
Abstract
For the last two decades, the development of substrates for Surface Enhanced Raman Spectroscopy (SERS) has been continuously pushed for better performances. In this research, we designed SERS substrates with better detection capability and more efficient sample collection. In particular, morphology-controlled Ag nanoparticles were deposited onto cotton swab (cotton Q-tip), followed by a thin cover layer of graphene oxide by the dip-coating method. The graphene oxide overlay was expected to not only protect Ag nanoparticles from degradation by the environment but also contribute to sample adsorption and signal enhancement. To demonstrate the performance of the SERS substrates, different concentrations of Rhodamine B as low as 10–10 M were successfully detected and the enhancement factors were estimated to be 3 × 1010–5 × 1010. It is our expectation that these cotton swab-based substrates will contribute to our ongoing efforts toward low-cost, high-efficiency and durable SERS substrate.
Export citation and abstract BibTeX RIS

Original content from this work may be used under the terms of the Creative Commons Attribution 4.0 licence. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
1. Introduction
Surface Enhanced Raman Spectroscopy (SERS) is a powerful spectroscopic sensing technique with the capability of detecting minute concentration of analyte in a sample by analyzing the molecular fingerprint of that analyte. Thus, SERS has been used in various fields of applications spanning from chemistry, food science, environmental science to biomedicine and so on [1]. Traditionally, coinage metals, such as Au, Ag and Cu, are used as SERS substrates due to their strong surface plasmon resonance (SPR) over most of the visible and infrared range [2]. These substrates were fabricated as colloidal suspensions [3], patterned surfaces [4], or as coating layers on top of glass [5] or Si [6, 7]. While these substrates showed good performances, they lacked portability, flexibility, stability, and recyclability. One method to improve the flexibility of SERS substrates was to use soft materials, such as plastics [8], textile [9], paper [9], or cotton swab [10–14]. Compared with other flexible substrates, cotton swab is more convenient to handle by hand without having to touch the substrate. It also allows effective collection of analytes within narrow, hollow, or deep regions. Nevertheless, the use of cotton swab as SERS substrate can only be found in a handful of published articles. For example, some research groups reported the fabrication of SERS substrate by immersing cotton swab in the suspension of Ag nanoparticles (Ag NPs) [10–12]. SERS substrates were also fabricated by growing Ag NPs in situ directly on the cotton swab. For example, Liu et al described the growth of Ag NPs on polymer-coated cotton swab [13] while Gong et al seeded the cotton swab with small Ag NPs to grow a dense layer of bigger Ag NPs [14]. In those reports, SERS substrates based on cotton swab were able to detect toxic plant-protection chemicals on agricultural products at as low concentration as ∼0.1 ng cm−2 or trace explosives at ∼1.2 ng cm−2 in fingerprints.
Among the three coinage metals, Cu is used the least due to its reactive nature in air. Au is the most stable in air and chemically inert, but it poorly adsorbs analytes leading to low sensitivity. Ag is the most popular substrate material for SERS due to its high sensitivity, but it is less stable than Au in air [15]. Thus, one way to improve SERS performance of coinage metals is covering them with a layer that protects them from the damaging effects of air and other corrosion reagents. This overlay must be sufficiently thin so that it does not diminish the effect of SPR from the metallic surface below.
Graphene is a one-atom thick, two-dimensional form of carbon with atoms arranged in a honeycomb lattice. In addition to its unique electronic and photonic properties, graphene possesses mechanical stability, high biocompatibility, chemical inertness and impermeability to all gases and liquids [16, 17]. Graphene oxide (GO) has all the properties known to graphene but its surface structure includes several oxygen-containing groups. Thus, it absorbs aqueous solutions and adsorbs analytes better [18]. However, graphene is a non-plasmonic material and, therefore, not ideal for SERS. Ling et al was the first authors to explore the applicability of graphene for SERS and they reported that concentration of protoporphyin IX as low as 10–8 M was detected by monolayer graphene [19]. Other variations of graphene substrates for SERS can also be found in the literature, such as large-scale UV/ozone oxidized graphene [20], reduced graphene oxide [21, 22] or graphene nanomesh [23], all of which showed better performance than pristine graphene. The observed enhancement of Raman signals by graphene was attributed to the chemical mechanism in which the ease of light-induced charge transfer from the graphene substrate to the adsorbed molecules might alter the geometrical or electronic structure of the adsorbed molecules leading to spectral shifts and intensity changes [21]. In addition, graphene offers stronger molecular interactions and adsorption of more analytes, which also contributed to the signal enhancement [24]. Nevertheless, the enhancement due to chemical mechanism is still modest when compared with that due to electromagnetic mechanism.
Preferably, graphene would be combined with plasmonic material and the resulting nanocomposite would take advantage of the electromagnetic and chemical mechanisms. For example, Au NPs, Ag NPs or Cu NPs were synthesized in situ directly on reduced GO [25–27] or on epoxy resin reinforced with graphene nanosheets [28], and the prepared nanocomposites improved the sensitivity significantly when compared with individual components alone. More complex, multidimensional nanocomposites, such as graphene sandwiched between plasmonic materials, were also proposed for SERS substrates [29, 30]. In these structures, graphene acted as nanospacer not only to synergistically boost Raman signal enhancement by plasmonic materials but also to create nanogap that facilitated strong coupling between surface plasmons. As mentioned above, due to its thinness and mechanical-chemical stability, graphene appeared to be an ideal overlay material to protect the plasmonic metals below and, indeed, such role was explored. In most reports, Ag or Cu substrates covered with graphene were selected for testing their performances over time. For example, graphene/GO covering Ag NPs [31–33], Ag nanoantenna [34], Ag nanopopcorn [35], large-area Ag-coated nanospheres arrays [36], Au-on-Ag bimetallic layer [37], and Cu and Ag films [17], or Cu core-graphene shell NPs [38] are some of the successful applications thus far. In these reports, the protected plasmonic metals were able to serve as SERS substrates for several months with minor degradation as compared with their unprotected counterparts.
In this work, we fabricated SERS substrate based on cotton swab. Morphology-controlled Ag NPs were synthesized via a green and simple method, which were then deposited by a reproducible dip-coating method onto cotton swab. We then further deposited a layer of GO on top of the Ag coating (GO/Ag NPs/cotton swab). We selected Rhodamine B (RhB) as probe molecules to test the performance of our substrates. A detection limit of 10–10 M and an enhancement factors of 3 × 1010–5 × 1010 were achieved. Our results also indicated that when having GO as a protection layer over Ag NPs-covered cotton swab (Ag NPs/cotton swab), the intensity of Raman signals clearly improved. This research, therefore, contributes additional scientific information to support the use of cotton swab for developing low-cost, flexible SERS substrate. It is the first report about cotton-swab based SERS substrate that explores the role of GO to boost performance and the first part of our development series toward a low-cost, high-efficiency and durable SERS substrate.
2. Materials and methods
2.1. Materials
The chemicals used for the synthesis of Ag NPs included silver nitrate (AgNO3, 99.8%) purchased from VWR (Radnor, PA, USA) and sodium citrate dihydrate (Na3C6H5O7.2H2O, ≥ 99%) purchased from Merck (Darmstadt, Germany). The chemicals used for the synthesis of GO included graphite flakes (∼5 μm, 99.8%), sulfuric acid (H2SO4, 98%), phosphoric acid (H3PO4, 85%), potassium permanganate (KMnO4, 98%), hydrochloric acid (HCl, 5%) and hydrogen peroxide (H2O2, 30%). All of these were purchased from Merck (Darmstadt, Germany). Commercial cotton swabs were purchased from a local supermarket. The cotton swabs were activated by (3-aminopropyl)trimethoxysilane (APTMS, 97%) obtained from Energy Chemical (Zhejiang, China) and cleaned by absolute ethanol and acetone. Rhodamine B (RhB) dye from Acros Organics (Waltham, MA, USA) was used as probe molecule for SERS experiments. All reagents were used as received and all solutions were prepared using deionized (DI) water.
2.2. Characterization
The size and morphology of the nanostructures were studied by scanning electron microscopy (SEM) using FESEM S4800 instrument (Hitachi, Japan) at 10 kV accelerating voltage. The SEM instrument was equipped with an Emax energy dispersive x-ray (EDX, Horiba, UK) system, which was used to conduct elemental analyses of the deposited nanostructures. Prior to SEM study, each cotton-based sample was coated with a thin layer of Pt to increase the conductivity of cotton and avoid charges built up on the surface. The sizes and size distribution of Ag NPs were also characterized by dynamic light scattering (DLS) method using the nanoPartica SZ-100V2 nanoparticle analyzer (Horiba, UK) performed at 25 °C in DI water. The result was represented by scattering light intensity. To analyze and compare the crystalline structures of Ag NPs/cotton swab and GO/Ag NPs/cotton swab, x-ray diffraction (XRD) was performed on a LabX XRD-6100 diffractometer (Shimadzu, Japan) equipped with a LYNXEYE XE-T detector, a vertical goniometer and a sealed Cu tube. UV–vis absorption spectroscopy for measuring optical property of Ag NPs was recorded by a Cary 60 (Agilent, USA) spectrophotometer. Raman spectroscopy and SERS measurements were performed at room temperature on an XploRA Plus Raman Spectrometer (Horiba, UK) with a laser's wavelength of 638 nm, using 10x objective lens for 3 accumulations, each of which for 35 s. SERS spectra were collected from several random regions on each sample to confirm homogeneity.
2.3. Synthesis of Ag NPs
Ag NPs were synthesized by the reduction method as reported by Lee and Meisel [39]. To a 150 ml Erlenmeyer flask, 36 mg AgNO3 was dissolved in 100 ml DI water to obtain 3.6 × 10–2% (w/w) AgNO3 solution. Then, 40 mg Na3C6H5O7.2H2O powder was dissolved in 3.96 ml DI water to obtain 1% (w/w) citrate solution. The synthesis of Ag NPs started by heating AgNO3 solution to the boiling temperature under vigorous stirring at 550 rpm. Then, citrate solution was added dropwise to the AgNO3 solution, turning it from colorless to clay bank to brownish gray. After that the reaction mixture was stirred vigorously for additional 30 min to ensure complete reaction. To stop the synthesis, the Erlenmeyer flask was removed from the hot plate and the suspension of Ag NPs was allowed to cool in air to room temperature. Then, the suspension of Ag NPs was centrifuged at 4000 rpm for 15 min three times. The supernatant was discarded and the settled Ag NPs were redispersed in 1.5 ml DI water before being transferred to an amber vial and stored at 4 °C in the dark.
2.4. Synthesis of GO nanosheets
In this work, GO nanosheets were fabricated from graphite flakes using the modified Hummers' method as described in a previous report [40]. Specifically, 12 ml H3PO4, 36 ml H2SO4 and 1 g graphite flakes were mixed and stirred at ∼5 °C in an ice bath for 15 min. Then, 12 g KMnO4 was added slowly to the previous mixture under continuous stirring and the reaction mixture was transferred to a water bath at ∼35 °C and stirred for another 4 h until a thick paste was observed. The paste was diluted by adding 90 ml DI water dropwise and the suspension was stirred for 5 h at ∼90 °C. After that, another 90 ml DI water was added, followed by the slow addition of 30 ml H2O2 until the suspension turned bright yellow, indicating the oxidation of graphite. The oxidized graphite was collected by centrifugation at 7000 rpm for 30 min, then cleaned with 200 ml HCl and DI water several times until reaching a pH of ∼6–∼7. The clean oxidized graphite was then redispersed in 500 ml DI water under mild sonication for 5 h to obtain exfoliated GO nanosheets. Finally, the thick, multilayered GO sheets were separated from the ultra-thin GO sheets by centrifugation at 10000 rpm for 3–5 min until all the visible particles settled down. The supernatant was collected and further centrifuged at 15000 rpm for 45 min to remove water-soluble byproducts.
2.5. Fabrication of SERS substrates
Before use, the cotton swab was thoroughly cleaned by immersing it in acetone, followed by ethanol and DI water for 10 min each in an ultrasonic bath. Between each immersion, the cotton swab was rinsed with DI water.
To coat it with APTMS, the cotton swab was immersed into a solution of 2% APTMS for 30 min, then dried in an oven at 120 °C for 30 min. To immobilize Ag NPs onto APTMS-coated cotton swab (Ag NPs/cotton swab), the suspension of Ag NPs was first sonicated and the APTMS-coated cotton swab was soaked into this suspension for 10 min at 60 °C. This process was repeated two more times as the suspension was reused. Finally, the Ag NPs/cotton swab was dried in an oven at 120 °C for 30 min before being stored in a black plastic food bag in the dark at 4 °C. Prior to use, a small portion of GO stock suspension was taken out and diluted to 7 times its original volume by DI water to obtain 0.3 mg/ml GO suspension. Then the GO suspension was sonicated for 1 h in an ice bath. In order to coat the Ag NPs/cotton swab with GO (GO/Ag NPs/cotton swab), the Ag NPs/cotton swab was soaked into GO suspension momentarily then dried in an oven at 120 °C for 30 min before being stored in a black plastic food bag in the dark at 4 °C.
To test the substrates for SERS performance, Ag NPs/cotton swab or GO/Ag NPs/cotton swab were momentarily soaked into 2 ml ethanolic RhB solution prepared at the desired concentration and dried at room temperature for 15 min. Such process was repeated four more times and the substrate was stored in a black plastic food bag in the dark at 4 °C. Figure 1 outlines the fabrication process.

Figure 1. Schematic illustration of the SERS substrate fabrication process. Some components of the substrate were not shown for simplicity.
Download figure:
Standard image High-resolution image3. Results and discussions
3.1. Characterization of cotton-swab based SERS substrates
Figure 2 shows a digital photograph of GO/Ag NPs/cotton swab substrate. The cotton part of the swab was completely covered by GO and Ag NPs indicated by its dark appearance. The synthesized Ag NPs had the morphology and size distribution as shown in figure 3. The SEM image (figure 3(a)) indicates that most Ag NPs are quasi-spherical in shape while others appear to be elongated or rod-like. We also manually measured sizes of Ag NPs using ImageJ software to quantitatively determine their size distribution. 283 particles were selected randomly for the measurements and the size of each particle was recorded based on the length of its longer side. To avoid outliers, we excluded any particles whose sizes surpassed 100 nm. The average size of Ag NPs thus was estimated to be 65.76 ± 11.67 nm or 65.76 nm ± 17.75%. This value shows relatively uniform Ag NPs and is supported by the histogram in figure 3(b) which assumes a nearly bell-shaped curve of a Gaussian distribution. We also used DLS as another means to examine size distribution of Ag NPs. The estimated mean size was 137.6 ± 35.7 nm or 137.6 ± 25.9%, which was two times larger than that estimated manually by ImageJ. The discrepancy was expected because there was a small number of rod-like Ag particles in our sample and these elongated particles increased light scattering, leading to a shift toward larger sizes [41]. Nevertheless, a Gaussian distribution was also observed by DLS, confirming the presence of relatively monodisperse Ag NPs.

Figure 2. Digital photograph showing the cotton swab after deposition of GO and Ag NPs (GO/Ag NPs/cotton swab).
Download figure:
Standard image High-resolution image
Figure 3. (a) SEM image of synthesized Ag NPs. Histograms showing size distributions of Ag NPs estimated by (b) ImageJ software and (c) DLS method.
Download figure:
Standard image High-resolution imageOptical response of Ag NPs was measured by UV–vis spectrophotometer. The resulting spectrum in figure 4 shows a single absorption peak at the wavelength of ∼423 nm, which matched that of spherical Ag NPs. The absence of any other peaks in the spectrum is an evidence that the elongated Ag NPs existed in small number that their effect on the optical property of the Ag NPs sample was unseen [42]. The absorption peak is sharp and tall which gives another indication of the relative uniformity of our NPs.

Figure 4. UV-Vis absorption spectrum of as-synthesized Ag NPs.
Download figure:
Standard image High-resolution imageUpon deposition of Ag NPs on cotton swab, several SEM images were taken at different locations on the sample to examine how the particles were distributed on cotton swab, one of which is shown in figure 5. Prior to deposition, the cotton strands appear to have smooth surfaces (figure 5(a)). After the deposition, much rougher surfaces were clearly observed (figure 5(b)). A close-up view of the deposition layer shows that Ag NPs did not fuse to form a flat, dense layer. Rather, a continuous, porous layer was formed by the binding of adjacent particles to cotton swab, facilitated by APTMS. The roughness of the layer was seen as other individual Ag NPs aggregating onto the bound particles or as the binding of particle aggregations to the cotton swab. Furthermore, nearly the entire surfaces were covered with Ag NPs, indicating that the deposition method used was simple but efficient. The particles were bound firmly to cotton swab even after several sample handling procedures and transportation to the characterization facility while the loosely bound particles were detached from the surface, leaving uncoated areas scattering throughout the surface. The chemical composition of Ag NPs/cotton swab substrate was determined by EDX and presented in figure 5(c). Clearly, Ag was the only metallic peak detected. C and O appeared to be the other two major peaks because they were the two major elements that made up the cotton swab. The XRD pattern obtained from the Ag NPs/cotton swab sample is presented in figure 5(d). The peaks were clearly seen and indexed. The diffraction peaks at 2θ values of ∼38°, ∼44°, ∼65°, ∼78° and ∼82° were identified as constructive interferences of the reflected x-rays from the (111), (200), (220), (311) and (222) planes of the fcc crystalline Ag NPs, respectively. The highest-intensity (111) peak and the broadening of the peaks reveal that a majority of crystal facets in Ag nanostructures was (111) and the sizes of particles were in the nanoscale. The non-indexed peaks, marked with (*) in figure 5(d), were identified as the peaks from the cotton strands whose composition included crystalline cellulose [43, 44].

Figure 5. SEM images of cotton swab (a) before and (b) after deposition of Ag NPs. The inset shows the surface of Ag NPs/cotton swab at higher magnification. (c) EDX spectrum of (b). The red mark on the inset of (b) is the location where EDX data were recorded. (d) XRD of Ag NPs/cotton swab.
Download figure:
Standard image High-resolution imageSurface morphology of the as-synthesized GO nanosheets was also characterized by SEM. As seen in figure 6(a), the SEM image indicates that GO material was layered and composed of individual GO sheets loosely linked with each other producing a three-dimensional network structure. From figure 6(b), it is obvious that single and/or few GO sheets with typical wrinkle characteristics were produced during sonication process. The edges of GO sheets slightly curved because of surface tension and/or the presence of oxygen-containing functional groups on their surfaces and edges.

Figure 6. SEM images at (a) low and (b) high magnifications of as-synthesized GO nanosheets.
Download figure:
Standard image High-resolution imageFigure 7 shows the surface of Ag NPs/cotton swab upon the deposition of GO. By relating to figures 5 and 6, the thin layer that covers most of the Ag NPs should be GO and the layer appears to be thin enough so the Ag NPs are seen underneath. We also obtained chemical composition of the GO/Ag NPs/cotton swab sample by EDX and the data are presented in table 1. Table 1 also includes chemical composition data of the Ag NPs/cotton swab sample for comparison. Prior to the deposition of GO, Ag NPs/cotton swab has, in terms of atomic percentage, 60.44% of C and 9.33% of Ag on its surface or the atomic amount of C is ∼6 times greater than that of Ag. On the other hand, the surface of GO/Ag NPs/cotton swab shows an increase in the relative amount of C to 77% while that of Ag decreases to 1.44%, equivalent to atomic amount of C ∼53 times greater than that of Ag. A significant increase in the atomic amount of C is a clear indication of the existence of the GO overlay. In addition, Raman spectroscopy is another effective method that has been used widely to characterize carbon-based materials. In figure 7(d), the two prominent peaks at ∼1343 and ∼1591 cm−1 can be attributed to the D and G peaks of GO, respectively. The G peak was generated by the bond stretching of the sp2 C atoms while the D peak was generated by the breathing mode of aromatic rings. The intensity of the G peak is nearly 3 times stronger than that of the D peak, indicating that the structure of GO nanosheets used in this current report had substantially fewer defects [45]. In addition, we observed a small peak at ∼470 cm−1 and would assign it to the stretching vibration of C-N-C, likely from the organic remnants on the surfaces of Ag NPs after the synthesis [46]. One might notice in figure 7(a) the presence of some Ag NPs above, instead of below, the GO overlay. To explain this observation, we consider the fact that upon soaking Ag NPs/cotton swab into GO suspension, some loosely bound Ag NPs fell off the surface to the GO suspension. Then when the substrate was lifted up, some of these Ag NPs re-attached to the surface and stayed on top of GO. While avoiding these re-attached Ag NPs is the subject of our on-going research, we believe that most of these loosely bound particles will soon detach again and their presence on the surface may not cause any substantial negative effect to the substrate performance.

Figure 7. (a), (b) SEM images of GO/Ag NPs/cotton swab taken at two different locations and magnifications. (c) EDX spectrum of (b). The red mark on (b) is the location where EDX data were recorded. (d) Raman spectrum of GO/Ag NPs/cotton swab.
Download figure:
Standard image High-resolution imageTable 1. EDX data of Ag NPs/cotton swab and GO/Ag NPs/cotton swab obtained from figures 5(c) and 7(c).
| Element | C | O | Ag | Pt | Total | |
|---|---|---|---|---|---|---|
| Weight % | Ag NPs/cotton swab | 32.76 | 21.83 | 45.42 | 100.00 | |
| GO/Ag NPs/cotton swab | 63.39 | 23.44 | 10.66 | 2.51 | 100.00 | |
| Atomic % | Ag NPs/cotton swab | 60.44 | 30.23 | 9.33 | 100.00 | |
| GO/Ag NPs/cotton swab | 77.00 | 21.37 | 1.44 | 0.19 | 100.00 | |
3.2. Cotton-swab based SERS substrates for the detection of RhB
To evaluate Ag NPs/cotton swab and GO/Ag NPs/cotton swab as possible platforms for SERS application, Rhodamine B (RhB) was chosen as probe molecule. It is an organic dye that has been widely used for SERS detection in the laboratories due to its well-defined vibrational features. A series of RhB concentrations from 10–3 M to 10–10 M was prepared for testing. Figure 8 shows the resulting Raman spectra obtained when Ag/cotton swab substrates were soaked in these RhB concentrations. Several prominent peaks at 620 cm−1, 932 cm−1, 1191 cm−1, 1281 cm−1, 1357 cm−1, 1505 cm−1, 1531 cm−1, 1649 cm−1 were easily observed and they were identified as the fingerprint vibrational modes attributed to RhB according to table 2. The peak intensities are strongest when RhB concentration is 10–3 M and gradually decrease with concentrations. We have currently achieved a detection limit of 10–10 M, which is comparable or better than those reported in some other publications using cotton swabs or flexible substrates [8–10, 13]. Additionally, plain cotton swab, without any Ag NPs or GO deposition, was also experimented as SERS substrate. No obvious peak was detected with this substrate even with RhB concentration of 10–3 M, thus confirming that cotton swab alone was not suitable for SERS and that the amplification of Raman signals solely was a result of the local electromagnetic enhancement induced by the strong SPR. Furthermore, as the concentrations of RhB decreased, we observed some gradual changes to the Raman spectra as well. For example, the peak at 932 cm−1 has low intensity compared to other peaks at high concentrations. However, its intensity rapidly increases at lower concentrations then exceeds other peaks at very low concentrations. In other instances, we observed some peak shifts, peak disappearance such as those at 1505 and 1531 cm−1, or new peak appearance such as the double peaks around 1400 cm−1. A possible explanation for those changes in Raman spectra as RhB concentrations decrease was proposed by Wang et al in which varying molecular interactions at different concentrations might promote the redistribution of charges and alter the electronic molecular structure of the RhB-Ag complex [47]. However, a more thorough understanding of the behaviors of RhB's Raman spectra at low concentrations is needed and requires more research.

Figure 8. Raman spectra of (a) 10–3 M – 10–6 M and (b) 10–7 M – 10–10 M RhB recorded from Ag NPs/cotton swab substrate. The bottom spectrum was recorded from plain cotton swab for comparison.
Download figure:
Standard image High-resolution imageTable 2. Raman mode assignments for RhB [48].
| Raman shift (cm−1) | Assignment |
|---|---|
| 1644 | Aromatic C-C stretch |
| 1591 | C-H stretch |
| 1551 | Aromatic C-C stretch |
| 1528 | C-H stretch |
| 1508 | Aromatic C-C stretch |
| 1426 | C-H stretch |
| 1360 | Aromatic C-C stretch |
| 1284 | Aromatic C-C stretch |
| 1199 | C-H in-plane bend |
| 1130 | C-H stretch |
| 932 | C-H stretch |
| 773 | C-H stretch |
| 622 | C-C-C stretch |
| 355 | |
| 278 | |
| 240 | Ag-N stretch |
| 213 |
In the next set of testing, we evaluated SERS performance of GO/Ag NPs/cotton swab by soaking it into RhB solutions of concentrations ranging from 10–3 M to 10–10 M, similar to those used for testing Ag NPs/cotton swab. In figure 9, Raman spectrum of GO/Ag NPs/cotton swab is placed side-by-side with that of Ag NPs/cotton swab at each RhB concentration for comparison. Clearly, those Raman peaks that were obtained from GO/Ag NPs/cotton swab always had higher intensities than the corresponding peaks obtained from Ag NPs/cotton swab. Our results, thus, confirm some of the advantages of adding GO to the plasmonic materials including charge transfer, synergistic interactions with plasmonic materials and stronger adsorption of more probe molecules. Interestingly, the Raman peaks of GO itself, as shown in figure 7(d), did not seem to interfere with those of RhB, if any, even though all Raman spectroscopic measurements in this report were performed under the same settings. Thus, we assumed that the SERS response of GO was much weaker than that of RhB.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 9. SERS performance comparison between Ag NPs/cotton swab and GO/Ag NPs/cotton swab substrates at RhB concentrations of (a) 10−3 M – 10−6 M and (b) 10−7 M – 10−10 M.
Download figure:
Standard image High-resolution image{kind=link}
{kind=link}
Finally, we estimated the enhancement factor (EF) to quantitatively evaluate the effectiveness of our SERS substrates. The most widely used formula to estimate EF is described as [25]
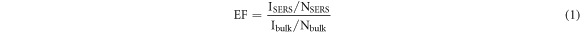
where ISERS and Ibulk are the SERS intensities that were recorded on the SERS-active substrate and on the bulk, respectively, while NSERS and Nbulk are the number of probe molecules illuminated by the laser light on the SERS-active substrate and on the bulk, respectively. We first estimated Nbulk using the formula [25]
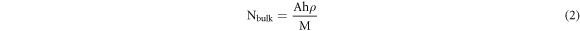
where A is the area of the laser spot (7.61 μm2), h is the laser penetration depth, presumably a few micrometers, say 3 μm [49, 50], ρ (0.79 g cm−3) and M (479.02 g mol−1) are the density and the molar mass of RhB, respectively. Thus, Nbulk can be estimated to be 3.77 × 10–14 mol. Next, to estimate NSERS, we chose the lowest concentration of RhB that was used to test our substrates for SERS performance, which is 10–10 M. We assumed that RhB would adsorb completely to the entire tip of the cotton swab. We also assumed that the cotton swab would absorb 103 μl of RhB after being soaked in RhB solution [51]. Thus, the total number of moles of RhB adsorbed to the substrate can be estimated as 1.03 × 10–14 mol. If we assume the geometrical shape of the cotton swab tip to be cylindrical, then the surface area of the cotton swab tip will be 56π and the RhB density on the substrate will be (1.03 × 10–14)/56π (mol/mm2). With the area of the laser spot to be 7.61 μm2,
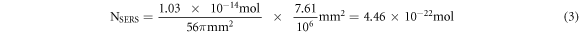
Regarding ISERS/Ibulk, since the Raman signal obtained from the bulk (the plain cotton swab in this case) was too weak to be detected by our instrument, we used the assumption made by Yamamoto et al in which the minimum detectable Raman signal intensity of 30 counts was considered for Ibulk [52]. Thus, ISERS/Ibulk with respect to Ag NPs/cotton swab and GO/Ag NPs/cotton swab were estimated to be ∼380 and ∼602, respectively. The peak intensity recorded for GO/Ag NPs/cotton swab is, therefore, more than 1.5 times higher than that recorded for Ag NPs/cotton swab. By substituting all estimated values of ISERS/Ibulk, Nbulk and NSERS to equation (1), we obtained EFs for Ag NPs/cotton swab and GO/Ag NPs/cotton swab substrates to be 3 × 1010 and 5 × 1010, which were comparable to those reported by others [8, 10–14].
4. Conclusion
In summary, SERS substrates based on Ag NPs/cotton swab and GO/Ag NPs/cotton swab were successfully fabricated using only the dip coating method. With RhB as probe molecule, we showed that our SERS substrates were capable of detecting RhB concentrations as low as 10–10 M with an enhancement factor of 3 × 1010–5 × 1010. In addition to functioning as a protection layer as we assumed, our results showed that a thin GO overlay on top of plasmonic material would also enhance Raman signal intensity, leading to possible lower detection limit. This is our first effort toward the development of a low-cost, high-efficiency and durable SERS substrate. In the coming weeks and months, we will continue to investigate the stability of our cotton-swab based SERS substrates as well as their recyclability. We will also look into applying these SERS substrates to detecting other organic molecules that contaminate agricultural products.
Acknowledgments
This research is funded by Murata Science Foundation under grant number 21VH08. We would like to thank Ho Chi Minh City University of Technology (HCMUT), VNU-HCM for the support of time and facilities for this study.
Data availability statement
All data that support the findings of this study are included within the article (and any supplementary files).