





Abstract
We present a phylogenetic hypothesis for 40 species in the bird family Paridae, based on comparisons of nucleotide sequences of the mitochondrial cytochrome-b gene. Parids, including tits and chickadees, are an older group than their morphological stereotypy suggests. The longest cytochrome-b distances between species reach 12% in uncorrected divergence. With the exception of one thrasher-like terrestrial tit species of the Tibetan plateau (Pseudopodoces humilis), morphological and ecological stasis have prevailed since the initial parid radiation in the Old World during the mid-Tertiary.
All trees support monophyly of the family Paridae, which includes Parus (sensu lato) and the monotypic Oriental genera Sylviparus, Melanochlora, and Pseudopodoces. Within the clade of chickadees and gray tits (Parus, subgenus Poecile), three Old World species, Parus lugubris of the eastern Mediterranean and Balkan regions, P. superciliosus of high elevations in the Himalayas of western China, and P. varius of the Orient are sisters to all other species. The Eurasian crested titmice (subgenus Lophophanes) and North American crested titmice (subgenus Baeolophus) are sister groups. Our data suggest two colonizations of the New World by parids in the late Tertiary. The ancestor of modern Baeolophus colonized North America 4 mya, and the ancestor of all North American chickadees colonized North America 3.5 mya.
Phylogénie chez la mésange (Paridés): II. Relations entre les espèces basées sur des séquences du gène mitochondrial cytochrome-b
Résumé
Nous présentons une hypothèse phylogénétique pour 40 espèces de la famille des Paridés basée sur des comparaisons de séquences de nucléotides du gène mitocochodrial cytochrome-b. Les Paridés constituent un groupe plus vieux que ce qui semble être suggéré par leur stéréotype morphologique. Les plus grandes distances de cytochrome-b entre les espèces atteignent 12% en divergence non corrigée. À l’exception d’une espèce terrestre de mésange provenant des plateaux tibétains (Pseudopodoces humilis), les stases morphologiques et écologiques ont prévalu depuis la radiation initiale des Paridés dans l’ancien monde au cours du mi-tertiaire.
Tous les arbres phylogénétiques appuient la monophylie de la famille des Paridés, qui inclut les Parus (sensu lato), les genres orientaux monotypiques Sylviparus, Melanochlora et Pseudopodoces. Dans le clade des mésanges et des mésanges «grises» (Parus, sous-genre Poecile), trois espèces de l’ancien monde, Parus lugubris, de l’est des régions méditerranéennes et balkaniques, P. superciliosus, vivant en haute altitude dans la chaîne himalayenne, et P. varius, de l’orient, sont apparentées avec toutes les autres espèces. Les mésanges «huppées» eurasiennes (Lophophanes) et les mésanges «huppées» de l’Amérique du Nord (Baeolophus) sont des groupes apparentés. Nos données suggèrent l’existence de deux colonisations de l’ancien monde par les Paridés à la fin du tertiaire. L’ancêtre moderne de Baeolophus a colonisé l’Amérique du Nord il y a environ 4 millions d’années et l’ancêtre de toutes les mésanges d’Amérique du Nord a colonisé l’Amérique du Nord il y a environ 3.5 millions d’années.
Members of the family Paridae exhibit substantial molecular divergences between species (Slikas et al. 1996), which is consistent with a hypothesis of Tertiary speciation. The family consists of 51 species of familiar, small, arboreal songbirds, called “tits” in Europe and “chickadees” or “titmice” in North America. Because of their overall morphological similarity, all but three species have been assigned to the genus Parus. Despite the outward similarity of parid species, however, the molecular divergences among many species within Parus are comparable to the molecular divergences among genera in other songbird families (Sheldon and Gill 1996, Slikas et al. 1996). Similarly, the divergences among some conspecific parid populations are comparable to those among full species in other songbird families (Gill et al. 1993). As a consequence of both morphological similarity among many species and the complex patterns of genetic divergence, parid taxonomy is confused, and we have a poor understanding of phylogenetic relationships. Our poor phylogenetic perspective prevents us from exploiting copious volumes of published data on parid ecology and behavior that otherwise could be used in comparative studies of their evolutionary history (e.g. Sheldon and Whittingham 1997).
Here we present a phylogenetic hypothesis for 40 parid species, including 39 traditional species of the family Paridae, plus Pseudopodoces humilis, previously misclassified as a corvid (James et al. 2003). Our phylogenetic estimates are based on comparisons of nucleotide sequences of the mitochondrial cytochrome-b gene. Our data complement and extend previous studies based on alternative molecular data sets, specifically allozyme comparisons (Gill et al. 1989), mitochondrial restriction fragment-length polymorphism (RFLP) data (Gill and Slikas 1992, Gill et al. 1993), cytochrome-b sequences (Kvist et al. 1996), and DNA-DNA hybridization distances (Sheldon et al. 1992, Slikas et al. 1996). In addition, comparison of parid cytochrome-b data with earlier molecular data sets permits an assessment of the evolutionary patterns of the cytochrome-b gene (e.g. as done by Slikas 1997; Sheldon et al. 1999, 2000).
Materials and Methods
Taxa
Our data set includes sequences from 44 species: 39 species commonly placed in the family Paridae (subfamily Parinae of Sibley and Monroe 1990); Ps. humilis, an aberrant species recently revealed to be a member of the Paridae (James et al. 2003); and 4 species from other songbird families (Table 1). Within the traditional Paridae, we have sequences from 37 species in the genus Parus and two species representing monotypic genera, Melanochlora sultanea and Sylviparus modestus. The outgroup taxa include three species representing the penduline tits (Remiz pendulinus, Auriparus flaviceps, Anthoscopus minutus), the parid sister group (Sheldon and Gill 1996), and a more distantly related sylvioid songbird (Sylvia atricapilla).
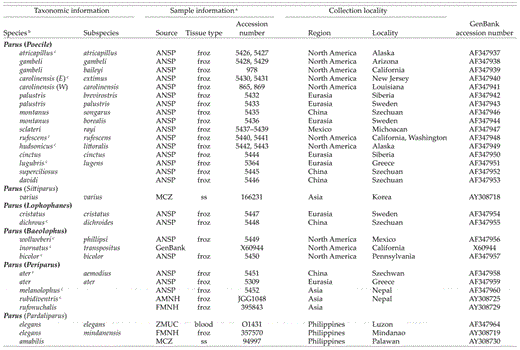
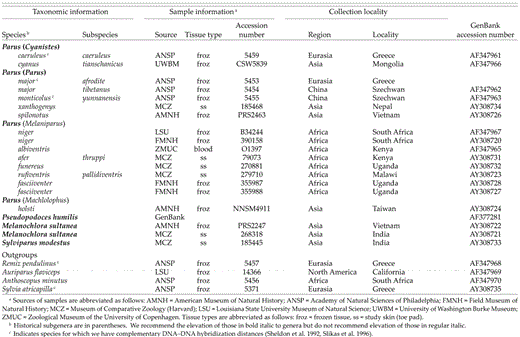
Table 1 includes subgeneric group names used by Thielcke (1968) and Harrap and Quinn (1995), corresponding to genera of Hellmayr (1903). Here, we employ those names as subgenera only, always preceded by the word “subgenus” or “subgenera,” to refer to groups of parid species. We address the validity of those groupings below. The American Ornithologists’ Union (AOU 1998) elevated the subgeneric names Poecile and Baeolophus to generic names for North American parid species. For clarity, however, we here use the generic name Parus sensu lato to include all species in the Paridae, except Sylviparus modestus, M. sultanea, and Ps. humilis. We do so to establish a comprehensive taxonomic perspective consistent with recent world checklists (e.g. Dickinson 2003) and to avoid prejudging or misrepresenting relations among species in the formal phylogentic analysis. In the end, we recommend adoption of a total of nine genera of parids, including Poecile and Baeolophus. Some subgenera included in Table 1 are in bold font, and others are not. We recommend elevation of those in bold to genera, but we do not recommend, at this time, the elevation of those in normal (italic) font.
We have sequences from most parid species of North America and Europe; the sampling of species from Africa and Asia is less complete. In particular, we have sequenced all known species of chickadees or “gray tits” (subgenus Poecile) and all but two species of New World parids. Not included are (1) the Juniper Titmouse (Parus ridgwayi), a species recently split from the Oak Titmouse (P. inornatus; Cicero 1996, AOU 1998) with correction of specific name from P. griseus to P. ridgwayi (AOU 2000); and (2) the Black-crested Titmouse (P. atricristatus), a species recently split from the Tufted Titmouse (P. bicolor; AOU 2002). For many taxa in the data matrix, we obtained at least partial sequences from additional individuals to control for identification or sequencing errors (Table 1). For some species, we included individuals from different populations to check for monophyly of the species.
Cytochrome-b DNA sequencing
We extracted DNA from tissue and blood samples using standard protocols. Samples were incubated overnight at 55°C in a buffer including STE (sucrose/Tris/EDTA), 1% sodium lauryl sulfate, and 1 mg mL-1 proteinase K. Following overnight digestion, samples were purified by phenolchloroform extraction. We precipitated DNA by adding to each sample 0.10 volume of 3M sodium acetate and 2 volumes of cold 95% ethanol. The DNA was pelleted by centrifugation, washed with 70% ethanol, air-dried, and resuspended in 0.1 × TE to a concentration of 1 mg mL-1. For each sample, concentration and 260:280 ratio were read on a spectrophotometer. We diluted an aliquot of each sample with H2O to a concentration of 25 ng μL-1 to serve as template for polymerase chain reaction (PCR) amplification. For eight individuals, we extracted DNA from a sliver (1 × 3 mm) of toepad from a museum study skin following protocols described in Joseph et al. (1999) and Slikas et al. (2000). We did not attempt to read the concentration of the latter extracts.
We amplified a 1,068-base-pair (bp) fragment of the mitochondrial genome, including most of the cytochrome-b gene, via PCR. The reaction mix included 1× buffer (Promega, Madison, Wisconsin), 200 μm of each dNTP, 0.8 μm of each primer, 1.5–3.0 mm MgCl2, 0.25 units of Taq polymerase (Promega), and 25 ng of wholegenomic DNA as template, in a 50-μL reaction volume. We amplified most samples with the primer pair: L14990 (5′-CCATCCAACATCTCA GCATGATGAAA-3′) and H16065 (5′-GGAGTC TTCAGTCTCTGGTTTACAAGAC-3′), modified from primers in Kocher et al. (1989). The PCR cycle included a denaturation step at 93°C, an annealing step at 45–52°C, and an extension step at 72°C. For each reaction, 35–38 cycles were run in an automated thermocycler (MJ Research, Waltham, Massachusetts). A 2-min denaturation at 94°C preceded the first cycle, and an 8-min extension at 72°C followed the last cycle.
Following amplification, we purified the double-stranded PCR product of excess nucleotides and primers by electrophoresis on a 1% agarose gel. We stained gel with ethidium bromide to visualize the PCR product. The product band was excised, and the amplified DNA was extracted using glassmilk (Geneclean, Bio 101, Vista, California). The double-stranded product was sequenced manually, using the dideoxy chain termination method (Sanger et al. 1977), with T7 DNA polymerase (SEQUENASE 2.0. United States Biochemical, Cleveland, Ohio) and P33-labeled nucleotides. Sequences were read manually from the autoradiographs and entered into the SEQED editing program (GCG Wisconsin Package; Accelrys, San Diego, California).
We obtained some sequences using automated sequencing methods. We cleaned double-stranded PCR products using a QIAquick PCR purification kit (Qiangen, Valencia, California). Sequences were generated by cycle sequencing with either dRhodamine or BigDye fluorescent Ready Reaction terminator mix (Applied Biosystems, Foster City, California). Sequences were visualized on an ABI 377 or 373 automated sequencer. We used SEQUENCHER 4.1 (Gene Codes Corporation, Ann Arbor, Michigan) to edit chromatograms, align sequences, and create NEXUS files.
DNA-DNA hybridization comparisons
In previous work, we used DNA-DNA hybridization of single-copy nuclear DNA to estimate phylogenetic relationships among selected parid species (Sheldon et al. 1999, 2000). Here, we use those distances as a measure of time-since-divergence to examine patterns and rates in the evolution of the cytochrome-b gene (Slikas 1997). The single-copy nuclear genome evolves much more slowly than the mitochondrial genome. As a result, DNA-DNA hybridization distances are not compressed by saturation over the range of divergence among parids (Sheldon and Bledsoe 1989), making them useful as a proxy for time. In addition, DNA-DNA hybridization distances are based on comparisons of single-copy nuclear genes, thus providing a measure of time that is independent of the mitochondrial cytochrome-b data. For 14 parid species and 2 outgroup species sequenced here (footnote c in Table 1), we have complementary DNA-DNA hybridization distances (Sheldon et al. 1992, Slikas et al. 1996). We plotted uncorrected percentage of difference between cytochrome-b sequences (y-axis) against uncorrected DNA-DNA hybridization distances (x-axis). For the cytochrome-b data, percentage differences were calculated and plotted for the following data partitions: (1) complete sequences; (2) transitions and transversions, each partitioned by codon position; and (3) transitions and transversions partitioned by codon position and by position along the cytochrome-b gene, as defined by Zhang et al. (1998). We also plotted transition-to-transversion ratios.
Phylogenetic analysis
The final data matrix included 1,068 base pairs of sequences from cytochrome b for each of 54 individuals representing 44 species (Table 1). Sequences for Ps. humilis and Parus inornatus were retrieved from GenBank; all other sequences were generated for the present study and deposited in GenBank (accession numbers AF347937–AF347970, AY308717–AY308735). We were able to align all sequences with each other and the Gallus gallus cytochrome-b sequence with no insertions or deletions; our sequences begin at position 14,894 in the Gallus gallus mitochondrial genome (Desjardins and Morais 1990). Amino acid translation was checked in MACCLADE 4.0 (Maddison and Maddison 2000); we also used MACCLADE to assign codon positions and to create step matrices. We used PAUP* (Swofford 2002) to calculate genetic distances, to search for optimal trees using maximum-parsimony (MP) and maximum-likelihood (ML) criteria, and to perform bootstrap analyses (MP only, 1,000 reps). All PAUP* searches were heuristic, with tree bisection reconnection (TBR) branch-swapping and the multiple parsimony (MULPARS) option in effect. Maximum-parsimony searches were run with a random addition-sequence of taxa (10 replicates), and ML searches were run with a simple addition sequence of taxa. All MP analyses imposed a step-matrix weighting transversions (Tv) over transitions (Ti) by 5:1 at first- and third-codon positions, to discount the more saturated transitional changes. This weighting falls within the range of observed Ti/Tv ratios between parid taxa.
We used MODELTEST 3.0 (Posada and Crandall 1998) to determine the optimal model of sequence evolution and parameter values for the ML analysis. The optimal model was general time-reversible, with gamma-distributed rate variation across sites and invariant sites (GTR + G + I). The parameter values for that model include a symmetric rate matrix specifying relative probabilities for all possible nucleotide changes (Rmatrix = 1.085700 [A-C], 13.511800 [A-G], 0.314600, [A-T], 1.336000 [CG], 10.341600 [C-T], 1.000000 [G-T]), the proportion of invariant sites (pinvar = 0.5125), and the shape parameter for the gamma distribution of rate variation (shape = 0.8614). The base frequencies were set as follows: A = 0.30410, C = 0.40820, G = 0.10080, T = 0.18690.
We used MRBAYES (Huelsenbeck and Ronquist 2001) to search for the best tree using Bayesian likelihood and to generate a posterior probability distribution of trees. The model selected for the MRBAYES search was a general time-reversible model with gamma-distributed rate variation across sites (GTR + G), with the following starting parameters: Rmatrix = 1.40881 [A-C], 13.00650 [A-G], 1.64341 [A-T], 0.33936 [C-G], 12.48968 [C-T], 1.00 [G-T], shape = 0.213875, and empirical base frequencies. A neighbor-joining tree, based on uncorrected sequence divergence, was generated in PAUP* and used as the starting tree in the MRBAYES search. The search was run with 4 chains for 500,000 generations, with a sampling frequency of 100 generations.
To test for rate heterogeneity among lineages, we employed a likelihood-ratio test. The test compares the log-likelihood for the ML tree to the log-likelihood for the same tree topology, but with a molecular clock enforced. We used a six-parameter substitution model with gammadistributed rate variation across sites; all parameters were optimized separately for clock and nonclock analyses. Twice the difference in log-likelihood is expected to have a chi-square distribution with df = n - 2, where n = number of taxa in the data matrix (Felsenstein 1988). With all taxa included, no significant rate heterogeneity was found with this test (P > 0.1).
Results
Sequence data characteristics
The sequence data matrix shows a skewed frequency distribution of nucleotides. Averaged across all taxa and codon positions, base frequencies are 27% for adenine, 36% for cytosine, 13% for guanine, and 23% for thymidine. Base frequencies do not differ significantly across taxa (chi-squared test in PAUP*, P = 1.00). The skew in base frequencies is not uniform across codon positions. At first-codon positions, base frequencies are relatively uniform; at second-codon positions, guanine (13%) and adenine (20%) are relatively uncommon, whereas thymine is over-represented (41%). At third-codon positions, guanine (2.6%) and thymine (8.4%) are markedly uncommon, and adenine (36%) and cytosine (53%) are over-represented. Across taxa, base frequency composition is most variable at third-codon positions. These patterns are typical for avian cytochrome-b sequences (e.g. Edwards et al. 1991, Helm-Bychowski and Cracraft 1993, Kornegay et al. 1993, Hackett 1996, Bloomer and Crowe 1999, Voelker 1999).
Among parid species, 405 (40%) of the 1,008 sites in the sequence data matrix are variable (19% at first-codon positions, 6% at second-codon positions, and 75% at third-codon positions) and 326 (32%) are potentially parsimony-informative (15% at first-codon positions, 3% at second-codon positions, and 82% at third-codon positions). With outgroups included, 437 (43%) of the nucleotide sites are variable (21% at first-codon positions, 8% at second-codon positions, 71% at third-codon positions) and 360 (36%) are parsimony-informative (17% at first-codon positions, 4% at second-codon positions, and 79% at third-codon positions). Among parids, 19% of the 336 amino acids in the data matrix are variable, and 10% of those are phylogenetically informative. With outgroups included, 24% of the amino acids are variable, and 13% are phylogenetically informative.
Uncorrected sequence divergences among species in the genus Parus range from 2.0% (between P. ater aemodius and P. melanolophus) to 12.0% (between P. fasciiventer and P. inornatus), with a median divergence of 8.73% (Table 2). Between taxa in Parus and the other three species in the Paridae, average pairwise divergences are as follows: 9.4% to Ps. humilis (SD = 0.7%), 10.4% to M. sultanea (SD = 0.7%), and 11.3% to S. modestus (SD = 0.8%). The average pairwise sequence divergence (uncorrected) from taxa in the family Paridae to the remizid outgroups is 13.8% (Remiz, Auriparus, Anthoscopus; SD = 1.0%) and to Sylvia atricapilla is 14.8% (SD = 0.7%).
Paridae cytochrome-b sequence divergence matrix. Upper = P distance; lower = maximum-likelihood distance.
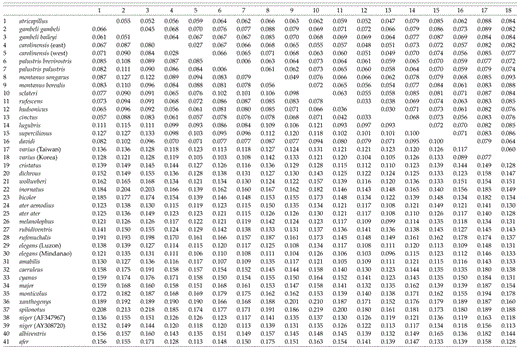
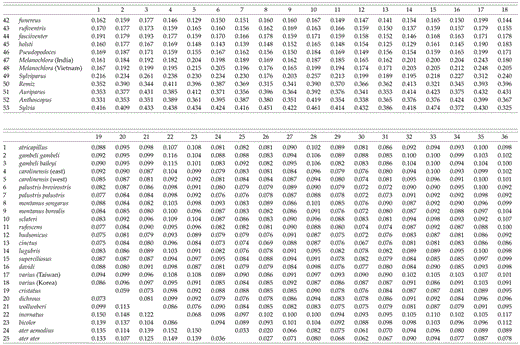
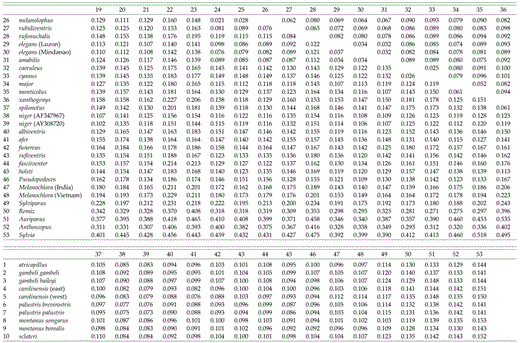
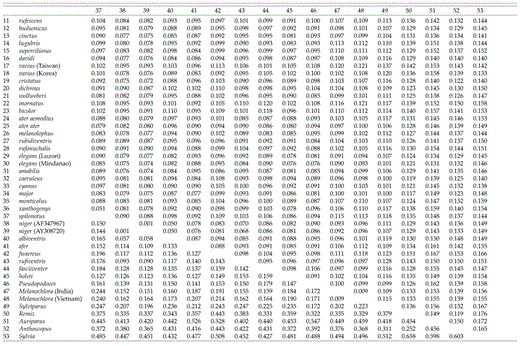
Paridae cytochrome-b sequence divergence matrix. Upper = P distance; lower = maximum-likelihood distance.
Within-taxa sequence divergences
Our sampling within species was not extensive. Within populations, divergence in cytochrome b was near zero. Complete sequences of two individuals of P. sclateri rayi from Michoacan (Mexico) were identical; the two differed by 0.6% from a third individual from the same locality. Two individuals of P. rufescens rufescens from California yielded identical sequences, and two individuals of P. hudsonicus littoralis from Alaska differed by 0.3%. Two individuals of P. niger niger from South Africa differed by 0.1%, and two individuals of P. fasciiventer from Uganda differed by 0.8%. For some species, we also found little divergence between populations. An individual of P. palustris from Siberia differed from an individual from Sweden by 0.60%. Two individuals of M. sultanea, one from India and one from Vietnam, differed by 0.9%.
Other parids displayed more substantial divergence between populations. An individual of P. montanus from Sweden (borealis) differed by 4.9% from an individual from China (songarus). Parus ater aemodius (China) differed by 3.3% from an individual of P. ater ater (Greece), and an individual of P. elegans from Luzon differed from an individual from Mindanao by 3.4%. For both P. ater and P. elegans, the individuals representing different subspecies are not sister taxa in the optimal trees (Fig. 1). Individuals of P. carolinensis from the east (P. c. extimus) differed by 2.7% from two individuals (P. c. carolinensis) from the west (Louisiana). Two individuals of P. gambeli from Arizona (P. g. gambeli) were identical in sequence, but differed by 4.5% from an individual from California (P. g. baileyi). The substantial genetic divergence between eastern and western populations of P. carolinensis and between Arizona and California populations of P. gambeli was reported previously on the basis of mitochondrial RFLP data (Gill et al. 1993, 1999). Finally, a sequence obtained from a feather of P. varius castaneoventris from Taiwan differed by 6% from a (toepad) sequence of P. varius varius from Korea. We included only the latter in our analyses, but point this out as a result that requires both confirmation and sampling of other island populations of this species.
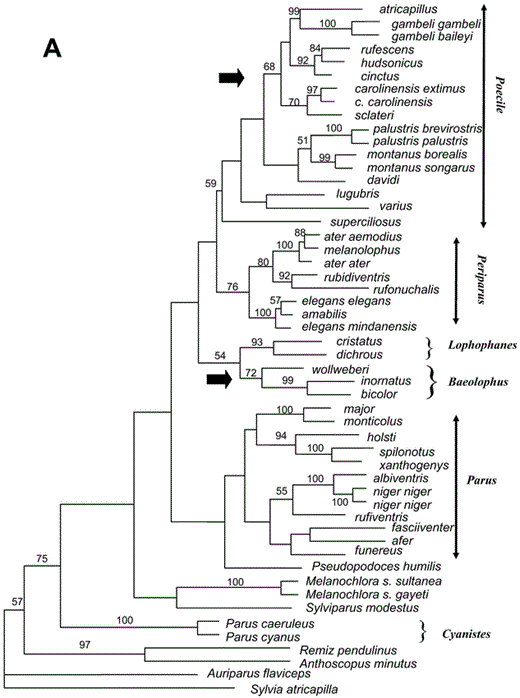
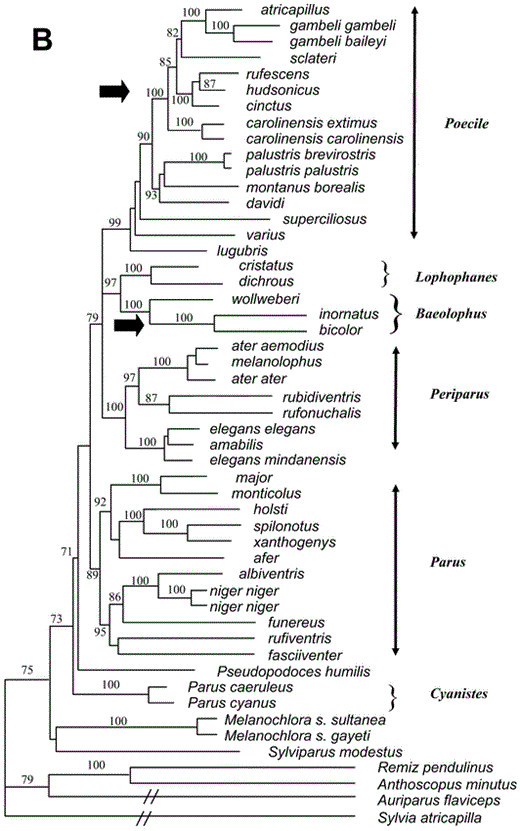
Relationships among species of Paridae. (A) Single most-parsimonious tree from a heuristic search in PAUP* (Swofford 2002), with TBR branch swapping and random addition-sequence of taxa (10 repetitions). Transversions were weighted 5:1 over transitions at first- and third-codon positions. Numbers on the branches are bootstrap percentages from 1,000 replicates. Hypothesized invasions of North America are indicated by black arrows. Recommended genera are indicated on right side of figure. (Continued on next page)
Phylogeny
A heuristic MP search yielded a single most-parsimonious tree (Fig. 1), rooted with Sylvia atricapilla. That tree is identical to the ML tree in most nodes. Differences are limited to a few weakly supported nodes, including (1) the position of sclateri within the clade of chickadees and gray tits (subgenus Poecile) and (2) the relationships among the African tits (e.g. Parus afer, P. funereus, P. rufiventris). The consensus tree from the Bayesian analysis is identical to the ML tree, except that a few nodes that are resolved in the ML tree are polytomies in the Bayesian consensus tree.
In all optimal trees, the family Paridae— including Pseudopodoces, Melanochlora, and Sylviparus—is monophyletic (bootstrap support = 75%, Bayesian posterior probability = 75%). Sylviparus and Melanochlora position as sister species to the rest of the Paridae, the latter including Pseudopodoces and species in the genus Parus. Within Parus, six clades corresponding to named subgenera appear in all optimal trees, with good support in the Bayesian tree and weaker support in the MP tree. Relationships among major clades are the same in all optimal trees, but the nodes defining those relationships have weak support. One exception is the pairing of the European and North American crested tit clades (subgenera Lophophanes and Baeolophus, respectively), which is well supported in the Bayesian tree. That pairing also was found in trees based on nuclear DNA-DNA hybridization data, though with weak support (Sheldon et al. 1992, Slikas et al. 1996).
Previously published phylogenies based on nuclear DNA-DNA hybridization data showed a basal split in the parid family, with P. caeruleus and P. major forming a clade that is the sister group of all other parids (Sheldon et al. 1992, Slikas et al. 1996). In the optimal trees based on the cytochrome-b sequence data, P. caeruleus, P. major, and their close relatives do not form a monophyletic group. Parus caeruleus and P. cyanus pair together with strong support; that pair is the sister clade to all other parids, excluding Sylviparus and Melanochlora. Pseudopodoces and Parus major and its close relatives branch off next. In the MP tree, Pseudopodoces is sister to the great tit clade (subgenus Parus); whereas in the consensus tree from the Bayesian analysis, Pseudopodoces and the great tit clade (subgenus Parus) branch off sequentially.
Relationships within subgenera
Chickadees and gray tits (subgenus Poecile) are monophyletic in all optimal trees (Bayesian posterior probability [BPP] = 99%, bootstrap [BOOT] = 59%). Three Eurasian and Oriental species— superciliosus, lugubris, and varius—branch off sequentially as sisters to the remaining chickadees, though the branching order differs between the MP and ML-Bayesian trees. The remaining chickadees form a clade with relatively weak support (BPP = 90%, BOOT < 50%). Within that clade, most relationships are consistent across optimal trees. Parus palustris, P. montanus, and P. davidi group together with weak support. North American chickadees (including P. cinctus) form a clade (BPP = 100%, BOOT = 68%). Within that clade, P. atricapillus is sister to P. gambeli. The brown-backed chickadees are monophyletic: P. rufescens is sister to P. hudsonicus, and that pair is sister to P. cinctus. Those latter relationships have strong support. Parus sclateri pairs differently in the two trees—that is, with P. atricapillus and P. gambeli in the ML-Bayesian tree and with P. carolinensis in the MP tree.
The Eurasian crested tits, P. dichrous and P. cristatus, pair as sister species (BPP = 100%, BOOT = 93%). In all optimal trees, the North American crested tits form a clade: P. bicolor pairs with P. inornatus (BPP = 100%, BOOT = 99%), and P. wollweberi is their sister (BPP = 100%, BOOT = 72%). The Eurasian and North American crested tit clades are sister groups in all optimal trees (BPP = 97%, BOOT = 54%).
Among the coal tits, P. ater is paraphyletic with respect to P. melanolophus. In all optimal trees, P. ater aemodius (China) pairs with P. melanolophus, and P. ater ater (Greece) is sister to that pair. According to Martens and Eck (1995), P. ater and P. melanolophus are a single species, because the two forms hybridize extensively in westcentral Nepal. In all optimal trees, the sister to the ater-melanolophus clade is a pair of species from the Himalayas, P. rufonuchalis and P. rubidiventris. That clade of Eurasian tits (P. ater, P. melanolophus, P. rufonuchalis, P. rubidi-ventris) is sister to a pair of species endemic to the Philippines, P. elegans and P. amabilis. The optimal trees suggest that the latter two species are paraphyletic, but the node suggesting paraphyly has weak support.
The great tit clade includes both Eurasian and African taxa. Parus major and P. monticolus pair as sister taxa with strong support. Parus holsti, a species endemic to Taiwan, pairs with P. spilonotus and P. xanthogenys of the mainland Orient, also with strong support. Five African species (P. albiventris, P. niger, P. funereus, P. rufiventris, and P. fasciiventer) form a clade in all optimal trees; in the MP tree, that clade also includes P. afer, another African species. In the ML tree, however, P. afer pairs weakly with P. holsti, P. spilonotus, and P. xanthogenys. Parus niger and P. albiventris pair with strong support, but other relationships among the African taxa have weak support. Parus caeruleus and P. cyanus pair as sisters, with strong support.
Saturation
When uncorrected cytochromeb sequence divergences were plotted against DNA hybridization distances (Fig. 2), sequences exhibited a fast initial rate of change (10× the scnDNA rate) up to 7% divergence, after which the rate slowed as a result of saturation by back mutations. Saturation is partly responsible for the packing of uncorrected distances at about 8– 10% (range of genetic distances among subgenera), making it difficult to resolve phylogenetic relationships. The packing effect of saturation can be ameliorated by correcting the cytochrome-b distances using the HKY95 model, but the corrections introduce another problem. Instead of falling out in a linear array with respect to relative time, HYK95 distances show considerable scatter, reflecting an increased variance as compared with uncorrected distances. An increase in variance is expected when relatively short sequences are corrected using a parameter-rich method (Swofford et al. 1996). The lack of linearity with time also may suggest the formation of (subgeneric) lineages in a short time.
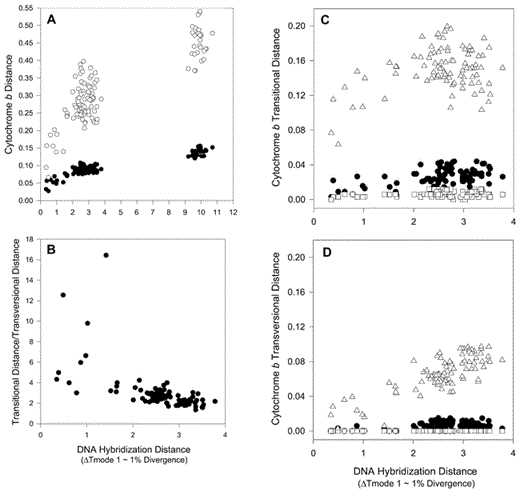
Cytochrome-b sequence data compared with DNA hybridization distance. (A) Uncorrrected cytochrome-b sequence distances versus DNA hybridization distances. Closed circles = uncorrected proportional distances; open circles = HKY95 distances (GTR rates: 1.7351446, 11.368306, 1.0266284, 0.32832423, 10.329434; codon site rates: 0.319910, 0.066472, 2.613617). (B) Cytochrome-b Ti/Tv values, based on uncorrected Ti and Tv distances, versus DNA hybridization distances. Uncorrected cytochrome-b distances, partitioned as (C) transitions and (D) transversions and by codon site positions, versus DNA hybridization distances. Closed circles = first position distances; open squares = second position distances; open triangles = third position distances
The plot of transition to transversion ratio (Ti/Tv) against DNA hybridization distance (Fig. 2) indicates large variance in Ti/Tv near the origin, a pattern that apparently results from a combination of small sample size and relatively few transversions between closely related species. At longer distances (2–4% scnDNA divergence), the Ti/Tv settles at about two.
Plots of partitioned cytochrome-b sequence distances versus DNA hybridization distances (Fig. 2) show the expected patterns with respect to relative rates and saturation (Irwin et al. 1991, Hackett 1996). Third-position transitions have an initial rate of evolution 20× that of single-copy nuclear DNA (scnDNA) before rapid saturation. The relatively unsaturated partitions display the following rate patterns vis-à-vis nuclear DNA: first-position transitions have about the same rate as scnDNA; second-position transitions and first- and second-position transversions have slower rates than scnDNA; and third-position transversions change at about twice the scnDNA rate.
When sequences are further partitioned according to protein structural regions (Fig. 3; Griffiths 1997; Sheldon et al. 1999, 2000), third-position transversions are again the least saturated, most rapidly changing subset of the data. First-position transitions and transversions from the transmembrane region also display fairly consistent, though slower, increases over time.
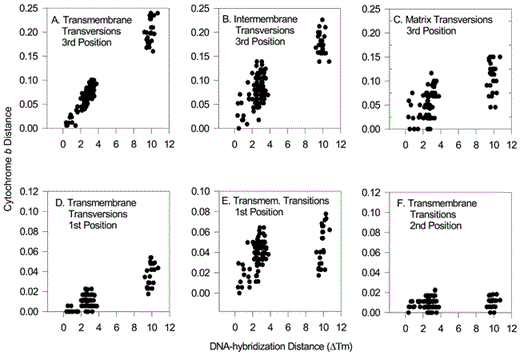
Distances computed for cytochrome-b protein regions versus DNA hybridization distances. Cytochrome-b values are uncorrected proportional distances. Only those cytochrome-b data partitions that increased with DNA hybridization distance are shown—with the exception of (F)—for comparison with Griffiths (1997)
Discussion
Phylogenetic history
The family Paridae comprises a mixture of regionally endemic species groups and widely distributed species and species groups. Our phylogenetic analyses support the traditional view that the family Paridae has an Old World center of origin (Mayr and Short 1970). Areas of high diversification and endemism are the Himalayas, Africa, and North America. Some of the older lineages, such as the blue tits (subgenus Cyanistes) and the great tits (subgenus Parus), show no particular regional pattern of diversity and endemism. Both are widespread and ubiquitous in the Old World. Extinction, dispersal, and other agents of time have obscured the trail of their diversification. An invasion of Africa and subsequent radiation there of one lineage of great tits seems apparent, as does vicariant evolution in the Himalayas. Trees based on both cytochrome-b and DNA hybridization data strongly indicate that parids invaded and radiated in the New World from the Old World on at least two occasions.
North American crested titmice (subgenus Baeolophus) form the sister group of the Old World crested tits (subgenus Lophophanes). Linking those two groups morphologically and vocally is the Bridled Titmouse (P. wollweberi) (e.g. Thielcke 1968, Hailman 1989). Previously, on the basis of its similarity to Old World species in plumage pattern, P. wollweberi was considered a North American representative of the subgenus Lophophanes, but phylogenetically it is clearly sister to the New World species of subgenus Baeolophus. That conclusion is supported by DNA hybridization as well as cytochrome-b evidence. The similarity in plumage pattern between P. wollweberi and the Old World Crested Tit (P. cristatus) may be the result of symplesiomorphy.
Some parid relationships were not resolved either by the cytochrome-b or by DNA hybridization studies—specifically, the positions of the chickadees (subgenus Poecile), coal tits (subgenus Periparus), and the crested tits (subgenera Baeolophus + Lophophanes) in relation to one another. That trichotomy likely reflects a rapid timing of the divergence among those groups. Resolution of divergence events is impossible if the time between successive splits is too short for the diverging clades to acquire many synapomorphies (Lanyon 1988). For DNA-DNA hybridization distances, if the length of an internode is less than the average standard error in distance (0.20; Slikas et al. 1996), then resolution is also impossible.
History of Old World taxa
Supporting an Old World center of origin are the patterns of taxonomic diversity, endemism, and ecological observations in the family. For example, members of all subgenera but one (subgenus Baeolophus) are found in the Old World. The oldest lineages in the family are Old World: Melanochlora, Sylviparus, Pseudopodoces, subgenus Cyanistes, and subgenus Parus. Also supporting the conclusion of an Old World origin are the positions of Old World and New World species of chickadees and gray tits (subgenus Poecile). The oldest lineages are three poorly known, widely disjunct Eurasian species, P. lugubris, P. superciliosus, and P. varius. Previous speculations about the affinity between P. lugubris and P. davidi (Eck 1988, 1994) are not supported here. Similarly, speculations about the affinity of P. superciliosus and P. gambeli of North America, based on their white eyebrow lines (Eck 1988, Cramp and Perrins 1993), are not supported.
The number of genetically divergent subgenera in the Old World bears directly on Lack’s hypothesis (1969, 1971) that the speciose guilds of Old World tits reflected a more ancient evolutionary history than the smaller guilds in the New World. The trees based on our molecular data support that ecological hypothesis. Old World species have diverged over longer periods of time, evolving morphological and behavioral traits that facilitate sympatry of species from phylogenetically divergent subgenera (McCallum et al. 2001).
History of New World taxa
The nearly complete DNA samples of New World parid species provide insights as to the timing of their evolution. Given a rough calibration of 2% divergence per million years (Arbogast and Slowinski 1998, Voelker 1999), we hypothesize that parids colonized the New World on two separate invasions in the late Tertiary. First, a colonization by the ancestor of modern North American crested tits (subgenus Baeolophus) occurred 4 mya, on the basis of the estimate of 8.5% sequence divergence between the subgenera Baeolophus and Lophophanes. Second and soon thereafter (3.5 mya), the ancestor of all North American chickadees (subgenus Poecile) colonized the continent, based on the average divergences of 7% (6.81 ± 0.76% [mean ± SD]) between North American species in Poecile and the two Eurasian species, P. montanus and P. palustris, the sister group to North American Poecile in our optimal trees.
Soon after their invasion, the North American crested titmice occupied the Madro Tertiary flora of the continent (Cicero 1996). They also underwent at least two successive cycles of speciation. First, the separation of P. inornatus and P. bicolor from wollweberi (and loss of black and white face pattern) occurred. Then, during the mid- to late Pliocene, P. inornatus and P. bicolor split. Pre-inornatus evolved “in conjunction with increased segregation and impoverishment of western sclerophyllous woodlands in the late Pliocene and early Pleistocene” (Cicero 1996). More recently, bicolor fragmented into the distinct P. atricristatus in Texas-Mexico and P. bicolor in the eastern half of North America. That split is hypothesized to have taken place in the late Pleistocene, 250,000 years ago, by Dixon (1978) and Gill and Slikas (1992). Similarly, P. ridgwayi, of the western interior, separated from P. inornatus of the west coast, as a result of regional desertification in southeastern California during the late Pleistocene and Holocene transition (Cicero 1996).
Our comparisons of cytochrome-b sequences include all currently recognized New World species of chickadees (subgenus Poecile). Several of those sort into two well-defined groups: ([hudsonicus, rufescens], cinctus) and (atricapillus, gambeli). But the positions of those groups visà-vis one another, and relative to P. carolinensis and P. sclateri, remain unclear. The relationships of P. sclateri of the Mexican highlands and southwestern U.S. remain unresolved. On the basis of cytochrome-b evidence, P. sclateri paired inconsistently with P. carolinensis and (atricapillus, gambeli). Parus sclateri associated weakly with (hudsonicus, rufescens) in a previous study of mtDNA restriction sites (Gill et al. 1993).
The present study confirmed the sister relationship between P. atricapillus and P. gambeli reported earlier by Gill et al. (1993). Parus atricapillus speciated from P. gambeli 2.5 mya. Parus atricapillus is not most closely related to P. carolinensis, even though the two morphologically similar species hybridize in a narrow zone of secondary contact in the eastern United States (Brewer 1963, Gill et al. 1993, E.D. Sattler unpubl. data 1996).
Three species of chickadees of northwestern North America (P. hudsonicus, P. rufescens, P. cinctus) traditionally have been affiliated with each other (Mayr and Short 1970). Parus hudsonicus and P. rufescens clearly are sister species, despite speculation that P. cinctus of the Old World and P. hudsonicus of the New World were conspecific (Eck 1980). Those three species formed early in the Quaternary, 1.5 mya, on the basis of their sequence divergence of 3%. Given the placement of P. cinctus well within the clade of North American chickadees in the subgenus Poecile, we hypothesize that its immediate ancestor colonized Eurasia from North America. The small modern population of cinctus in Alaska likely represents either a remnant of that ancestral population or a subsequent recolonization of North America.
Comparative divergence of parid taxa
Initially, we noted that the parids exhibit a mosaic pattern of population divergence within species and larger-than-expected divergences between species. Our comparisons of parid cytochome b addressed mainly the second issue. To investigate the patterns of species divergence further, we plotted cytochrome-b divergence within and between species and within and between genera of parids in Figure 4. The figure may be compared to figure 1 in Johns and Avise (1998) to obtain a relative impression of divergence in parids versus other groups of birds.
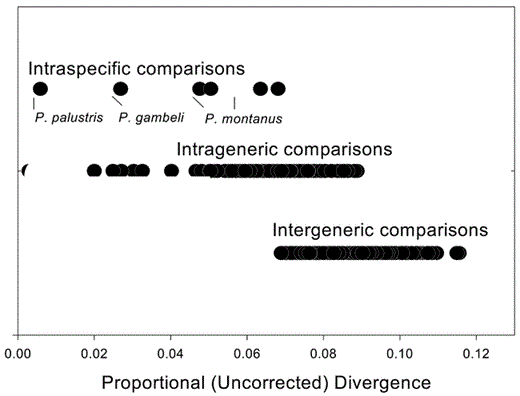
Relative ranges of divergence for intraspecific, intrageneric, and intergeneric comparisons of cytochrome b among parids. “Intrageneric comparisons” are between species in the same parid subgenus and “intergeneric comparisons” are between parid species in different parid subgenera
Parids are remarkably similar to one another in morphology and behavior and are distinct from other groups of passerines. As such, they appear to be a closely related, recently evolved group, and for that reason all species have been considered congeneric. However, the longest cytochrome-b distances between parid species reach 12% in uncorrected divergence, which suggests that parids are an older group than previously realized. Their relative age is evident when parid divergence is compared to intrageneric distances plotted by Johns and Avise (1998). They examined 88 genera, of which 70 (80%) exhibit less pairwise divergence than parids. Even if we consider only those genera with multiple species comparisons, 31 of 48 (65%) have lower between-species divergence than the most distantly related parids. Although such comparisons assume a molecular clock and are biased by the subjectivity of generic allocations, it is clear that the titmice are a relatively old group of songbirds characterized by morphological stasis, with one stunning exception.
The enigmatic Tibetan Ground-Jay (Ps. humilis) was long thought to be a corvid, though with doubt (Borecky 1978, Hope 1989). This terrestrial, small thrasher-like species is restricted to a limited region of the high Tibetan plateau. It builds nests in crevices and rodent burrows and in cavities that it excavates in banks (Schäfer 1938). On the basis of separate parsimony analyses of 55 osteological traits and a limited set of DNA sequences (James et al. 2003), Pseudopodoces—it turns out—is an unconventional titmouse. Phylogenetic analyses based on nuclear (c-myc) and mitochondrial (cytochromeb) gene sequences place Pseudopodoces within the Paridae, not the Corvidae. In all optimal trees based on the cytochrome-b sequences of this study, as in James et al. (2003), Pseudopodoces falls within the family Paridae, sister to the great tits (subgenus Parus). The cytochrome-b divergence between Pseudopodoces and other species of titmice and chickadees is 9.4% (uncorrected), roughly the level of divergence among parid subgenera. Thus, Pseudopodoces is the most morphologically and ecologically divergent of all tit species. Such divergence contrasts with the morphological stasis that is the surface hallmark of the Paridae. Ground Tit would be the appropriate English name for this unusual species.
Adaptive radiation of the Paridae
The 51 species of titmice and chickadees constitute a well-defined, monophyletic assemblage of sylvioid passerine birds on the basis of both morphological and molecular characters (Moreno 1985, Harrap and Quinn 1995, Sheldon and Gill 1996, James et al. 2003). Their feeding behavior of hammering open seeds with their short bills is associated with diagnostic skull osteology (James et al. 2003).
More than in their morphology, parids exhibit substantial diversification in key features of behavior and in their vocalizations. Two of the oldest clades (subgenera Parus and Cyanistes) do not cache and recover seeds as the majority of parid species do. That difference in behavior is correlated with differences in brain morphology and social systems (Ekman 1989, Sherry 1992, Slikas et al. 1996). Even more striking is the diversification of vocal repertoires among clades (Thielcke 1968, Hailman 1989).
Parids generally have elaborate vocal repertoires with dozens of elements that include differentiated whistled songs, “gargles,” alarm calls, and a combinatorial “chick-a-dee” call complex. Hailman (1989, fig. 4) suggested that the primary evolutionary direction of the repertoires was toward increasingly complex vocalizations, from unit vocal patterns, to contextual use of songs, to repertories of equivalent songs, followed by reduced songs in chickadees and gray tits (subgenus Poecile). Our phylogeny of parid taxa suggests different polarities of change among the major features of vocal organization, because at least three taxa with complex repertoires (i.e. Sylviparus, Parus, and in the genus Parus, the subgenera Cyanistes) are among the oldest lineages in our trees.
Blue tits of the subgenus Cyanistes produce many of the note types found in other parid species (but not the whistled songs of species of the subgenus Poecile; J. Martens unpubl. data), which implies that the entire note repertoire of the Paridae was present early in the diversification of the family. Blue tits also employ combinatorial syntax (Hailman 1989), which we project has been a feature of vocal behavior since the early origin of Parus sensu lato. Thus, certain repertoires (i.e. that of hudsonicus) are secondarily simplified to isolated notes and unit vocal patterns. We defer the formal analysis of vocal repertoire changes, including additions and deletions of whistled songs, to a separate paper (McCallum et al. unpubl. data). We hypothesize here that cultural restructuring and simplification of complex vocal repertoires may have played a major role in parid speciation. Our phylogenetic framework of parid taxa will help guide future decisions about the polarities of change among the major features of vocal organization.
Taxonomy
To be descriptively useful, a genus should be monophyletic, reasonably compact, and ecologically, morphologically, or biogeographically distinct. Thus, Sheldon and Winkler (1993) advocated the splitting of the large swallow genus Hirundo into monophyletic groups, such as cliff swallows (Petrochelidon), house martins (Delichon), crag martins (Ptyonoprogne), and so forth, on the basis of distinctive nesting habits. Given that logic, it may be desirable to split the genus Parus, which currently consists of 51 species and is one of the largest genera of birds. The AOU (1998) started the process by elevating the North America crested titmice (subgenus Baeolophus) and chickadees (subgenus Poecile) to genera. As mentioned above, we have attempted a comprehensive review of relations among species based on the traditional primacy of the large genus Parus, with explicit and secondary use of historical subgeneric names. On the basis of the results presented here, we now recommend the elevation of five subgenera to genera, and restricted use of Parus for members of the clade of great tit species only. Those changes would result in the adoption of six genera of parids in addition to continued recognition of the monotypic Melanochlora, Sylviparus, and Pseudopodoces. Restructuring the traditional genus Parus into six clades of generic status should facilitate future analyses of evolution among parids. Each of the six recommendend genera is a monophyletic group that displays distinctive behavioral and morphological characteristics. Those six genera elevated from subgenera are:
(1) Poecile. The familiar chickadees of North America and gray tits of Eurasia also include P. varius of the Orient. Hellmayr (1903), Thielcke (1968), and Harrap and Quinn (1995) separated varius as the monotypic genus-subgenus Sittiparus.
(2) Baeolophus. We support recognition of this genus, as adopted by the AOU (1998), for the North American crested titmice, including wollweberi.
(3) Lophophanes. We recommend recognition of this strictly Eurasian genus to include P. cristatus and P. dichrous, but not P. wollweberi.
(4) Periparus. In addition to the traditional species of coal tits, two Oriental species are members of this group. Parus elegans and P. amabilis were previously separated as Pardaliparus. Löhrl (1988) also allied P. venustulus to P. amabilis as the third member of Pardaliparus. We recommend including all three species in Periparus and would not recognize Pardaliparus at this time.
(5) Parus. In addition to the core species (P. major, P. monticolus), the great tits include the Oriental species, P. xanthogenys and P. spilonotus, as proposed by Hellmayr (1903) and Thielcke (1968); the African tits (Melaniparus, Harrap and Quinn 1995); and P. holsti, previously assigned to the monotypic Machlolophus. We do not have any insight into the relationship of P. semilarvatus, which Harrap and Quinn (1995) placed in the Sittiparus with P. holsti, so we retain it in Parus next to P. holsti.
(6) Cyanistes. The blue tits include the traditional widespread P. caeruleus and the north Asian species P. cyanus (including flavipectus).
Acknowledgments
We are grateful to the following colleagues who helped us obtain or loaned us tissues and sequences for this study: P. Arctander, G. Barrowclough, K. Bryant, P. Escalente, D. Gibson, S. Hackett, W. Howe, T. Huels, J. Ligon, R. Moldenhauer, A. Navarro, J. Pitochelli, J. Remsen, M. Robbins, S. Rohwer and S. Birks, S. Russell, B. Silverin, and E. Zhao. Extraction and amplification of DNA from museum study skins was performed in the Molecular Genetics Laboratory (MGL) at the National Zoo, Smithsonian Institution, with the generous permission of R. Fleischer, director of the MGL. For assistance in Greece, we thank G. I. Handrinos (Ministry of Agriculture) and A. Kyrkos and P. Pergantis. S.L.L. Gaunt and D.A. McCallum were great field companions in China and shared their wealth of knowledge of the Paridae, especially vocal repertoires. C. Spolsky, M. Kinnarney, and B.-A. Rechard assisted in the lab. H. James and P. Ericson kindly provided us with unpublished data on Pseudopodoces. S. Hackett, J. Martens, A. McCallum, and C. Perrins provided helpful comments on early drafts of the manuscript. This research was supported by National Science Foundation grants BSR 88-06647 to F.B.G., BSR 90-20183 to F.H.S., and BSR 92-07991 to F.B.G. and F.H.S.
Literature Cited

{kind=link}
{kind=link}
{kind=link}
{kind=link}