inhibition; P < 0.001).
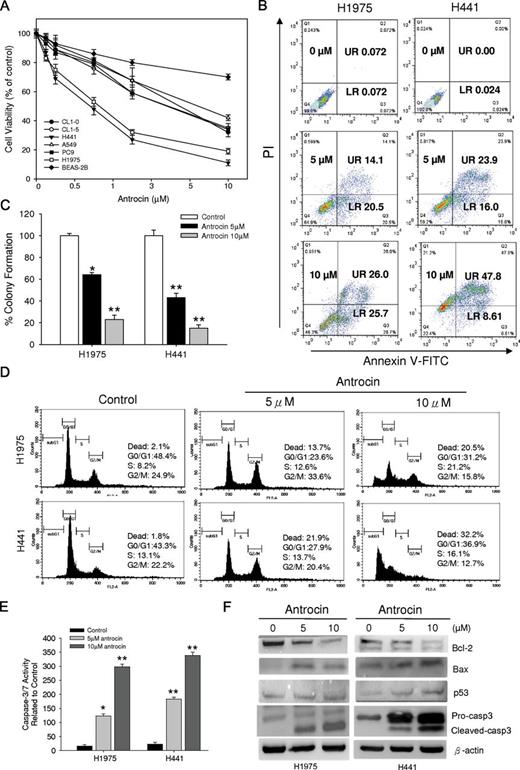
Antrocin inhibits the proliferation potential and induces apoptosis in human NSCLC cell lines. ( A ) Human NSCLC cells were treated with the indicated doses of antrocin or vehicle for 2 days and growth was measured using an SRB assay. Growth of the vehicle-treated cell was set to 100%. Results are the mean percent inhibition compared with vehicle-treated cells ( n = 3). ( B ) Antrocin induces apoptosis in H1975 and H441 cells. The lower right and upper right quadrant indicates the percentage of early and late apoptotic cells, respectively. ( C ) H1975 and H441cells were treated with the indicated concentrations of antrocin, and the clonogenicity was assessed. H1975 and H441 cells were plated in soft agar, and 1 day later, both cells were treated with 5 or 10 µM antrocin or vehicle. Media with or without antrocin was replaced every 3–4 days for a total of 20 days. Number of colonies was counted using the ImageJ software. Colony formation of the vehicle-treated cells was set to 100%. Percent colony formation of the antrocin-treated cells compared with vehicle is shown. Results are represented as the mean + standard deviation. Significant differences were set at * P < 0.05 and ** P < 0.001 compared with DMSO-treated control. ( D ) Cell-cycle analyses. H1975 and H441cells were treated with antrocin for 24h and examined by flow cytometry following PI staining for DNA content. Representative data are shown from three independent experiments. ( E ) Antrocin induces caspase-3/7 activity. H1975 and H441 cells were incubated with antrocin for 24h and analyzed for caspase-3/7 activity. Antrocin increased the number of apoptotic cells compared with untreated controls. Significant differences were set at * P < 0.05 and ** P < 0.001 compared with DMSO-treated control. ( F ) Cell lysates from H1975 and H441 cells incubated with the antrocin were analyzed by western blotting. Caspase-3 (both proform and cleaved form), Bcl-2, Bax and p53 protein expression levels were compared in vehicle- and antrocin-treated H1975 and H441 cells.
Antrocin induces cell death by apoptosis
We subsequently carried out cell-cycle analysis to further characterize antrocin’s growth-inhibitory effect. Antrocin (24h treatment) induced cell death in both H1975 and H441 cells ( Figure 1D ). At 48h, there was a significant increase of cell death in both H1975 and H441 cells (data not shown). We therefore determined the mechanism of cell death. Increased activation of caspase-3/7 was observed within 24h in both H1975 and H441 cells treated with antrocin ( Figure 1E ). This was further confirmed by western blot analyses of H1975 and H441 cell lysates, which showed a significant increase in activated caspase-3 in cells treated with antrocin. In addition, antrocin inhibited the expression of antiapoptotic genes Bcl-2 protein while increasing the expression of apoptosis-promoting Bax and p53 ( Figure 1F ). These data suggested that antrocin is a potent inducer of apoptosis of lung cancer cells.
Microarray profiling in antrocin-treated H441 cancer cells
To further explore the mechanism of antrocin-mediated cell death, we treated H441 lung cancer cells with antrocin (5 µM) for 12h, isolated mRNA and subsequently analyzed gene expression using an Illumina oligonucleotide array. Genes with a P -value of <0.05 were considered differentially expression genes relative to vehicle-treated controls. After removing unannotated and repeated entities, there was a total of 1643 differentially expression genes modulated by antrocin treatment in H441 cells compared with untreated control cells ( P < 0.05). Among these, 54 genes were upregulated and 207 were downregulated > 2-fold. The top 20 upregulated and top 20 downregulated genes were shown in Supplementary Table 1 , available at Carcinogenesis Online. Several genes known to be involved in the cell response to fatty acid catabolism were significantly upregulated, including SC4MOL (3.4), ALDOC (3.1), ACSS2 (3.09), FASN (2.69), MT1F (2.44), and FDFT1 (2.37). Interestingly, the genes related to JAK2/STAT3 signaling, including STAT3 transcript variant 3 (−3.36), STK36 (−2.05), as well as targets BIRC3 (−2.33) and CSF2RA (−2.26), were all decreased. To investigate the biological function of genes differentially modulated by antrocin exposure, the list of genes changed >1.5-fold was uploaded into the Ingenuity Pathway Analysis tool. As shown in Figure 2A , the top two biological function categories are “molecular mechanism of cancer” and “death receptor signaling” suggesting that one of the early events in antrocin-treated cells was a change in cell growth property. Genes associated with top biological functions include STAT3, STK36, CSF2RA, etc. Furthermore, we further confirmed that the STAT3-dependent transcriptional activity was affected by a luciferase analysis. Antrocin inhibited STAT3-dependent transcriptional activity in a dose-dependent manner ( Figure 2B ). To further analyze the impact of antrocin on the inhibition of STAT3, we examined the transcription profile of STAT3 downstream target genes by RT–PCR. We treated H441 lung cancer cells with antrocin (5 or 10 μM) or dimethyl sulfoxide for 24h. We found that antrocin treatment resulted in an inhibition of the transcription of STAT3-regulated genes such as cyclin D1, survivin, and Mcl-1 ( Figure 2C ).
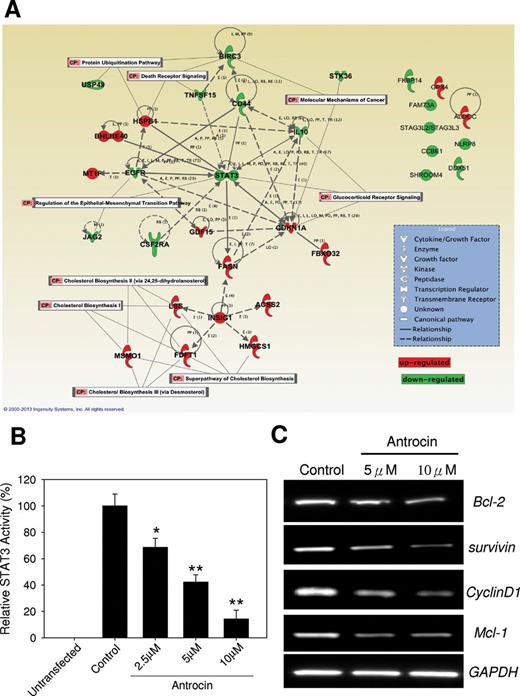
Antrocin modulates STAT3 pathway. ( A ) Top represented gene network identified by Ingenuity Pathway Analysis. The color of each node indicates the regulation of gene expression. Red: upregulation; green: downregulation. Total RNA from H441 cells treated with antrocin (5 µM) for 12h was hybridized using Illumina oligonucleotide array. Data show significant effect on the STAT3 pathway by antrocin. ( B ) Antrocin inhibited STAT3-dependent transcriptional activity in a dose-dependent manner. ( C ) Antrocin attenuated the STAT3-regulated genes Bcl-2, survivin, cyclin D1 and Mcl-1. Significant differences were set at * P < 0.05 and ** P < 0.001 compared with control.
Antrocin inactivates STAT3 and prevents its nuclear localization in H441 cells
To gain insight into inhibition of the JAK/STAT pathway in H441cells, first we tested the effect of antrocin on the constitutively activated STAT3 in H441 cells (a cell line known to possess high basal level of STAT3). As shown in Figure 3A , antrocin inhibited the constitutive phosphorylation of STAT3 on Tyr705 site in a dose-dependent manner, without altering the overall STAT3 protein level. The activation state of other tyrosine kinases p-TYK, p-JAK1 (Try1022), JAK2 (Try1007) and p-Akt (Ser473), which are considered to be responsible for phosphorylation of STAT3 Tyr705, was also significantly inhibited by the presence of 5 and 10 μM antrocin, as determined by western blot analysis using phospho-specific antibodies against tyrosine residues that are believed to be autophosphorylated by the respective kinases themselves ( Supplementary Figure 1 , available at Carcinogenesis Online). We then examined the protein level of gp130 following antrocin treatment in H441 cells to determine whether antrocin has an effect on gp130-related cytokine signaling. We found that antocin dose dependently attenuated the gp130 expression in H441 cells. Interleukin (IL)-6 has been shown to be a major cytokine involved in STAT3 activation. To further examine whether antrocin may inhibit exogenous IL-6-induced STAT3 phosphorylation in lung cancer cells, we used PC9 pulmonary adenocarcinoma cell lines, which is known to possess low basal level of STAT3 activation and IL-6 mRNA level ( Supplementary Figure 2 , available at Carcinogenesis Online). PC9 cells were pretreated with antrocin followed by IL-6. As shown in Figure 3B , IL-6 stimulation on PC9 cells was associated with marked increases of p-STAT3 levels, which was effectively abolished by antrocin treatment. It is known that under resting conditions, STAT3 is retained in the cytoplasm, and it translocates to the nucleus once phosphorylated and activated. Here, we also analyzed the effect of antrocin on STAT3 nuclear translocation. In the untreated control cells, STAT3 resided predominantly in cytoplasm and p-STAT3 was barely detectable. Upon IL-6 stimulation, p-STAT3 level substantially increased in both cytoplasmic and nuclear fractions, especially the nuclear fraction. Consistently, pretreatment of antrocin almost completely abolished the p-STAT3 nuclear translocation ( Figure 3B ). We next investigated the effect of gp130 in cells being stimulated with IL-6. As shown in Figure 3B , gp130 expression was also suppressed by antrocin in PC9 cells. Accordingly, antrocin affected upstream gp130 and JAK/STAT3 in the IL-6-triggered signaling cascade. Electrophoretic mobility shift assay experiment further performed on nuclear lysates obtained from starved PC9 cells preincubated with 10 μM of antrocin and subsequently stimulated with IL-6 showed that STAT3 DNA-binding activity had been inhibited ( Figure 3C ). Because tyrosine phosphorylation and dimerization of STATs is a prerequisite for their cytokine-induced nuclear translocation, we would expect that the antrocin inhibit IL-6-induced STAT3 nuclear translocation. We then examined whether antrocin pretreatment may block IL-6-induced p-STAT3 nuclear accumulation and STAT3 nuclear translocation using immunofluorescence staining. PC9 cells were pretreated with antrocin followed by IL-6. Then, immunofluorescence was performed to analyze the localization of p-STAT3 and STAT3. IL-6 induced the accumulation of p-STAT3 in the nucleus, whereas the pretreatment with antrocin blocked this process ( Figure 3D ).
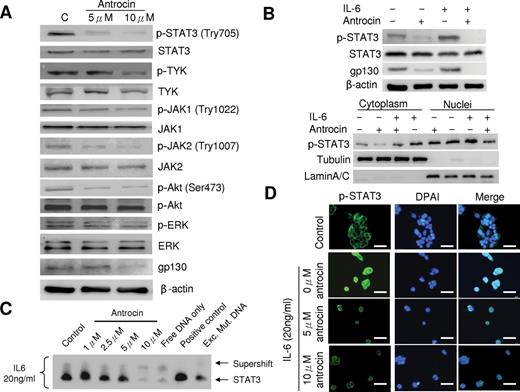
Antrocin inhibits constitutive and IL-6-induced STAT3 signaling pathway. Immunobloting assays were performed with specific antibodies. Antrocin inhibits phosphorylation of STAT3 (Tyr705), TYK, JAK1 (Try1022), JAK2 (Try1007), AKT (Ser473), ERK and gp130 expression after 24h treatment in H441 cells. ( B ) Antrocin blocks IL-6-induced STAT3 phosphorylation and nucleus translocation. PC9 cells were pretreated with antrocin (10 μM) for 3h and then stimulated with IL-6 (20ng/ml) for 30min. Whole cell lysates were immunoblotted with the indicated antibodies. Tubulin and lamin A/C were used as loading controls for cytosol and nucleus subfractions, respectively. IL-6 downstream molecule, gp130 expression was also suppressed by antrocin treatment. ( C ) Antrocin Inhibits STAT3 dimerization and DNA binding in vitro. PC9 cells were treated as described in (A), nuclear lysates were prepared and analyzed in electrophoretic mobility shift assay using the same DNA probe for STAT3. ( D ) Antrocin blocks IL-6-induced p-STAT3 nuclear accumulation. PC9 cells were cultured on coverslips in serum-free medium overnight and were pretreated with 5 and 10 μM of antrocin for 3h followed by 20ng/ml of IL-6 for 30min. After treatment, the localization of p-STAT3 was analyzed by immunofluorescence.
Antrocin treatment led to the upregulation of the Let-7c and consequently downregulation of the expression of c-myc, Kras and IL-6R in H441 cells
MicroRNA let-7 has been shown as a negative regulator in the IL-6/STAT signaling cascade and a tumor suppressor ( 22 ). Here, we examined whether antrocin-mediated suppression of STAT3 also functioned through let-7 modulation. Ectopic expression of let-7c was performed in both H441 and IL-6-induced PC9 cells. STAT3 activity was significantly downregulated in both H441 and IL-6-induced PC9 cells overexpressing let-7c. A similarly decreased STAT3 activity was observed in both cells in the presence of antrocin ( Figure 4A ). Next, we examined the let-7c level in both cell lines. Both H441 and IL-6 induced PC9 cells without any treatment demonstrated a relatively low let-7c expression level. The addition of antrocin increased let-7c level in both cell lines ( Figure 4B ). Transfection of cells with let-7c precursor showed increased expression of let-7c as determined by quantitative RT–PCR compared with control ( Figure 4C ), and these results were correlated with decreased expression of c-myc and Kras ( Figure 4D ). Transfection of cells with let-7c precursor led to a significant inhibition of colony formation ( Figure 4E ), and these effects were much more pronounced after antrocin treatment. Interestingly, the let-7c binding site in the 3′UTR of IL-6R mRNA is shown in Figure 4F , we also demonstrated that let-7c appeared to target IL-6R and functioned as a suppressor of IL-6R. Re-expression of let7c decreased the mRNA of IL-6R in H441 cells. The results suggest that let-7c suppressed colony-forming ability of lung cancer cell possibility via the suppression of two key oncogenes, c-Myc and Kras.
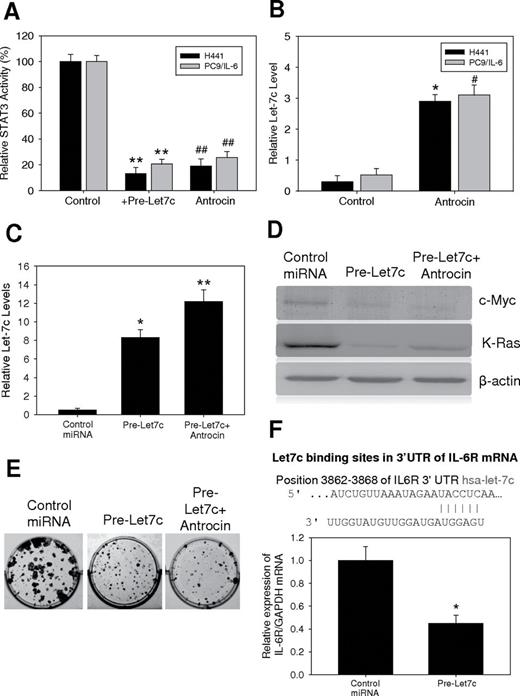
Antrocin treatment modulates microRNA Let-7c/STAT3 signaling pathway. ( A ) H441 and IL-6-induced PC9 cells were examined for their STAT3 activity in relationship with Let-7c. When Let-7c was transiently overexpressed in H441 and IL-6-induced PC9 cells (lanes marked by Let-7c), STAT3 activity was downregulated by approximately 4-fold and 3-fold respectively. **Compared with the H441 control group, P < 0.001; *compared with the H441 control group, P < 0.05; ##compared with the PC9/IL 6 control group, P < 0.001; #compared with the PC9/IL 6 control group, P < 0.05. ( B ) MicroRNA Let-7c appeared to be negatively associated with STAT3 activity. Antrocin treatment led to an increased level of Let-7 in both H441 and IL-6-induced PC9 cells. ( C ) Let-7c regulated c-myc, Kras and IL-6R expression and inhibited clonogenic capacity of H441 cells. Re-expression of let-7c or antrocin treatment in H441 cells resulted in an increased expression of let-7c as assessed by quantitative-PCR technique. ( D ) Re-expression of let-7c or antrocin treatment in H441 cells led to decreased expression of c-myc and Kras compared with control cells. ( E ) Transfection of cells with let-7c precursor led to a significant inhibition of colony formation. ( F ) Using Target Scan software, let-7c was predicted to bind the 3′UTR of IL-6R mRNA. We found that the re-expression of let-7c led to a decrease in IL-6R mRNA in H441 cells, which suggests that let-7c targets key oncogenic markers such as c-myc, Kras and IL-6R.
Simultaneous interruption of JAK2/STAT3 signaling pathways results in the induction of apoptosis in H441 cells
Antrocin contributes to the induction of apoptosis in H441 cells; we depleted the cells of STAT3 by RNAi. Depletion of STAT3 resulted in the downregulation of ERK1/2 expression, and the combination of such depletion and antrocin treatment resulted in both downregulation of Mcl-1 and upregulation of cleavage caspase-3 ( Figure 5A ). The combination of STAT3 depletion and antrocin treatment also resulted in a markedly greater increase in the number of apoptotic cells compared with either approach alone. Similarly, the combination of JAK2 depletion by RNAi and antrocin treatment resulted in a markedly enhanced apoptotic response in H441 cells ( Figure 5B ). Collectively, these data suggested that simultaneous interruption of JAK2/STAT3 signaling pathways is required for the induction of apoptosis in H441 lung cancer cells.
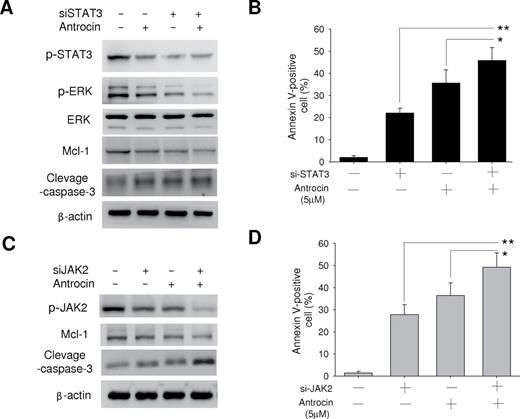
Effects of the combinations of the antrocin with either JAK2 depletion or STAT3 depletion on signal transduction and apoptosis in H441 lung cancer cells. ( A ) H441 cells were transfected with scramble or STAT3 siRNAs and incubated with or without antrocin (10 μM) for 48h, after which cell lysates were subjected to immunoblot analysis with antibodies to the indicated proteins (left). ( B ) Alternatively, the cells were transfected and treated with antrocin for 60h, after which the proportion of apoptotic cells was determined by staining with annexin V and PI followed by flow cytometry (right). ( C ) H441 cells were transfected with scramble or JAK2 siRNAs and incubated with or without antrocin (10 μM) for 48h, after which cell lysates were prepared and subjected to immunoblot analysis with antibodies to the indicated proteins (left). ( D ) Alternatively, the cells were transfected and treated with antrocin for 60h, after which the proportion of apoptotic cells was determined by staining with annexin V and PI followed by flow cytometry (right). All quantitative data are means ± standard error from three independent experiments. Significant differences were set at * P < 0.05 and ** P < 0.001 for the indicated comparisons.
Antrocin suppresses lung cancer growth in vivo
The antitumor effects of antrocin were further evaluated in vivo . H441-L2G cells expressing firefly luciferase and green fluorescent protein were implanted (intravenous injection via the lateral tail vein at 5.5 × 10 5 cells/100 μl PBS) into non-obese diabetic/severe combined immunodeficiency mice. Daily intraperitoneal injection of antrocin (two dosages: 5 and 10mg/kg) over a period of 4 weeks demonstrated a significant tumor inhibitory effect, as evident by the bioluminescence images ( Figure 6A and B ). The body weights of both low- and high-dose antrocin-treated mice were not significantly different from the control group (data not shown). The antrocin-treated animals exhibited a longer survival rates than those in vehicle-treated control counterparts ( Figure 6C ). A pronounced tumor inhibitory response was observed when antrocin was combined with JAK2 inhibitor ( Supplementary Figure 3 , available at Carcinogenesis Online).
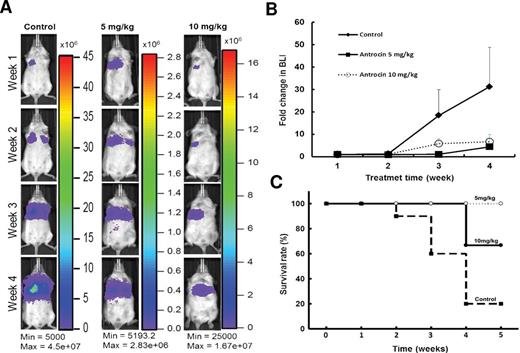
Antrocin inhibits lung tumorigenesis in vivo . H441-L2G cells were intravenously injected into the mice via lateral tail vein at a concentration of 5.5 × 10 5 cells/100 μl PBS. Antrocin treatments (two dosages 5 mg/kg and 10 mg/kg) were performed by daily intraperitoneal injection into tumor-bearing animals over a period of 4 weeks. ( A ) Representative non-invasive bioluminescent images are shown over the period of 4 weeks. ( B ) The tumor growth was quantitatively expressed in the fold change in bioluminescence intensity. ( C ) Kaplan–Meier survival curves were calculated and plotted.
Discussion
In our previous study, antrocin has been demonstrated as a novel class of small-molecule inhibitor of Akt/mTOR signaling in metastasis breast cancer cells ( 20 ). In this study, we demonstrated that antrocin had potent cytotoxicity against various lung cancer cells and can induce subG1 accumulation and apoptosis ( Figure 1 ). The complementary DNA microarray data showed that antrocin markedly suppressed the expression of JAK/STAT components ( Figure 2 ). We showed further that the key signaling pathways affected in lung cancer cell survival involved the inactivation of JAK2 and STAT3 ( Figures 3–5 ). When given alone, antrocin inhibited tumorigenesis in lung cancer xenograft mice in vivo ( Figure 6 ). Our data suggest that antrocin affects cell growth by inhibiting the JAK2/STAT3 signaling pathway.
Sesquiterpene lactones are a new class of anticancer agents because of their novel chemical structure combined with the unique spectrum of activity compared with current antitumor drugs, based on National Cancer Institute tumor cell panel studies ( 23 , 24 ). A related compound, germacranolides, failed in clinical trials because of central nervous system toxicity, investigators have since tried to develop derivatives that are not toxic to the central nervous system ( 25 ). Previously, we reported that the sesquiterpene lactone antrocin showed superior in vitro antiproliferative activity in MDA-MB-231 cells compared with doxorubicin and cisplatin, prevented colony formation and was non-toxic to non-tumorigenic MCF10A and HS-68 cells ( 20 ). The biological function and molecular mechanism responsible for the effect of antrocin on lung cell cytotoxicity merit investigation. Interestingly, H441 (EGFR wild type) and H1975 (EGFR mutant) cells exhibited the same sensitivity (IC 50 ) to antrocin, even though they have different intensities of EGFR expression ability. This suggests that the key mechanism responsible for antrocin’s ability to suppress cell growth is involved in tumorigenesis, as well as in the chemoresistance machinery.
JAK/STAT is a key oncogenic signaling cascade involved in tumorigenesis and found frequently deregulated in many types of cancer ( 2 ). Activation of JAK/STAT results in the expression of downstream genes that control major cellular responses (including cell proliferation and invasion, as well as cancer cell survival) such as cyclin D1, survivin and Bcl-2 ( 26 ). Suppression of JAK/STAT activity leads to induction of an apoptotic response. Thus, JAK/STAT is an important therapeutic target in cancer treatment. Studies with genetic approaches including antisense and RNA interference (siRNA) have demonstrated that inhibition of JAK2/STAT3 signaling suppresses tumor growth and induces apoptosis in vitro and in vivo ( 27 ). Moreover, pharmacological agents including pyridone 6 and AG490 have also been demonstrated to be potential anticancer drugs due to their inhibitory effects on STAT3 signaling ( 28 ). Antrocin was previously reported to possess potent antitumor activity through the inhibition of nuclear factor-kappaB (p65) ( 20 ), an important transcription factor playing major roles in inflammatory pathways, as well as tumorigenesis ( 29 ). Interestingly, some inhibitors of nuclear factor-kappaB pathway, such as phytochemicals including curcumin, resveratrol, ursolic acid, capsaicin and plumbagin could also suppress the JAK2/STAT3 pathway ( 30 ), suggesting an inherent link between these two pathways. But to date, there is no report of antrocin on JAK2/STAT3 signaling pathway.
By using microarray analysis, we identified gene expression profiles that contribute to the biological effects of antrocin ( Figure 2 ). Of the antrocin-suppressed genes, JAK/STAT, Mcl-1, cyclin D1, survivin and Bcl-2 were of particular interest because they are known tumor initiator and their expression induced overall cell viability and contributed to antrocin-induced cytotoxicity. This cell growth inhibition in both lung tumor cells is associated with inhibition of the STAT3 signaling pathway. Although there are different growth properties among these tumor cells, inhibition of phosphorylated STAT3 at Tyr705 is the majority to H441 cells. The formation of STAT3/DNA complexes in nuclei is consequently decreased by antrocin in this tumor cells ( Figure 3 ). As documented previously, the inhibition of the STAT3 phosphorylation is majorly achieved via the inhibition of upstream STAT3-activating receptor tyrosine kinases such as JAK1 and JAK2 ( 31 ). Furthermore, tyrosine residue in STAT3 is phosphorylated by JAK or Src family kinases in response to cytokines or growth factors ( 32 ). Our results show that antrocin inhibited the tyrosine phosphorylation and nuclear translocation of STAT3 ( Figure 3 ). Antrocin also abrogated the phosphorylation of JAK2 in tumor cells, both JAKs and protein tyrosine phosphatases are involved in the mechanisms of action of antrocin on dephosphorylation of STAT3. Activated EGFR signaling is associated with higher levels of activated STAT3. Our data demonstrated that H441 cells (with wild-type EGFR genotype) responded to antrocin treatment and exhibited suppressed STAT3 activity. In contrary, PC9 cells harboring L858R mutation in EGFR gene showed essentially no STAT3 activity measured either by immunoblot or by DNA-binding assay ( Figure 3 ). Similarly, H1299 or H460 cells expressing an activating mutant of EGFR did not show increase in activation of STAT3. Our results also suggest that while activating mutations of EGFR may enhance IL-6 production and autocrine stimulation of STAT3 activity; however, additional factors may also participate in modulating this pathway.
Of potential importance, the dual inhibition of STAT3/IL-6 and AKT/mTOR signaling pathways by antrocin reveals the complexity of lung carcinogenesis and novel targets for drug development. It has been established that IL-6 induced STAT3 activation and contributes to the lung carcinogenesis and metastasis ( 33 ). A cross-talk between IL-6/STAT3 and AKT/mTOR axes has been shown to provide survival advantage and contribute to drug resistance in multiple myeloma ( 34 ). The same study showed that targeting IL-6/STAT3 or MAPK independently did not induce apoptosis in myeloma cells while combined targeting of both pathways efficiently induced apoptosis. Furthermore, we revealed that antrocin treatment led to an increase in let-7c microRNA in lung cancer cells. Since let-7c has been indicated to target and suppress several oncogenes including Kras and c-Myc ( 35 ). Therefore, the suppression of these oncogenes was correlated to the decreased colony-forming ability. Interestingly, let-7c also appeared to target IL-6R, thereby disrupting IL-6/STAT3 axis ( Figure 4 ) as reported previously ( 22 ). Thus, antrocin may represent a new agent that can target multiple key oncogenic pathways to suppress lung tumorigenesis. Collectively, these observations underscore the fact that combinations of new drugs such as antrocin that disrupt multiple pathways will have direct implications in better efficacy.
In the present study, we found that antrocin inhibited IL-6-inducible STAT3 phosphorylation in PC9 cells in parallel with the suppression of JAK2 activation ( Figure 5 ). These results are consistent with previous reports that curcumin ( 36 ) and epigallocatechin gallate ( 37 ) inhibited the constitutive and the IL-6-inducible STAT3 signaling pathway. In particular, quercetin inhibited the phosphorylation of STAT3 induced by IL-6 without affecting the constitutive baseline level of phosphorylated STAT3 ( 38 ). All these results suggest that antrocin’s inhibitory effect on STAT3 activation could be mainly executed via inhibition of upstream STAT3-activating tyrosine kinase JAK2. This notion was supported by the observation where antrocin treatment increased the expression of let-7c microRNA, which has been demonstrated to inhibit IL-6/STAT3 cascade ( Figures 3 and 4 ). The anticancer effects mediated by antrocin appear to be similar to a number of phytochemicals that have been reported to inhibit JAK/STAT3 pathway, such as curcumin, ursolic acid and capsaicin ( 39 ). Furthermore, recently results showed that the expression of Bax and that of Mcl-1 are independently regulated by ERK and STAT3 signaling pathways in NSCLC cells ( 40 ). We further found that the combination of antrocin with depletion of either STAT3 or JAK2 also exhibited a pronounced proapoptotic effect in H441 cells, supporting the notion that simultaneous interruption of JAK2/STAT3 and ERK pathways mediates antrocin-induced apoptosis ( Figure 5 ). At present, the exact mechanisms by which JAK2 phosphorylation is abrogated by antrocin are still elusive and remain to be further investigated. Finally, to further substantiate the in vitro growth-inhibitory effects of antrocin on human H441 cells, we extended our studies to in vivo conditions by implanting H441 tumor xenografts in non-obese diabetic/severe combined immunodeficiency mice. Antrocin treatment in H441 cell xenografts model resulted in decreased tumor growth ( Figure 6 ). Notably, JAK2 and its associated pathways have been shown to contribute to drug resistance in lung cancer and IL-6/JAK2/STAT3 signaling to promote lung tumorigenesis and metastasis ( 31 , 41 , 42 ). Thus, combining antrocin (which suppresses IL-6/STAT3 signaling) and JAK2 inhibitor (specifically inhibits JAK2 pathway) may provide a more effective therapeutic regimen. We examined this possibility by treating H441-bearing mice with the combination of antrocin and JAK2 inhibitor. In agreement with our hypothesis, we observed a pronounced tumor inhibitory response using this approach ( Supplementary Figure 3 , available at Carcinogenesis Online). The hypothetical diaphragm describing the molecular mechanisms responsible for anticancer effects of antrocin on lung cancer cells was illustrated in Figure 7 .
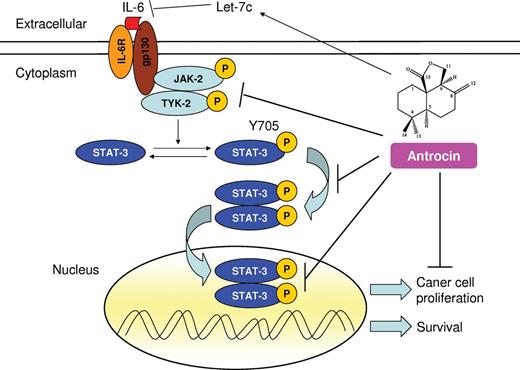
Proposed mechanism of action of antrocin in the suppression of NSCLC. IL-6 binds to the IL-6R and induces a cascade of phosphorylated JAK kinase that leads to the activation of STAT3. Activated STAT3 translocates into the nucleus where it targets genes involved in apoptosis, proliferation and survival. Treatment of cells with antrocin blocks the constitutive and IL-6-induced activation of JAK2 and TYK2, and STAT3 pathway results in apoptosis of human lung cancer cells. Antrocin also appears to increase the tumor-suppressor microRNA Let-7c. A blunt arrow denotes suppression, whereas an arrow represents induction in the signaling cascade.
In conclusion, our data indicate for the first time that treatment with antrocin produces both antiproliferative and proapoptotic effects. Antrocin could be a potential agent for targeting cancer cells with constitutively activated JAK2/STAT3 owing to its ability to modulate let-7c/JAK2/STAT3 signaling cascade and its potent growth-suppressive activity. Importantly, antrocin also significantly suppressed lung tumorigenesis in vivo . Thus, antrocin not only has the potential to be a therapeutic agent for the treatment of human lung cancer but also other cancer types with constitutively activated JAK2/STAT3 including breast, colon and liver.
Supplementary material
Supplementary Table 1 and Supplementary Data can be found at Supplementary Data
Funding
National Science Council of Taiwan (100-2113-M-324-001-MY3, 101-2811-M-324-001) to Y-M.T; National Science Council of Taiwan (99-2628-B-038-101-MY3) and National Research Program for Biopharmaceuticals of Taiwan (100-2325-B-010-009, 101CGH-TMU-06, 100TMU-TMUH-09, 101TMU-SHH-05) to A.T.H.W.; National Science Council of Taiwan (100-2313-B-038-001-MY3, 101-2325-B-038-005, 102TMU-SHH-02) to C-T.Y; Natural Science Foundation of China (Grant No. 91013004) to Z.Y.
Abbreviations:
-
EGFR
epidermal growth factor receptor
-
IL
interleukin
-
JAK/STAT
Janus kinase/signal transducer and activator of transcription
-
NSCLC
non-small-cell lung cancer
-
PBS
phosphate-buffered saline
-
PI
propidium iodide
-
SCLC
small-cell lung cancer
-
siRNA
small interfering RNA
-
SRB
sulforhodamine B
-
RT–PCR
reverse transcription–polymerase chain reaction.
Acknowledgements
The authors thank Po-Yang Huang for his technical support in animal imaging and tumor model establishments. The authors also thank Fang-Ting Kuo (Cell Sorter Core Facility Center, Office of Research and Development, Taipei Medical University) for her assistance with the molecular biology experiment and flow cytometry assay.
Conflict of Interest Statement: None declared.
References

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}