





Abstract
From recent research it has become clear that at least two different possibilities for anaerobic ammonium oxidation exist in nature. ‘Aerobic’ ammonium oxidizers like Nitrosomonas eutropha were observed to reduce nitrite or nitrogen dioxide with hydroxylamine or ammonium as electron donor under anoxic conditions. The maximum rate for anaerobic ammonium oxidation was about 2 nmol NH+4 min−1 (mg protein)−1 using nitrogen dioxide as electron acceptor. This reaction, which may involve NO as an intermediate, is thought to generate energy sufficient for survival under anoxic conditions, but not for growth. A novel obligately anaerobic ammonium oxidation (Anammox) process was recently discovered in a denitrifying pilot plant reactor. From this system, a highly enriched microbial community with one dominating peculiar autotrophic organism was obtained. With nitrite as electron acceptor a maximum specific oxidation rate of 55 nmol NH+4 min−1 (mg protein)−1 was determined. Although this reaction is 25-fold faster than in Nitrosomonas, it allowed growth at a rate of only 0.003 h−1 (doubling time 11 days). 15N labeling studies showed that hydroxylamine and hydrazine were important intermediates in this new process. A novel type of hydroxylamine oxidoreductase containing an unusual P468 cytochrome has been purified from the Anammox culture. Microsensor studies have shown that at the oxic/anoxic interface of many ecosystems nitrite and ammonia occur in the absence of oxygen. In addition, the number of reports on unaccounted high nitrogen losses in wastewater treatment is gradually increasing, indicating that anaerobic ammonium oxidation may be more widespread than previously assumed. The recently developed nitrification systems in which oxidation of nitrite to nitrate is prevented form an ideal partner for the Anammox process. The combination of these partial nitrification and Anammox processes remains a challenge for future application in the removal of ammonium from wastewater with high ammonium concentrations.
1 Introduction
The oxidation of ammonium has been investigated mainly in aerobic or oxygen-limited systems. In theory ammonium could also be used as an inorganic electron donor for denitrification. The free energy for this reaction (Table 1) is nearly as favorable as for the aerobic nitrification process. It was on the basis of these thermodynamic calculations that the existence of two chemolithoautotrophic microorganisms capable of oxidizing ammonium to dinitrogen gas was already predicted two decades ago [1]. The actual discovery of such a process was only recently described [2,3]. During experiments on a denitrifying pilot plant of a multi-stage wastewater treatment system at Gist-Brocades (Delft, The Netherlands) it was noted that ammonium disappeared from the reactor effluent at the expense of nitrate with a concomitant increase in dinitrogen gas production. A maximum ammonium removal rate of 1.2 mmol l−1 h−1 was observed. In continuous flow experiments the nitrogen and redox balances showed that ammonium really disappeared under anaerobic conditions, and that for every mol of ammonium consumed 0.6 mol of nitrate was required, resulting in the production of 0.8 mol of dinitrogen gas. This newly discovered process was named the Anammox (anaerobic ammonium oxidation) process.
| Reaction equation | ΔG°′ (kJ mol−1 NH+4 or NO−3) |
| 2NO−3+5H2+2H+→N2+6H2O | −560 |
| 8NO−3+5HS-+3H+→4N2+4H2O+5SO2−4 | −465 |
| 3NO−3+5NH+4→4N2+9H2O+2H+ | −297 |
| NO−2+NH+4→N2+2H2O | −358 |
| 2O2+NH+4→NO−3+H2O+2H+ | −349 |
| 6O2+8NH+4→4N2+12H2O+8H+ | −315 |
| Reaction equation | ΔG°′ (kJ mol−1 NH+4 or NO−3) |
| 2NO−3+5H2+2H+→N2+6H2O | −560 |
| 8NO−3+5HS-+3H+→4N2+4H2O+5SO2−4 | −465 |
| 3NO−3+5NH+4→4N2+9H2O+2H+ | −297 |
| NO−2+NH+4→N2+2H2O | −358 |
| 2O2+NH+4→NO−3+H2O+2H+ | −349 |
| 6O2+8NH+4→4N2+12H2O+8H+ | −315 |
| Reaction equation | ΔG°′ (kJ mol−1 NH+4 or NO−3) |
| 2NO−3+5H2+2H+→N2+6H2O | −560 |
| 8NO−3+5HS-+3H+→4N2+4H2O+5SO2−4 | −465 |
| 3NO−3+5NH+4→4N2+9H2O+2H+ | −297 |
| NO−2+NH+4→N2+2H2O | −358 |
| 2O2+NH+4→NO−3+H2O+2H+ | −349 |
| 6O2+8NH+4→4N2+12H2O+8H+ | −315 |
| Reaction equation | ΔG°′ (kJ mol−1 NH+4 or NO−3) |
| 2NO−3+5H2+2H+→N2+6H2O | −560 |
| 8NO−3+5HS-+3H+→4N2+4H2O+5SO2−4 | −465 |
| 3NO−3+5NH+4→4N2+9H2O+2H+ | −297 |
| NO−2+NH+4→N2+2H2O | −358 |
| 2O2+NH+4→NO−3+H2O+2H+ | −349 |
| 6O2+8NH+4→4N2+12H2O+8H+ | −315 |
2 Biological nature of the Anammox reaction
In a follow-up study, the biological nature of the Anammox process was investigated in more detail [4]. In anoxic batch experiments ammonium and nitrate were converted within 9 days of incubation when intact seed material (Anammox biomass) from the pilot plant was added. However, when the seed material was subjected to γ-radiation or sterilization at 121°C or when the seed material was omitted from the incubation no change in the concentration of nitrate and ammonium could be observed. Furthermore the addition of various inhibitors (2,4-dinitrophenol, carbonyl cyanide m-chlorophenylhydrazone or HgCl2) to the incubations resulted in a complete inhibition of the ammonium oxidation and nitrate reduction (Table 2). In these experiments with initial ammonium concentrations of 5 mM and higher, the rate of ammonium oxidation was proportional to the amount of biomass used. Taken together these experiments strongly suggested that the anaerobic ammonium oxidation was a biological process carried out by microorganisms. The specific rate of ammonium oxidation (0.08 nmol NH+4 min−1 (mg dry weight)−1) in these batch experiments was quite low compared to rates (1.2 nmol NH+4 min−1 (mg dry weight)−1) obtained in the pilot plant. This indicated that in the batch experiments the conversion was limited by diffusion of the substrates to the biomass granules. Labeling experiments with 15NH+4 in combination with 14NO−3 showed an almost exclusive production of 14-15N2 gas. This finding did not agree with the postulated overall reaction [3] (see Section 1) in which for every labeled ammonium, 0.6 nitrate would react to form 0.8 dinitrogen gas (i.e. 75% of the formed dinitrogen would be 15-14N2 and 25% would be 15-15N2). Only if nitrite rather than nitrate was assumed as the actual oxidizing agent the observed and calculated values would be in agreement [4].
| Treatment inhibitor/stimulator | Mode of action | Concentration or period tested | Effect |
| NH+4+NO−2 | activity test | 0–7 mM | normal activity |
| No biomass | none | 0 mg l−1 | no activity |
| Sterilization at 121°C | denaturation | 20–120 min | no activity |
| Gamma irradiation | inactivation | 60 min | no activtiy |
| Penicillin V | inhibition of cell wall synthesis of bacteria | 0–100 mg l−1 | none |
| Penicillin G | id. | 0–1000 mg l−1 | none |
| Bromoethane sulfonic acid | inhibition of methanogenesis | 0–20 mM | none |
| Na2SO4 | stimulation of sulfate reduction | 20 mM | none |
| Na2MoO4 | inhibition of sulfate reduction | 20 mM | none |
| Chloramphenicol | inhibition of protein synthesis | 0–400 mg l−1 | none |
| Hydrazine | inhibition of NH2OH oxidation | 0–3 mM | activation |
| Acetone | solvent for N-serve | 10 mM | none |
| N-serve | inhibition of nitrification | 0–50 mg l−1 | none |
| Allylthiourea | inhibition of nitrification | 0–10 mM | none |
| Acetylene | inhibition of nitrification and denitrification | 6 mM | inhibition |
| 2,4-Dinitrophenol | uncoupler | 0–400 mg l−1 | inhibition |
| Carbonyl cyanide m-chlorophenylhydrazone | uncoupler | 0–40 mg l−1 | inhibition |
| HgCl2 | cell damage | 0–300 mg l−1 | inhibition |
| Oxygen | oxidative stress | 0–0.2 mM | inhibition |
| Phosphate | chelating agent | <1 mM | none |
| Phosphate | chelating agent | >2 mM | inhibition |
| Treatment inhibitor/stimulator | Mode of action | Concentration or period tested | Effect |
| NH+4+NO−2 | activity test | 0–7 mM | normal activity |
| No biomass | none | 0 mg l−1 | no activity |
| Sterilization at 121°C | denaturation | 20–120 min | no activity |
| Gamma irradiation | inactivation | 60 min | no activtiy |
| Penicillin V | inhibition of cell wall synthesis of bacteria | 0–100 mg l−1 | none |
| Penicillin G | id. | 0–1000 mg l−1 | none |
| Bromoethane sulfonic acid | inhibition of methanogenesis | 0–20 mM | none |
| Na2SO4 | stimulation of sulfate reduction | 20 mM | none |
| Na2MoO4 | inhibition of sulfate reduction | 20 mM | none |
| Chloramphenicol | inhibition of protein synthesis | 0–400 mg l−1 | none |
| Hydrazine | inhibition of NH2OH oxidation | 0–3 mM | activation |
| Acetone | solvent for N-serve | 10 mM | none |
| N-serve | inhibition of nitrification | 0–50 mg l−1 | none |
| Allylthiourea | inhibition of nitrification | 0–10 mM | none |
| Acetylene | inhibition of nitrification and denitrification | 6 mM | inhibition |
| 2,4-Dinitrophenol | uncoupler | 0–400 mg l−1 | inhibition |
| Carbonyl cyanide m-chlorophenylhydrazone | uncoupler | 0–40 mg l−1 | inhibition |
| HgCl2 | cell damage | 0–300 mg l−1 | inhibition |
| Oxygen | oxidative stress | 0–0.2 mM | inhibition |
| Phosphate | chelating agent | <1 mM | none |
| Phosphate | chelating agent | >2 mM | inhibition |
| Treatment inhibitor/stimulator | Mode of action | Concentration or period tested | Effect |
| NH+4+NO−2 | activity test | 0–7 mM | normal activity |
| No biomass | none | 0 mg l−1 | no activity |
| Sterilization at 121°C | denaturation | 20–120 min | no activity |
| Gamma irradiation | inactivation | 60 min | no activtiy |
| Penicillin V | inhibition of cell wall synthesis of bacteria | 0–100 mg l−1 | none |
| Penicillin G | id. | 0–1000 mg l−1 | none |
| Bromoethane sulfonic acid | inhibition of methanogenesis | 0–20 mM | none |
| Na2SO4 | stimulation of sulfate reduction | 20 mM | none |
| Na2MoO4 | inhibition of sulfate reduction | 20 mM | none |
| Chloramphenicol | inhibition of protein synthesis | 0–400 mg l−1 | none |
| Hydrazine | inhibition of NH2OH oxidation | 0–3 mM | activation |
| Acetone | solvent for N-serve | 10 mM | none |
| N-serve | inhibition of nitrification | 0–50 mg l−1 | none |
| Allylthiourea | inhibition of nitrification | 0–10 mM | none |
| Acetylene | inhibition of nitrification and denitrification | 6 mM | inhibition |
| 2,4-Dinitrophenol | uncoupler | 0–400 mg l−1 | inhibition |
| Carbonyl cyanide m-chlorophenylhydrazone | uncoupler | 0–40 mg l−1 | inhibition |
| HgCl2 | cell damage | 0–300 mg l−1 | inhibition |
| Oxygen | oxidative stress | 0–0.2 mM | inhibition |
| Phosphate | chelating agent | <1 mM | none |
| Phosphate | chelating agent | >2 mM | inhibition |
| Treatment inhibitor/stimulator | Mode of action | Concentration or period tested | Effect |
| NH+4+NO−2 | activity test | 0–7 mM | normal activity |
| No biomass | none | 0 mg l−1 | no activity |
| Sterilization at 121°C | denaturation | 20–120 min | no activity |
| Gamma irradiation | inactivation | 60 min | no activtiy |
| Penicillin V | inhibition of cell wall synthesis of bacteria | 0–100 mg l−1 | none |
| Penicillin G | id. | 0–1000 mg l−1 | none |
| Bromoethane sulfonic acid | inhibition of methanogenesis | 0–20 mM | none |
| Na2SO4 | stimulation of sulfate reduction | 20 mM | none |
| Na2MoO4 | inhibition of sulfate reduction | 20 mM | none |
| Chloramphenicol | inhibition of protein synthesis | 0–400 mg l−1 | none |
| Hydrazine | inhibition of NH2OH oxidation | 0–3 mM | activation |
| Acetone | solvent for N-serve | 10 mM | none |
| N-serve | inhibition of nitrification | 0–50 mg l−1 | none |
| Allylthiourea | inhibition of nitrification | 0–10 mM | none |
| Acetylene | inhibition of nitrification and denitrification | 6 mM | inhibition |
| 2,4-Dinitrophenol | uncoupler | 0–400 mg l−1 | inhibition |
| Carbonyl cyanide m-chlorophenylhydrazone | uncoupler | 0–40 mg l−1 | inhibition |
| HgCl2 | cell damage | 0–300 mg l−1 | inhibition |
| Oxygen | oxidative stress | 0–0.2 mM | inhibition |
| Phosphate | chelating agent | <1 mM | none |
| Phosphate | chelating agent | >2 mM | inhibition |
3 Autotrophic growth during selective enrichment in continuous systems
Once it was realized that nitrite rather than nitrate might be the electron acceptor of autotrophic denitrification with ammonium as electron donor, a medium was composed for the selective enrichment of the microorganisms responsible for the Anammox process. This medium contained ammonium (5–30 mM), nitrite (5–35 mM), bicarbonate (10 mM), minerals and trace elements [5]. The phosphate concentration of the medium was kept below 0.5 mM and the oxygen concentration below detection levels (<1 μM) in order to avoid possible inhibitory effects (Table 2). Since the specific rate of ammonium oxidation in batch experiments was considerably lower than in perfusion systems a fluidized bed reactor was chosen to perform the enrichment. Using biomass from the original pilot plant as an inoculum, it was possible to obtain stable enrichment cultures within 3–4 months of operation. In total more than 20 reactor runs have been carried out with synthetic medium, the longest (Fig. 1) lasting more than 27 months [5–7]. So far only two runs failed in the enrichment mainly due to mechanical problems of the setup. After enrichment with synthetic medium the conversion rate in the reactor systems increased from 0.4 kg NH+4 N m−3 per day to about 3 kg NH+4 N m−3 per day. The maximum specific activity of the biomass in the fluidized bed reactor was about 25 nmol NH+4 min−1 (mg dry weight)−1. For every mol of CO2 incorporated into biomass 24 mol of ammonium had to be oxidized. When biomass from the reactors was used in batch experiments supplied with ammonium, nitrite and 14CO2, the cells became rapidly labeled. The incorporation of label was completely dependent on the combined presence of both nitrite and ammonium. The ribulose bisphosphate carboxylase activity of cell extracts was only 0.3 nmol CO2 min−1 (mg dry weight)−1 which is 3-fold lower than expected on the basis of the stoichiometry determined for ammonium and bicarbonate conversion (24:1). The estimated growth rate in the fluidized bed systems was 0.001 h−1, which is equivalent to 1 doubling time of about 29 days. The main product of the reaction was dinitrogen gas, but about 17% of the nitrite supplied could be recovered as nitrate. The overall nitrogen balance averaged over 15 runs showed a ratio of 1:1.31:0.22 for conversion of ammonium and nitrite to the production of nitrate. In the fluidized bed reactor no other intermediates like hydroxylamine, hydrazine, NO or nitrous oxide could be detected. The production of nitrate from nitrite was verified with 15N-NMR analysis [8]. Only when labeled nitrite was supplied to the cultures could formation of 15NO−3 be observed. In the presence of 15N labeled ammonium, no 15NO−3 was ever observed [8]. The function of this nitrate formation from nitrite is assumed to be the generation of reducing equivalents necessary for the reduction of CO2.
![Operation of a fluidized bed reactor for the enrichment and maintenance of anaerobic ammonium-oxidizing microorganisms. The medium was composed of ammonium sulfate, sodium nitrite, NaHCO3, minerals, trace elements [5,6]. The gray area represents the nitrite and ammonium load into the reactor; the black area is the nitrite or ammonium load out of the reactor. The average removal percentage (•) over 934 days was 99.5% for nitrite and 84.6% for ammonium.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/femsre/22/5/10.1111_j.1574-6976.1998.tb00379.x/1/m_FMR_421_f1.jpeg?Expires=1716343231&Signature=HHGeqLOlG731cTpQgc6-NkIulfTyx2aYm9MdxIc42VK9uAZMvfrsyop6uq1XsuJ42jmkciinCGiJOdvGvsYkH57SiF4HE9G72Sfbk3vYnU1aCOOmo1hM2Hu25bcLxlYzctecPpKUgHeY8mU4v1Cc3hBBFFcDk1s6mIEO56tcogieTdzpLOjtABIr0pWAuAPt7OmhRsDeCDvuCFsyf~dCaC92Fx6grjY0bPUwpJFcgRc~R6T1WvYx1rFTQCyg-wglrp6YYhvEYc4TEMWakLj7aKdG6zLPiiQr~I9uq3Yh~KD1N2PqE9umiBugQ23rCdIslTZlm3IXcQFdnOkQqbaeWQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Operation of a fluidized bed reactor for the enrichment and maintenance of anaerobic ammonium-oxidizing microorganisms. The medium was composed of ammonium sulfate, sodium nitrite, NaHCO3, minerals, trace elements [5,6]. The gray area represents the nitrite and ammonium load into the reactor; the black area is the nitrite or ammonium load out of the reactor. The average removal percentage (•) over 934 days was 99.5% for nitrite and 84.6% for ammonium.
4 Cultivation of Anammox biomass and determination of physiological parameters
Currently available microbiological techniques are not designed very well to deal with very slowly growing microorganisms such as the Anammox culture. In addition to the fluidized bed systems, a sequencing batch reactor (SBR) was applied and optimized for the quantitative study of the microbial community that oxidized ammonium anaerobically [9]. The SBR was a powerful experimental setup in which the biomass was retained very efficiently (>90%). Furthermore a homogeneous distribution of substrates, products and biomass aggregates over the reactor was achieved, and the reactor has been in operation reliably for more than 2 years under substrate-limiting conditions. Several important physiological parameters ([9] M. Strous, personal communication) such as the biomass yield (0.066±0.01 C mol (mol ammonium)−1), the maximum specific ammonium consumption rate (45±5 nmol min−1 (mg protein)−1) and the maximum specific growth rate (0.0027 h−1, doubling time 11 days) could now be determined more accurately than with the fluidized bed reactors. The temperature range for Anammox was 20–43°C (with an optimum at 40°C). The Anammox process functioned well at pH 6.7–8.3 (with an optimum at pH 8). Under optimal conditions the maximum specific ammonium oxidation rate was about 55 nmol min−1 (mg protein)−1. The affinity for the substrates ammonium and nitrite was very high (affinity constants≤5 μM) (M. Strous, personal communication). The Anammox process was inhibited by nitrite at nitrite concentrations higher than 20 mM but lower nitrite concentrations (>10 mM) were already suboptimal. When nitrite was present at high concentrations for a longer period (12 h), Anammox activity was completely lost. In addition, the persisting stable and strongly selective conditions of the SBR led to a high degree of enrichment (74% of the desired dominant peculiar microorganisms, see Section 5).
5 Characterization of the enriched microorganisms
5.1 Dominant cell type
The dominant microorganism of the enrichment cultures was a Gram-negative light-breaking coccoid cell (Fig. 2A,B) which showed an unusual irregular morphology under the electron microscope (Fig. 2C) when fixed with 2.5% glutaraldehyde in 20 mM K2HPO4/KH2PO4 buffer pH 7.4. Once the unusual morphology of the cells was recognized, an estimate of the enrichment from the original culture could be made by counting the cells in a large number of thin sections. After 177 days of enrichment 64% of all cells counted (7317 out of 11 433 total) were of the described type. This was a four-fold increase (16%; 1632 out of 10 200 total) compared to the numbers found in the biomass from the pilot plant. Together with the increase in cell numbers of this peculiar organism an increase in the percentage of ether-like lipids was observed. The amount of ester lipids typical for most Bacteria remained more or less constant [5]. The presence of the ether lipids seems to be confined to most ancient microorganisms such as the Archaea or very deep-branching Bacteria like Thermotoga and Aquifex. A detailed knowledge of the exact structure and composition of the lipids of the dominant cell type would be most helpful to find the taxonomic affiliation of these cells [5].
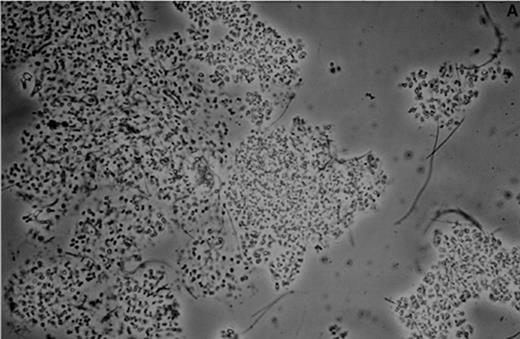
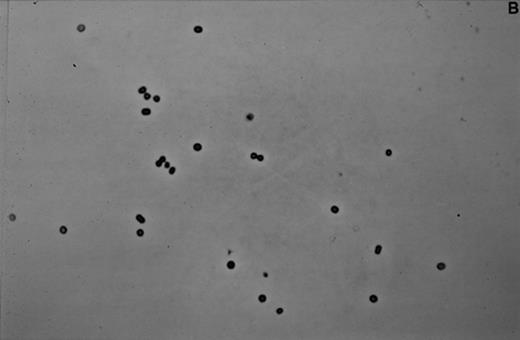
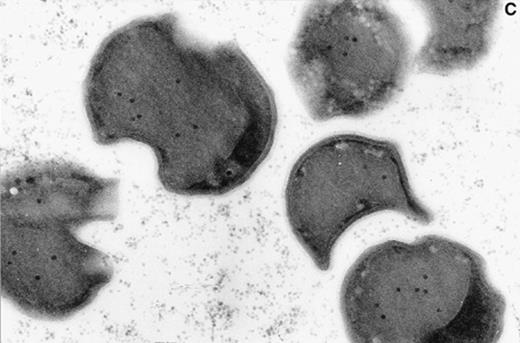
A: Micrograph of a biomass aggregate from an Anammox enrichment culture. The dominant coccoid cell is present in packages and microcolonies. B: Micrograph of the dominant coccoid cell present in the Anammox enrichment cultures. Preparation was obtained after sedimentation of suspended material from a fluidized bed reactor. C: Electron micrograph of suspended Anammox biomass fixed with 2.5% glutaraldehyde in 20 mM K2HPO4/KH2PO4 buffer pH 7.4. The micrograph was taken at the Department of Electron Microscopy (I. Keizer, K. Sjollema, M. Veenhuis), State University of Groningen, The Netherlands.
5.2 Cytochrome spectra
During the enrichment on synthetic medium, the color of the biomass changed from brown to deep red. Visible spectra of cells and cell extracts of the enriched culture showed a pronounced increase in the signal for cytochromes of the c type. Spectra of cells at 77 K revealed the absence of a-type, b-type and d1-type cytochromes. Very interestingly, during increase in Anammox activity of the biomass, gradually an increased signal was observed at 468 nm. In Fig. 3 a typical spectrum of an Anammox cell extract with the 468-nm feature is shown. This absorption peak at 468 nm disappeared irreversibly after treatment with carbon monoxide. A similar signal has been observed in aerobic ammonium-oxidizing bacteria at 463 nm [10–13]. This signal was attributed to one of the hemes present in the enzyme hydroxylamine oxidoreductase and is mostly referred to as cytochrome P460.
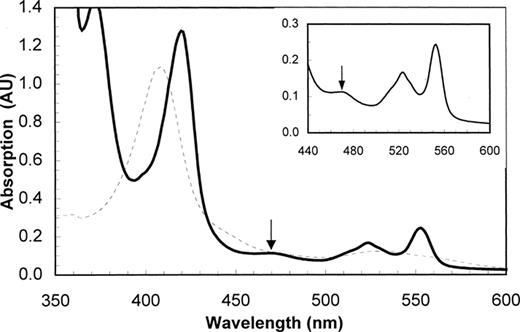
Cytochrome spectrum of cell extract from an Anammox enrichment culture. Dashed line, oxidized spectrum; solid line, spectrum after reduction with dithionite. Inset shows a close-up between 440 and 600 nm. The arrow indicates the typical peak at 468 nm.
5.3 Identification
So far many isolation methods including serial dilution, floating filters, and plating have been used to obtain the responsible microorganisms in pure culture. None of the isolates thus obtained is able to perform the Anammox reaction, but many of the isolates are novel denitrifying oligotrophic (proteo)bacteria. In addition to classical microbial techniques, the Anammox community was analyzed using modern molecular biological methods. The genomic DNA was extracted and amplified via PCR using (eu)bacterial primers 27f-BamHI (5′-CACGGATCCAGAGTTTGATMTGGCTTCAG-3′), and 1492r-HindIII (5′-TGTAAGCTTACGGYTACCTTGTTACGACT-3′). The PCR products were cloned, and 396 out of the more than 4000 clones obtained were screened. One dominant (28%) clone belonging to the Cytophaga/Flexibacter phylum was identified. However, in situ analysis with fluorescent probes specific for the Cytophaga phylum did not confirm the molecular identity of the dominant organism as a Cytophaga.
6 Aerobic versus anaerobic ammonium oxidation
The presence of a P460-like signal in the enriched Anammox biomass, and the recent reports on the metabolic versatility of aerobic ammonium oxidizers initiated a more detailed investigation. The studies were concentrated on three issues: the influence of oxygen on the Anammox process, the number of aerobic ammonium oxidizers present in the Anammox enrichment cultures and the (anaerobic) capabilities of ‘classical’ nitrifiers of the Nitrosomonas type.
6.1 Influence of oxygen on anaerobic ammonium oxidation
The influence of oxygen on the Anammox process was investigated in both batch and continuous systems. Initial batch experiments showed that oxygen completely inhibited the Anammox activity when it was deliberately introduced into the enrichment cultures [5,14]. In a follow-up study an intermittently oxic (2 h) and anoxic (2 h) reactor system was used to study the reversibility of the oxygen inhibition for 20 days [15]. From these studies it became clear that ammonium was not oxidized in the oxic periods, but that the Anammox activity in the anoxic periods remained constant throughout the experiment, indicating that the inhibitory effect of oxygen was indeed reversible. The sensitivity of the Anammox enrichment culture to oxygen was further investigated under various sub-oxic conditions. In four consecutive experiments, the oxygen tension was decreased stepwise from 2 to 0% of air saturation (Fig. 4). No ammonium was oxidized in the presence of 0.5, 1, or 2% of air. Only when all the air was removed from the reactor by vigorously flushing with argon gas, the conversion of ammonium and nitrite resumed, thus indicating that the Anammox activity in these enrichment cultures is only possible under strict anoxic conditions.
![The Anammox activity at four different air saturations (A 2%, B 1%, C 0.5%, and D 0%). Only when all oxygen was removed from the incubation by flushing with argon gas the disappearance of ammonium (•) and nitrite (▪) could be observed [15].](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/femsre/22/5/10.1111_j.1574-6976.1998.tb00379.x/1/m_FMR_421_f4.jpeg?Expires=1716343232&Signature=HJl6lwvzXll1kd22UWOkY~03JV~n4mZGHFuxxjIDWKuYKJ-lvR~KJN8CniC06zOvldarExbH7NPA23fgCM-85UbKKFpwIUSrAlNb8nJGmNKJdlmmTKEdV1jjh2tdwTlDYGw8VQQiIyoC5KzfCk5apWz~Orz7zweyesdOoWfampDwUVlXRwHQfQSYCKBnwNvw3oSbi~OulQ-16s-ZMCEeBeKYHnpG-5NbERimlB9X37e2Tq-ZFsl8FIszq4Rw2~CigwEFKPF5sX4mTQcmDRLqbaCKWVZs9ZsgkUxl3ZZDhfKRwJLfgSR8re5LUQKKc3zEu1fBOwdr06GrDgbKnstchA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
The Anammox activity at four different air saturations (A 2%, B 1%, C 0.5%, and D 0%). Only when all oxygen was removed from the incubation by flushing with argon gas the disappearance of ammonium (•) and nitrite (▪) could be observed [15].
6.2 Aerobic ammonium oxidizers
The second question which was addressed in these studies concerned the number of aerobic ammonium-oxidizing bacteria present in the Anammox enrichment cultures. Using standard, aerobic most probable number (MPN) methods, the number of nitrifiers was estimated to be 9±5×103 cells of ammonium oxidizers per milliliter of biomass sample. Electron micrographs of the MPN cultures showed the characteristic cytoplasmic membrane structures reported for several aerobic ammonium-oxidizing bacteria [16]. The consistent presence of these aerobic nitrifiers in the Anammox biomass confirmed that they can survive very long periods of anaerobiosis as was previously shown by Abeliovich [17]. Furthermore, it was possible to enrich such organisms in a repeated fed-batch reactor when oxygen was continuously supplied at 50–80% of air saturation [15]. The enriched aerobically nitrifying community grew exponentially with a doubling time of 1.2 days. Interestingly, not all of the 27 mM of ammonium supplied could be recovered as nitrite. Since nitrite was not further oxidized to nitrate, the remainder of the nitrogen might have been lost as gaseous nitrogen compounds (NO, N2O or N2) during aerobic denitrification. After enrichment of the nitrifiers, also this culture was subjected to the same alternating 2 h oxic/2 h anoxic regime to verify if these nitrifiers were capable of an anaerobic conversion of ammonium or nitrite. During 20 days, the fate of the supplied 30 mM ammonium was followed and it was observed that only in the aerobic periods oxidation of ammonium to nitrite occurred. This indicated that this community of aerobic ammonium-oxidizing bacteria was not able to use nitrite as an electron acceptor in this case.
This is in contrast to observations made with several pure cultures of different Nitrosomonas strains. Poth showed that a new Nitrosomonas isolate was able to produce dinitrogen gas under anaerobic conditions [18,19], while Abeliovich and Vonshak demonstrated the reduction of nitrite with pyruvate as electron donor by N. europaea[20].
6.3 Metabolic versatility of Nitrosomonas strains
More recently substantial N losses have been reported for both mixed and pure cultures of N. eutropha grown under oxygen limitation [16,21,22], and for pure cultures of N. europaea in anoxic batch experiments [23]. When molecular hydrogen was used as an electron donor for nitrite reduction, growth of N. eutropha was stoichiometrically coupled to nitrite reduction with dinitrogen gas and nitrous oxide as end products. In mixed cultures of N. eutropha and Enterobacter aerogenes 2.2 mM of ammonium and nitrite were consumed during 44 days of incubation, but no cell growth was observed [16]. In a follow-up study the rate of anaerobic ammonium oxidation by N. eutropha could be estimated at 0.08 nmol NH+4 min−1 (mg protein)−1 which is equivalent to about 0.2 nmol NH+4 min−1 (mg dry weight)−1. However, when the nitrogen atmosphere of the incubations was supplemented with 25 ppm nitrogen dioxide, the rate increased 10-fold to 2.2 nmol NH+4 min−1 (mg protein)−1[21]. It was estimated that 40–60% of the formed nitrite (and NO) was denitrified to dinitrogen gas while N2O and hydroxylamine were detected as intermediates. The source of oxygen for the oxidation of ammonia under these anoxic conditions remained unknown, but could theoretically be derived from either NO, NO2 or nitrite. Very recently it was shown that N. eutropha also exhibited denitrifying capabilities in the presence of NO2 when the dissolved oxygen concentration was maintained at 3–4 mg l−1[22]. In these experiments with complete biomass retention 50% of the produced nitrite was aerobically denitrified to dinitrogen gas. NO gas was much less efficient in stimulating this aerobic denitrification than NO2 and became toxic above 25 ppm. Furthermore, an eight-fold increased aerobic nitrification rate and higher cell numbers were observed when the air was supplemented with 25–50 ppm NO2. In Table 3 a summary is presented of the reported rates of anaerobic ammonium oxidation in various experiments with Nitrosomonas and/or Anammox cultures. From this table it is evident that the specific rates for anaerobic ammonium oxidation of the classical nitrifiers are 25-fold lower than the rates observed in the Anammox process studied in Delft. Furthermore, aerobic ammonium oxidizers prefer to use oxygen as the terminal electron acceptor, whereas this compound completely inhibits the Anammox process. Taken together these examples showed that further research to elucidate the role of nitrogen oxides during (an)aerobic ammonium oxidation is necessary.
Rates of anaerobic oxidation (nmol min−1 (mg protein)−1) of ammonium, hydroxylamine and hydrazine by various cultures in batch experiments
| Culture | Compounds tested | NO−2 conversion rate | NH2OH/NH+4/N2H4 conversion rate | Products | Reference |
| N. europaea | NH2OH+NO−2 | 2 | 3 | N2O | [23] |
| N. europaea | NH+4+NO−2 | 2 | 3 | N2O | [23] |
| N. eutropha | H2+NO−2 | 7 | not applicable | N2O, N2 | [16] |
| N. eutropha | NH+4+NO−2 | <1 | <1 | N2O | [16] |
| N. eutropha | NH+4+NO−2 | 0.9 | 1.1 | NO, N2O | [21] |
| Anammox | NH+4+NO−2 | 12 | 9 | N2 | [5] |
| Anammox | NH2OH | n/a | 12 | N2 | [8] |
| Anammox | N2H4+NO−2 | 13 | 11 | NH+4, N2 | [8,24] |
| Anammox | NH+4+NO−2 | 68 | 55 | N2 | [9], M. Strous, personal communication |
| Culture | Compounds tested | NO−2 conversion rate | NH2OH/NH+4/N2H4 conversion rate | Products | Reference |
| N. europaea | NH2OH+NO−2 | 2 | 3 | N2O | [23] |
| N. europaea | NH+4+NO−2 | 2 | 3 | N2O | [23] |
| N. eutropha | H2+NO−2 | 7 | not applicable | N2O, N2 | [16] |
| N. eutropha | NH+4+NO−2 | <1 | <1 | N2O | [16] |
| N. eutropha | NH+4+NO−2 | 0.9 | 1.1 | NO, N2O | [21] |
| Anammox | NH+4+NO−2 | 12 | 9 | N2 | [5] |
| Anammox | NH2OH | n/a | 12 | N2 | [8] |
| Anammox | N2H4+NO−2 | 13 | 11 | NH+4, N2 | [8,24] |
| Anammox | NH+4+NO−2 | 68 | 55 | N2 | [9], M. Strous, personal communication |
Rates of anaerobic oxidation (nmol min−1 (mg protein)−1) of ammonium, hydroxylamine and hydrazine by various cultures in batch experiments
| Culture | Compounds tested | NO−2 conversion rate | NH2OH/NH+4/N2H4 conversion rate | Products | Reference |
| N. europaea | NH2OH+NO−2 | 2 | 3 | N2O | [23] |
| N. europaea | NH+4+NO−2 | 2 | 3 | N2O | [23] |
| N. eutropha | H2+NO−2 | 7 | not applicable | N2O, N2 | [16] |
| N. eutropha | NH+4+NO−2 | <1 | <1 | N2O | [16] |
| N. eutropha | NH+4+NO−2 | 0.9 | 1.1 | NO, N2O | [21] |
| Anammox | NH+4+NO−2 | 12 | 9 | N2 | [5] |
| Anammox | NH2OH | n/a | 12 | N2 | [8] |
| Anammox | N2H4+NO−2 | 13 | 11 | NH+4, N2 | [8,24] |
| Anammox | NH+4+NO−2 | 68 | 55 | N2 | [9], M. Strous, personal communication |
| Culture | Compounds tested | NO−2 conversion rate | NH2OH/NH+4/N2H4 conversion rate | Products | Reference |
| N. europaea | NH2OH+NO−2 | 2 | 3 | N2O | [23] |
| N. europaea | NH+4+NO−2 | 2 | 3 | N2O | [23] |
| N. eutropha | H2+NO−2 | 7 | not applicable | N2O, N2 | [16] |
| N. eutropha | NH+4+NO−2 | <1 | <1 | N2O | [16] |
| N. eutropha | NH+4+NO−2 | 0.9 | 1.1 | NO, N2O | [21] |
| Anammox | NH+4+NO−2 | 12 | 9 | N2 | [5] |
| Anammox | NH2OH | n/a | 12 | N2 | [8] |
| Anammox | N2H4+NO−2 | 13 | 11 | NH+4, N2 | [8,24] |
| Anammox | NH+4+NO−2 | 68 | 55 | N2 | [9], M. Strous, personal communication |
7 Possible reaction mechanisms for Anammox
The possible metabolic pathway for anaerobic ammonium oxidation was investigated using 15N labeling experiments. These experiments showed that ammonium was biologically oxidized with hydroxylamine as the most probable electron acceptor [8]. The hydroxylamine itself is most likely derived from nitrite. In batch experiments with excess hydroxylamine and ammonium, a transient accumulation of hydrazine was observed (Fig. 5). The conversion of hydrazine to dinitrogen gas is postulated as the reaction generating the electron equivalents for the reduction of nitrite to hydroxylamine. Whether the reduction of nitrite and the oxidation of hydrazine occur at different sites of the same enzyme (Fig. 6A) or the reactions are catalyzed by different enzyme systems connected via an electron transport chain (Fig. 6B) remains to be investigated. The occurrence of hydrazine as an intermediate in microbial nitrogen metabolism is rare [24]. Hydrazine has been proposed as an enzyme-bound intermediate in the nitrogenase reaction [25]. Furthermore, the purified hydroxylamine oxidoreductase (HAO) of N. europaea is capable of catalyzing the conversion of hydrazine to dinitrogen gas [12]. The finding of high HAO activity in cell extracts of the Anammox culture indicated that a similar enzyme might be operative in the Anammox process. Indeed very recently a novel type of HAO was purified from the Anammox community via anion exchange and gel filtration chromatography (J. Schalk, personal communication). Native PAGE showed that the Anammox enzyme had a smaller molecular mass than the enzyme of Nitrosomonas. Furthermore, the amino acid sequence of several HAO peptide fragments was unique, without any homologue in the databases. Similar to the Nitrosomonas hydroxylamine oxidoreductase, the enzyme from Anammox contained several c-type cytochromes. The special spectroscopic P460-like feature was found at 468 nm in the Anammox enzyme. The enzyme was able to catalyze the oxidation of both hydroxylamine and hydrazine. Although hydroxylamine was the preferred substrate the rate of hydrazine oxidation in cell extracts (150 nmol min−1 (mg protein)−1) was high enough to sustain a growth rate of 0.003 h−1. In Table 4 some properties of the two HAO enzymes are summarized (J. Schalk, personal communication).
![Concentration profile of ammonium (•), nitrite (▪), hydroxylamine (▴) and hydrazine (♦) during anaerobic batch experiments with an Anammox culture supplemented with 3 mM hydroxylamine [8].](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/femsre/22/5/10.1111_j.1574-6976.1998.tb00379.x/1/m_FMR_421_f5.jpeg?Expires=1716343232&Signature=aqcHfgIwdD0rJ0C2ohJQQU9MXmid-N66SADA1vwvyNbb8G32hNj9pKfyUF92sIQGbqa7Sfw0~N8fKqytJfsNU5enppFDd5ux9IAwFmq7B4Suhn9-RwSS1a58l7p-qLZIFODNGfJLEPcocTGCfeWViNQTpXh24iqgVJc9KL7unO3icRbRgkk47HM-7YexR9yrVWhw~ScRUEyBhwOxLveWSkWUUZE3z3Md3Jstfi9h-ZdsQc7L~15~Rwv8IoODidEAzEY0zUQOBtSp5IlOdQT1zmit1fzairDbhGcULND33c84dNpgY5e84ukIY4FNgRhI2WRKciRgTCP5jYGhAd~~bA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Concentration profile of ammonium (•), nitrite (▪), hydroxylamine (▴) and hydrazine (♦) during anaerobic batch experiments with an Anammox culture supplemented with 3 mM hydroxylamine [8].
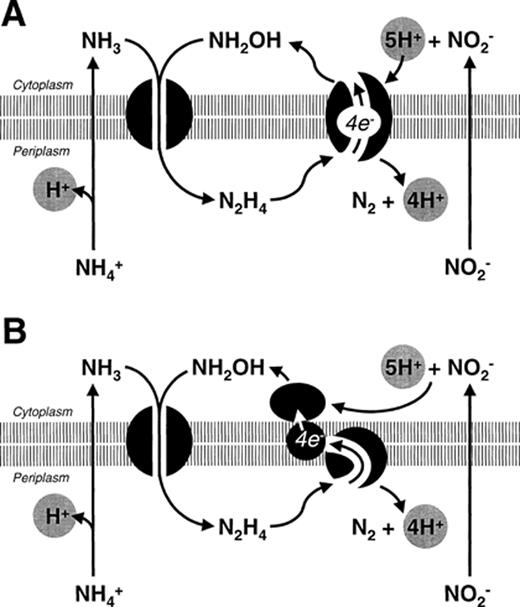
Possible reaction mechanisms and cellular localization of the enzyme systems involved in anaerobic ammonium oxidation. A: Ammonium and hydroxylamine are converted to hydrazine by a membrane-bound enzyme complex, hydrazine is oxidized in the periplasm to dinitrogen gas, nitrite is reduced to hydroxylamine at the cytoplasmic site of the same enzyme complex responsible for hydrazine oxidation with an internal electron transport. B: Ammonium and hydroxylamine are converted to hydrazine by a membrane-bound enzyme complex, hydrazine is oxidized in the periplasm to dinitrogen gas, the generated electrons are transferred via an electron transport chain to nitrite reducing enzyme in the cytoplasm.
Comparison of the properties of the hydroxylamine oxidoreductase (HAO) enzyme isolated from Anammox (J. Schalk, personal communication) and Nitrosomonas europaea[12]
| HAO of Anammox | HAO of Nitrosomonas | |
| Molecular mass | 150 kDa | 125–175 kDa |
| Subunit | 60–95 kDa | 63 kDa |
| Composition | α2–α3 | α2–α3 |
| Ratio 410/280 | 4.5 | 3.3 |
| Heme | 22±4/150 kDa | 24/α3 |
| Active center | P468 | P463 |
| Vmax | 21 U mg−1 | 75 U mg−1 |
| Km | 26 μM | not reported |
| pH optimum | 8 | 8 |
| pI | 5.5 | 5.3 |
| N-terminus | blocked | DISTV |
| HAO of Anammox | HAO of Nitrosomonas | |
| Molecular mass | 150 kDa | 125–175 kDa |
| Subunit | 60–95 kDa | 63 kDa |
| Composition | α2–α3 | α2–α3 |
| Ratio 410/280 | 4.5 | 3.3 |
| Heme | 22±4/150 kDa | 24/α3 |
| Active center | P468 | P463 |
| Vmax | 21 U mg−1 | 75 U mg−1 |
| Km | 26 μM | not reported |
| pH optimum | 8 | 8 |
| pI | 5.5 | 5.3 |
| N-terminus | blocked | DISTV |
Comparison of the properties of the hydroxylamine oxidoreductase (HAO) enzyme isolated from Anammox (J. Schalk, personal communication) and Nitrosomonas europaea[12]
| HAO of Anammox | HAO of Nitrosomonas | |
| Molecular mass | 150 kDa | 125–175 kDa |
| Subunit | 60–95 kDa | 63 kDa |
| Composition | α2–α3 | α2–α3 |
| Ratio 410/280 | 4.5 | 3.3 |
| Heme | 22±4/150 kDa | 24/α3 |
| Active center | P468 | P463 |
| Vmax | 21 U mg−1 | 75 U mg−1 |
| Km | 26 μM | not reported |
| pH optimum | 8 | 8 |
| pI | 5.5 | 5.3 |
| N-terminus | blocked | DISTV |
| HAO of Anammox | HAO of Nitrosomonas | |
| Molecular mass | 150 kDa | 125–175 kDa |
| Subunit | 60–95 kDa | 63 kDa |
| Composition | α2–α3 | α2–α3 |
| Ratio 410/280 | 4.5 | 3.3 |
| Heme | 22±4/150 kDa | 24/α3 |
| Active center | P468 | P463 |
| Vmax | 21 U mg−1 | 75 U mg−1 |
| Km | 26 μM | not reported |
| pH optimum | 8 | 8 |
| pI | 5.5 | 5.3 |
| N-terminus | blocked | DISTV |
The formation of hydroxylamine via an ammonium monooxygenase seems highly improbable, since the Anammox reaction is strongly but reversibly inhibited by oxygen. An alternative mechanism for the formation of hydroxylamine might be the incomplete reduction of nitrite by a cytochrome c-type nitrite reductase. However, it will be very difficult to obtain direct evidence for this mechanism in Anammox. Hydroxylamine is the compound most rapidly metabolized by Anammox, and a selective inhibitor has not yet been discovered.
A possible role of NO or HNO in (an)aerobic ammonium oxidation was proposed by Hooper [12] to be a condensation of NO or HNO and ammonia on an enzyme related to the ammonium monooxygenase family (Table 5). The formed hydrazine or imine could thereafter be converted by the enzyme hydroxylamine oxidoreductase into dinitrogen gas and the reducing equivalents required to combine NO or HNO and ammonia or to reduce nitrite to NO.
| NO as intermediate | ||
| NO+NH3+3H++3e− | →N2H4+H2O | (ammonia monooxygenase-like enzyme) |
| N2H4 | →N2+4H++4e− | (hydroxylamine oxidoreductase-like enzyme) |
| NO−2+2H++e− | →NO+H2O | (nitrite reductase) |
| NH3+NO−2+H+ | →N2+2H2O | |
| HNO as intermediate | ||
| HNO+NH3 | →N2H2+H2O | (ammonia monooxygenase-like enzyme) |
| N2H2 | →N2+2H++2e− | (hydroxylamine oxidoreductase-like enzyme) |
| NO−2+2H++2e− | →HNO+OH− | (nitrite reductase) |
| NH3+NO−2 | →N2+H2O+OH− |
| NO as intermediate | ||
| NO+NH3+3H++3e− | →N2H4+H2O | (ammonia monooxygenase-like enzyme) |
| N2H4 | →N2+4H++4e− | (hydroxylamine oxidoreductase-like enzyme) |
| NO−2+2H++e− | →NO+H2O | (nitrite reductase) |
| NH3+NO−2+H+ | →N2+2H2O | |
| HNO as intermediate | ||
| HNO+NH3 | →N2H2+H2O | (ammonia monooxygenase-like enzyme) |
| N2H2 | →N2+2H++2e− | (hydroxylamine oxidoreductase-like enzyme) |
| NO−2+2H++2e− | →HNO+OH− | (nitrite reductase) |
| NH3+NO−2 | →N2+H2O+OH− |
| NO as intermediate | ||
| NO+NH3+3H++3e− | →N2H4+H2O | (ammonia monooxygenase-like enzyme) |
| N2H4 | →N2+4H++4e− | (hydroxylamine oxidoreductase-like enzyme) |
| NO−2+2H++e− | →NO+H2O | (nitrite reductase) |
| NH3+NO−2+H+ | →N2+2H2O | |
| HNO as intermediate | ||
| HNO+NH3 | →N2H2+H2O | (ammonia monooxygenase-like enzyme) |
| N2H2 | →N2+2H++2e− | (hydroxylamine oxidoreductase-like enzyme) |
| NO−2+2H++2e− | →HNO+OH− | (nitrite reductase) |
| NH3+NO−2 | →N2+H2O+OH− |
| NO as intermediate | ||
| NO+NH3+3H++3e− | →N2H4+H2O | (ammonia monooxygenase-like enzyme) |
| N2H4 | →N2+4H++4e− | (hydroxylamine oxidoreductase-like enzyme) |
| NO−2+2H++e− | →NO+H2O | (nitrite reductase) |
| NH3+NO−2+H+ | →N2+2H2O | |
| HNO as intermediate | ||
| HNO+NH3 | →N2H2+H2O | (ammonia monooxygenase-like enzyme) |
| N2H2 | →N2+2H++2e− | (hydroxylamine oxidoreductase-like enzyme) |
| NO−2+2H++2e− | →HNO+OH− | (nitrite reductase) |
| NH3+NO−2 | →N2+H2O+OH− |
8 Substrate spectrum
Aerobic ammonium and methane oxidizers are able to catalyze both the oxidation of ammonium and methane [26] albeit at different rates. The ability of the Anammox culture to use methane or other substrates was tested in batch experiments. In Fig. 7 it is shown that addition of methane to the argon/CO2 head space of the incubations did not lead to an inhibition of ammonium and nitrite conversion. This indicated that the enzyme responsible for anaerobic ammonium conversion is different from the aerobic ammonia or methane monooxygenases. In longer experiments it could also be shown that methane itself was not converted by the Anammox biomass (Fig. 8). In addition to methane also hydrogen was tested in batch incubations (Fig. 9). The addition of hydrogen to the argon/CO2 head space showed a clear stimulation of the anaerobic ammonium oxidation in short-term experiments. However, hydrogen could not replace ammonium as electron donor in these experiments. Addition of various organic substances (pyruvate, methanol, ethanol, alanine, glucose, casamino acids) in short-term batch experiments led to a severe inhibition of the Anammox activity. Thus the substrate spectrum seems to be restricted to ammonium, nitrite and the intermediates hydrazine and hydroxylamine. However, supplementation with 1 mM hydrazine could not sustain growth of the Anammox culture for longer periods [24].
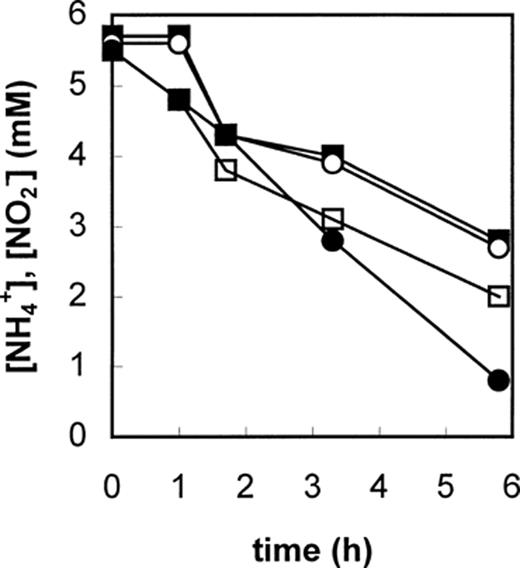
Concentration profile of ammonium (•) and nitrite (▪) in the absence of methane in the head space, and of ammonium (◯) and nitrite (□) in the presence of 50% methane in the argon/CO2 head space during anoxic batch experiments with Anammox biomass.
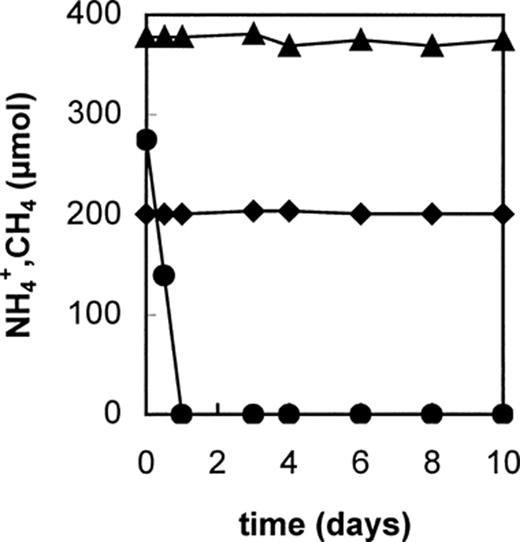
The absence of methane conversion by Anammox biomass at two different methane concentrations in the head space. Closed triangles (▴, 380 μmol methane) and diamonds (♦, 195 μmol methane) represent incubations with Anammox biomass in the presence of 300 μmol nitrite as electron acceptor. The closed circles represents a control incubation of Anammox biomass with 280 μmol ammonium (•), and 300 μmol nitrite as electron acceptor. In the control all nitrite was converted, in the incubations with methane the nitrite was not converted.
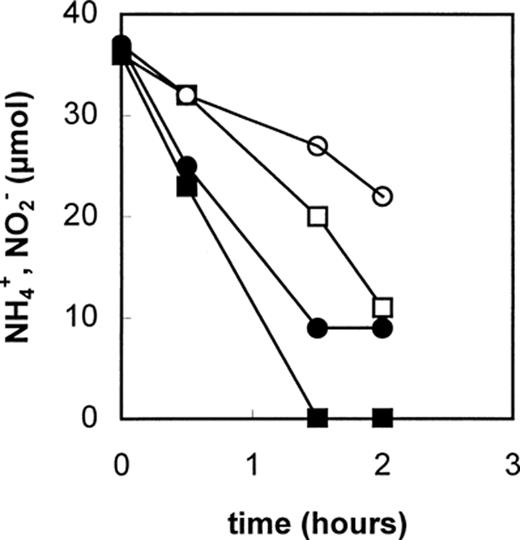
Concentration profile of ammonium (•) and nitrite (▪) in the presence of 95% hydrogen and 5% CO2 gas in the head space, and of ammonium (◯) and nitrite (□) in the presence of 95% argon and 5% CO2 gas in head space during anoxic batch experiments with Anammox biomass.
9 Ecological habitats
The discovery of the Anammox process in a denitrifying pilot plant has raised the question as to where else such organisms would occur in nature. Already in 1932 it was reported that dinitrogen gas was generated via an unknown mechanism during fermentation in the sediments of Lake Mendota (USA) [27]. Also in sediments of Lake Kizakiko (Japan) indications were found for the direct formation of dinitrogen gas from ammonium [28]. Very recently these observations were confirmed in studies with freshwater sediments [29]. One prerequisite for the occurrence of anaerobic ammonium oxidation via an Anammox mechanism would be the simultaneous presence of both ammonium and nitrite (or nitrate) and the absence of oxygen. The nitrite could be formed either in ecosystems in which oxygen (diffusion) limits nitrification or in places with a limited supply of electron donor (sulfide or organic substances) for denitrification of nitrate. The oxic/anoxic interface of many sediments would thus be an ideal habitat for anaerobic ammonium-oxidizing microorganisms. Micro-electrode studies have revealed overlapping profiles of nitrate and ammonium in a stratified zone of the Black Sea [30,31] indicating that a habitat for Anammox really exists. More recently after the development of nitrite microsensors, also overlapping profiles of nitrite and ammonium have been reported in oxygen-limited nitrifying activated sludge flocs [32–34]. Indeed man-made ecosystems like wastewater treatment plants could create a habitat for the Anammox organisms. The abundant supply of ammonium via the wastewater in combination with a limited oxygen availability would provide conditions in which both ammonium and nitrite could occur. High nitrogen losses (70–90%) in the form of dinitrogen gas have been reported in two rotating biological contractor systems [35–37] treating landfill leachate with about 200–400 mg ammonium per liter. Comparison of the microorganisms in these systems with the Delft Anammox culture would give more insight into the biodiversity of the anaerobic ammonium oxidation.
10 Application of the Anammox process
In a recent study [7,38] the feasibility of the Anammox process for the removal of ammonium from sludge digester effluents was evaluated. The results of this study showed that the Anammox biomass was not negatively affected by the digester effluent. The pH (7.0–8.5) and temperature (30–37°C) optima for the process were well within the range of the values found in digester effluents. Experiments with a laboratory-scale (2-l) fluidized bed reactor showed that the Anammox biomass was capable of removing ammonium and (externally added) nitrite efficiently from the sludge digester effluent (Table 6). The nitrogen load of the Anammox fluidized bed reactor could be increased from 0.46 kg Ntot m−3reactor per day to 2.6 kg Ntot m−3reactor per day. Due to the nitrite limitation, the maximum capacity was not reached. The nitrogen conversion rate during the experiment with sludge digester effluents increased from 0.05 kg Ntot kg−1 SS per day to 0.26 kg Ntot kg−1 SS per day. The Anammox sludge biomass removed 88% of the ammonium and 99% of the nitrite from the sludge digester effluent (Table 6). In these studies nitrite was supplied from a concentrated stock solution. However, for application in real wastewater practice, a suitable system for biological nitrite production has to be developed. One such system is the SHARON (single reactor high activity ammonium removal over nitrite) process [38].
| SHARON | Anammox | ||
| Ammonium load | 0.63–1.0a | 0.24–1.34 | kg NH+4 N m−3reactor day−1 |
| Nitrite load | not applicable | 0.22–1.29 | kg NO−2 N m−3reactor day−1 |
| Nitrogen load | 0.63–1.0 | 0.46–2.63 | kg Ntot m−3reactor day−1 |
| NH+4 N effluent | 199 | 27±85 | mg N l−1 |
| NO−2 N effluent | 469 | 3±3 | mg N l−1 |
| Efficiency NH+4 N | 76–90 | 88±9 | % |
| Removal | |||
| Efficiency NO−2 N | n/a | 99±2 | % |
| Removal | |||
| Sludge load | 10.3 | 0.05–0.26 | kg Ntot kg−1 SS per day |
| SHARON | Anammox | ||
| Ammonium load | 0.63–1.0a | 0.24–1.34 | kg NH+4 N m−3reactor day−1 |
| Nitrite load | not applicable | 0.22–1.29 | kg NO−2 N m−3reactor day−1 |
| Nitrogen load | 0.63–1.0 | 0.46–2.63 | kg Ntot m−3reactor day−1 |
| NH+4 N effluent | 199 | 27±85 | mg N l−1 |
| NO−2 N effluent | 469 | 3±3 | mg N l−1 |
| Efficiency NH+4 N | 76–90 | 88±9 | % |
| Removal | |||
| Efficiency NO−2 N | n/a | 99±2 | % |
| Removal | |||
| Sludge load | 10.3 | 0.05–0.26 | kg Ntot kg−1 SS per day |
aThis value is linearly proportional to the influent concentration.
| SHARON | Anammox | ||
| Ammonium load | 0.63–1.0a | 0.24–1.34 | kg NH+4 N m−3reactor day−1 |
| Nitrite load | not applicable | 0.22–1.29 | kg NO−2 N m−3reactor day−1 |
| Nitrogen load | 0.63–1.0 | 0.46–2.63 | kg Ntot m−3reactor day−1 |
| NH+4 N effluent | 199 | 27±85 | mg N l−1 |
| NO−2 N effluent | 469 | 3±3 | mg N l−1 |
| Efficiency NH+4 N | 76–90 | 88±9 | % |
| Removal | |||
| Efficiency NO−2 N | n/a | 99±2 | % |
| Removal | |||
| Sludge load | 10.3 | 0.05–0.26 | kg Ntot kg−1 SS per day |
| SHARON | Anammox | ||
| Ammonium load | 0.63–1.0a | 0.24–1.34 | kg NH+4 N m−3reactor day−1 |
| Nitrite load | not applicable | 0.22–1.29 | kg NO−2 N m−3reactor day−1 |
| Nitrogen load | 0.63–1.0 | 0.46–2.63 | kg Ntot m−3reactor day−1 |
| NH+4 N effluent | 199 | 27±85 | mg N l−1 |
| NO−2 N effluent | 469 | 3±3 | mg N l−1 |
| Efficiency NH+4 N | 76–90 | 88±9 | % |
| Removal | |||
| Efficiency NO−2 N | n/a | 99±2 | % |
| Removal | |||
| Sludge load | 10.3 | 0.05–0.26 | kg Ntot kg−1 SS per day |
aThis value is linearly proportional to the influent concentration.
This SHARON process is ideally suited to remove nitrogen from waste streams with a high ammonium concentration (>0.5 g N l−1). The SHARON process is performed in a single, stirred tank reactor without any sludge retention. At temperatures above 25°C it is possible to effectively outcompete the nitrite oxidizers. This results in a stable nitrification with nitrite as end-product [38]. When combined with the Anammox process only 50% of the ammonium needs to be converted to nitrite. This implies that no extra addition of base is necessary, since most of the wastewater resulting from anaerobic digestion will contain enough alkalinity (in the form of bicarbonate) to compensate for the acid production if only 50% of the ammonium needs to be oxidized. The SHARON process has been extensively tested at the laboratory scale for the treatment of sludge digester effluents (Table 6) and is currently under construction at two Dutch wastewater treatment plants.
11 The combined SHARON-Anammox process
The combination of the Anammox process and SHARON process has been tested in our laboratory using sludge digester effluent (Fig. 10). The SHARON reactor was operated without pH control with a total nitrogen load of about 0.8 kg N m−3 per day [38]. The ammonium present in the sludge digester effluent was converted to nitrite and a small amount of nitrate (11%) (Table 7). The nitrate formation was due to biofilm wall growth, which was not always regularly removed. In large-scale applications this will be significantly lower because of the larger volume to surface ratio. In this way an ammonium-nitrite mixture suitable for the Anammox process was generated. The effluent of the SHARON reactor was used as influent for the Anammox fluidized bed reactor. In the nitrite-limited Anammox reactor all nitrite was removed, the surplus ammonium remained. During the test period the overall ammonium removal efficiency was 83%. In Table 7 the nitrogen balances of the two systems are summarized. The optimization and application of the combination of these two processes on pilot plant and full scale remain challenges towards implementations in a future wastewater treatment plant.
![Ammonium removal from sludge digester effluent with the combined SHARON-Anammox system. Ammonium (♦) or nitrite (▪) load in the effluent of the SHARON reactor is used as the ammonium or nitrite load into the Anammox reactor. The ammonium or nitrite load in the effluent of the Anammox is represented by open diamonds and open squares, respectively. The pH value in the Anammox reactor was stable at 7.8 [38].](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/femsre/22/5/10.1111_j.1574-6976.1998.tb00379.x/1/m_FMR_421_f10.jpeg?Expires=1716343232&Signature=nqtA~PZCqGNs05dleTZZRl3nkGszZLsgnrBqosa0eQqYoEsjKHWsOU6p0dUX2IM0aw2~0UqU9AlErr1uPkOWs43aGSsFUONrzlZUbkVo067~Mu5jtJJd~gO~qRyQVjDwVqDKppFbukDj~G-J58cAlMy2PynKjUCDCYjgIeShsmI6f0KpIuAZ1CoGY1kMJi7azY7ghgZkoLqEVMQWY4mIv95ZQveyOmOFVjllhMaQKeVPtGr5KCFrGpxy9ZhEuNrmok451U8acS2fYc2O13rnLZr0rRLQHqftXRGTTN8016BAc8xHxm0yl9QS43GKkhPETZ3AGKo8LBnrGc7ACCe9bA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Ammonium removal from sludge digester effluent with the combined SHARON-Anammox system. Ammonium (♦) or nitrite (▪) load in the effluent of the SHARON reactor is used as the ammonium or nitrite load into the Anammox reactor. The ammonium or nitrite load in the effluent of the Anammox is represented by open diamonds and open squares, respectively. The pH value in the Anammox reactor was stable at 7.8 [38].
Nitrogen balances in the combined SHARON-Anammox system
| SHARON | Anammox | ||
| Influent | Effluent/Influent | Effluent | |
| NH+4 | 584 (100%) | 267(46%) | 29 (5%) |
| NO−2 | <1 | 227 (39%) | 1.4 |
| NO−3 | <1 | 64 (11%) | 83 (14%) |
| N2Oa | <1 | 4 | <1 |
| N2a | <1 | <1 | 476b (82%) |
| SHARON | Anammox | ||
| Influent | Effluent/Influent | Effluent | |
| NH+4 | 584 (100%) | 267(46%) | 29 (5%) |
| NO−2 | <1 | 227 (39%) | 1.4 |
| NO−3 | <1 | 64 (11%) | 83 (14%) |
| N2Oa | <1 | 4 | <1 |
| N2a | <1 | <1 | 476b (82%) |
Results are mg N l−1, percentages are given in parentheses.
aConcentration relative to the influent flow.
bDetermined as the difference between dissolved and gaseous nitrogen compounds.
Nitrogen balances in the combined SHARON-Anammox system
| SHARON | Anammox | ||
| Influent | Effluent/Influent | Effluent | |
| NH+4 | 584 (100%) | 267(46%) | 29 (5%) |
| NO−2 | <1 | 227 (39%) | 1.4 |
| NO−3 | <1 | 64 (11%) | 83 (14%) |
| N2Oa | <1 | 4 | <1 |
| N2a | <1 | <1 | 476b (82%) |
| SHARON | Anammox | ||
| Influent | Effluent/Influent | Effluent | |
| NH+4 | 584 (100%) | 267(46%) | 29 (5%) |
| NO−2 | <1 | 227 (39%) | 1.4 |
| NO−3 | <1 | 64 (11%) | 83 (14%) |
| N2Oa | <1 | 4 | <1 |
| N2a | <1 | <1 | 476b (82%) |
Results are mg N l−1, percentages are given in parentheses.
aConcentration relative to the influent flow.
bDetermined as the difference between dissolved and gaseous nitrogen compounds.
Acknowledgements
The research on anaerobic ammonium oxidation was financially supported by the Foundation for Applied Sciences (STW), the Foundation of Applied Water Research (STOWA), the Netherlands Foundation for Life Sciences (NWO-SLW), the Royal Netherlands Academy of Arts and Sciences (KNAW), Gist-Brocades, DSM, and Grontmij consultants. The contributions of various co-workers and students over the years are gratefully acknowledged.
References
Van de Graaf, A.A., Mulder, A., Slijkhuis, H., Robertson, L.A. and Kuenen, J.G. (1990) Anoxic ammonium oxidation. In: Proceedings of the 5th European Congress on Biotechnology (Christiansen, C., Munck, L. and Villadsen, J., Eds.), Vol. I, pp. 338–391. Munksgaard, Copenhagen.
Schoonen, K.T., de Bruyn, A., Strous, M., Kuenen, J.G. and Jetten, M.S.M. (1997) Anaerobic ammonium oxidation. In: International Symposium on Environmental Biotechnology (Verachert, H. and Verstraete, W., Eds.), Vol. II, pp 53–57. Technological Institute, Gent.
Strous, M., Heijnen, J.J., Kuenen, J.G. and Jetten, M.S.M. (1998) The sequencing batch reactor as a powerful tool to study very slowly growing micro-organisms. Appl. Microbiol. Biotechnol. (in press).
Koyama, T. (1965) Formal discussion of paper III-10. Discussion of paper by Richards. In: Advances in Water Pollution Research (Pearson, E.A., Ed.), Vol. 3, pp. 234–242. Pergamon Press, Tokyo.
Author notes
Netherlands Institute for Sea Research NIOZ, Den Burg, Texel, The Netherlands.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}